

Abstract
To evaluate the potential of using the heme containing lipocalin nitrophorin 1 as a template for protein engineering, we have replaced the native axial heme coordinating histidine residue with glycine, alanine and cysteine. We report here the characterization of the cysteine mutant H60C_NP1 by spectroscopic and crystallographic methods. The UV/Vis, resonance Raman, and magnetic circular dichroism spectra suggest weak thiolate coordination of the ferric heme in the H60C_NP1 mutant. Reduction to the ferrous state resulted in loss of cysteine coordination, while addition of exogenous imidazole ligands gave coordination changes that varied with the ligand. Depending on the substitution of the imidazole, three heme coordination states could be distinguished: 5-coordinate mono-imidazole, 6-coordinate bis-imidazole and 6-coordinate imidazole/thiolate. Ligand binding affinities were measured and found to be generally 2-3 orders of magnitude lower for the H60C mutant relative to NP1. Two crystal structures of the H60C_NP1 in complex with imidazole and histamine were solved to 1.7 Å and 1.96 Å resolution, respectively. Both structures show that the H60C mutation is well tolerated by the protein scaffold and suggest that heme-thiolate coordination in H60C_NP1 requires some movement of the heme within its binding cavity. This adjustment may be responsible for the ease with which the engineered heme-thiolate coordination can be displaced by exogenous ligands.
Keywords: Nitrophorin 1, heme protein, crystal structure, resonance Raman spectroscopy, magnetic circular dichroism, axial heme ligand
Introduction
Heme enzymes catalyze a broad range of chemical transformations, and the identity of the axial heme ligand is believed to influence the stability and reactivity of the critical intermediates involved in catalysis. Many efforts have been reported to alter the coordination and catalytic properties of heme enzymes that naturally contain histidine (1-6) or cysteine heme coordination (7-13). In addition, efforts to introduce catalytic behavior into oxygen binding proteins have also been published (14-17). A central goal in these efforts has been an understanding of how the oxidized heme intermediates are stabilized by the various heme environments of these enzymes. The histidine coordination of peroxidases requires additional proximal and distal influences to promote formation and stabilization of the oxidized Compound I intermediates, while cysteine coordination of P450s provides electron donation to assist formation of such intermediates, but may be insufficient for their stabilization (18-21). The ferryl porphyrin cation radical of Compound I (Fe(IV)=O, P+.) is thought to be a common intermediate in heme mediated oxidation and oxygenation reactions. P450 enzymes are proposed to access this intermediate by reduction of the ferrous-dioxygen complex, while peroxidases can produce the Compound I state through reaction of the ferric heme with hydrogen peroxide, organic peroxides or peracids (22). Thiolate coordination is believed to be important for efficient O-O bond cleavage, and may also contribute to the basicity of the ferryl oxygen atom during oxygen transfer (23,24). Thus, several groups have pursued the engineered introduction of thiolate ligation into heme proteins in an effort to modify its catalytic properties (25-27). Thus, cysteine coordination has been introduced into Mb (25,28), cytochrome b5 (29), heme oxygenase (26) and CcP (27). In many of these cases, cysteine coordination is weakened or lost upon reduction, while protonation of the cysteinate ligand to give a neutral iron-coordinated thiol has been proposed in others (30). However, most binding sites for heme are rich in α-helical structure, so past efforts to engineer axial ligand replacements in heme proteins have been subject to the unique abilities of α-helical architectures to adapt to the structural challenges imposed by the introduced ligands. In addition, it is possible that the protein scaffolds of natural heme enzymes possess an intrinsic bias to support a particular catalytic function and it may be difficult to overcome this solely by re-engineering the immediate heme environment. Thus, it would be of interest to introduce cysteine coordination into a heme protein with a fold that is distinct from that of a globin to better understand the linkage between protein architecture and heme coordination properties.
Nitrophorins are a group of heme containing lipocalins, derived from a blood sucking insect (Rhodnius prolixus) (31-33). They are small (20 kDa), monomeric, extra-cellular transport proteins for nitric oxide and histamine. They have been proposed to suppress wound closure and blood clotting while the insect extracts blood from its prey. Nitrophorins are structurally defined by a β-barrel formed from a single, highly twisted, eight stranded β-sheet, possessing a typical lipocalin fold (34,35). The heme binds within one end of the barrel and is coordinated to a single histidine, leaving the distal face of the heme free for exogenous ligands (i.e. NO and histamine). Notably, the β-barrel architecture (36) differs significantly from most heme protein scaffolds used previously for ligand replacement. Thus, it is of interest to examine the ability of these proteins to tolerate axial heme ligand replacement. Such studies may allow one to address structural and mechanistic questions of heme enzyme catalysis from a novel perspective.
We report here studies of the nitrophorin I (NP1) mutant in which the axial histidine ligand is replaced by cysteine (H60C_NP1). The state of heme coordination and ligand binding in solution is described, and the crystal structures of H60C_NP1 in complex with histamine and imidazole are presented. The results illustrate similarities and differences with previous efforts to engineer thiolate coordination in heme proteins.
Material and methods
Mutation, expression, purification, refolding and reconstitution of nitrophorin
A plasmid based on the pET17b vector containing the NP1 gene was obtained as a kind gift from A. Walker (University of Arizona). Site directed mutagenesis was used to introduce the H60C mutation using the PCR based QuickChange methodology (Qiagen) and confirmed by DNA sequencing. All chemicals were from Sigma-Aldrich, biochemical enzymes from New England BioLabs, growth media from Difco or Fisher Scientific, materials for gel electrophoresis were from Invitrogen. Expression and purification of NP1 and the H60C_NP1 mutant followed the methodology described by Anderson et al. (37) with the following modifications: Inclusion bodies were washed three times with 150 mM NaCl, 50 mM Tris, 2 M urea, 1% Triton X100, pH 7.4, then dissolved in 150 mM NaCl, 50 mM Tris, 6M guanidinium chloride, pH 8.0, affording enrichment of NP1 to more than 80%. Refolding of guanidinium solubilized protein was found to be most efficient by rapid drop-wise dilution into a 25-fold volume of high-salt refolding buffer (0.4 M arginine, 0.2 M Tris, 0.1 M ammonium sulfate, 2 mM EDTA, pH 8.4, 1mM oxidized glutathione, 0.2 mM reduced glutathione). Refolding was usually allowed to proceed overnight and no significant precipitation occurred upon concentrating the refolded sample to about 1/5 of the starting volume over an ultrafiltration membrane. However, subsequent lowering of the ionic strength and pH by dialysis against 50 mM NaOAc pH 5.2 led to precipitation of mis-folded nitrophorin, and co-precipitation of almost all pertinacious contaminants. After clarification by centrifugation, the remaining soluble protein was further purified by ion-exchange (SP-Sepharose) and size exclusion chromatography (S-100 Sephacryl). The protein could be purified, concentrated and stored as apo-protein. Incorporation of the heme group was usually done prior to ion-exchange purification on SP-sepharose. An Akta FPLC chromatography system from Amersham-Pharmacia was used for protein purification. MALDI-MS analysis of apoand heme reconstituted H60C_NP1 demonstrated that no modification, especially oxidation of Cys-60, had occurred. Protein samples were concentrated to 1-2 mM (20-40 mg/ml) and stored at -70°C. Yields of purified protein were typically 50-100 mg per liter of bacterial culture.
UV/Vis spectroscopy
UV/Vis absorption spectra were recorded on a HP8453 UV-visible spectrophotometer. Heme protein concentration was determined measuring absorbance at the Soret peak and a molar extinction coefficient of 82 mM-1cm-1 at 389 nm for H60C_NP and 159 mM-1cm-1 for NP1 at 404 nm was determined by the reduced pyridine hemochrome method (38). Ligand titrations were done in PBS buffer (50mM Na-phosphate, 100 mM NaCl, pH 7.4) in 3 ml sample volume with incremental addition of ligand in PBS buffer. The total added volume did not exceed 3 % and the spectral data were corrected for dilution. Samples were allowed to equilibrate for at least 3 min following ligand addition prior to measuring absorbance spectra. Equilibrium binding constants were calculated using Specfit (39), a global fitting program.
Resonance Raman
NP1 and H60C_NP1 samples (~120 μL, ~50 μM in heme, pH 7) were prepared in a septum-sealed, cylindrical quartz cell. Samples were reduced to the deoxy form by first flushing the sample with argon and then injecting a molar excess of buffered sodium dithionite solution. Deoxy samples were flushed briefly with carbon monoxide gas to form the ferrous-CO complex. 12C16O and 13C18O gases were purchased from Airgas Specialty Gases and Icon Isotopes (Mt Marion, NY), respectively. The rotating sample cell was irradiated with 4 mW of 413.1 nm light using a mixed krypton/argon ion laser (Spectra Physics, Beamlok 2060). The spectral acquisition time was 5 min. The scattered light was collected at right angles to the incident beam and focused onto the entrance slit (125 μm) of a 0.8 m spectrograph, where it was dispersed by a 600 groove/mm grating and detected by a liquid-N2-cooled CCD camera (Horiba-JY). Spectral calibration was performed against the lines of mercury and indene.
Magnetic circular dichroism
MCD spectra were measured in a 0.2 cm cuvette with a Jasco J600A spectropolarimeter equipped with a Jasco MCD-1B electromagnet operated at a magnetic field strength of 1.41 T. Data acquisition and manipulation were done as reported previously (40). Sample integrity was checked by recording the absorption spectra of samples before and after each MCD/CD measurement. All spectra were recorded at 4°C.
Crystallization, data collection and structure determination
Crystals were grown by vapor diffusion from hanging drops, obtained by mixing equal volumes of protein solution (1 mM nitrophorin, 50 mM potassium phosphate, pH 7.4) and reservoir solution (0.1 M Na-cacodylate pH 5.3, 2.6-2.9 M ammonium phosphate and 5 mM imidazole or histamine). Single crystals usually formed within two days. Cryo-protection of crystals for freezing was achieved by adding glycerol to the mother liquor reservoir solution to a final concentration of 16% and briefly soaking the crystal in mother liquor. Diffraction data for the H60C_NP1 imidazole complex were collected at 100K on a MAR research image plate at SSRL beam line 7.1 at a wavelength of 1.08 Å. Data were processed using Denzo (HKL Research Inc. Charlottesville, VA, USA) and Scala (41). Data for the histamine complex were obtained at 100K using a Rigaku X-ray generator equipped with a rotating copper anode, graphite monochromator, confocal mirrors and a Raxis IV image plate. Data were reduced with CrystalClear and d*trek (42). Both structures were solved by molecular replacement using wild type nitrophorin 1 (pdb code 1NP1) as starting model and refined using the CNS (43) and CCP4 (44) software packages. Superposition of protein chains was performed using the McLachlan algorithm (45) as implemented in the program ProFit (Martin, A.C.R., www.bioinf.org.uk/software/profit/). Prior to fitting, amino acid side chains (except glycine) were truncated to an “all alanine” model. Secondary structure analysis was done using the program Promotif v3.0 (46).
Results
UV/Vis spectroscopic properties
UV/Vis absorption spectra suggest that H60C_NP1 contains either a thiolate or bis-aquo coordinated heme. In the absence of exogenous ligands, wild-type NP1 exists in a 6-coordinate high-spin state, with His-60 and water as the axial ligands (37). However, as seen in Figure 1, replacement of the axial histidine ligand with cysteine results in a broadened and blue-shifted Soret band with a peak at 389 nm, consistent with a high-spin 5-coordinate thiolate complex. Indeed, the spectroscopic properties of H60C_NP1 are similar to those of substrate bound P450cam and other heme proteins with a proximal cysteine ligand (27,48,49). However, the H175G mutant of cytochrome c peroxidase (H175G_CCP) also shows a similar Soret band, and has been shown to contain a bis-aquo coordinated heme (50,51). Thus, the UV/Vis absorbance spectra alone do not exclude the possibility of a bis-aquo complex in which a water, and not Cys-60, is coordinated to the proximal heme face.
Figure 1.
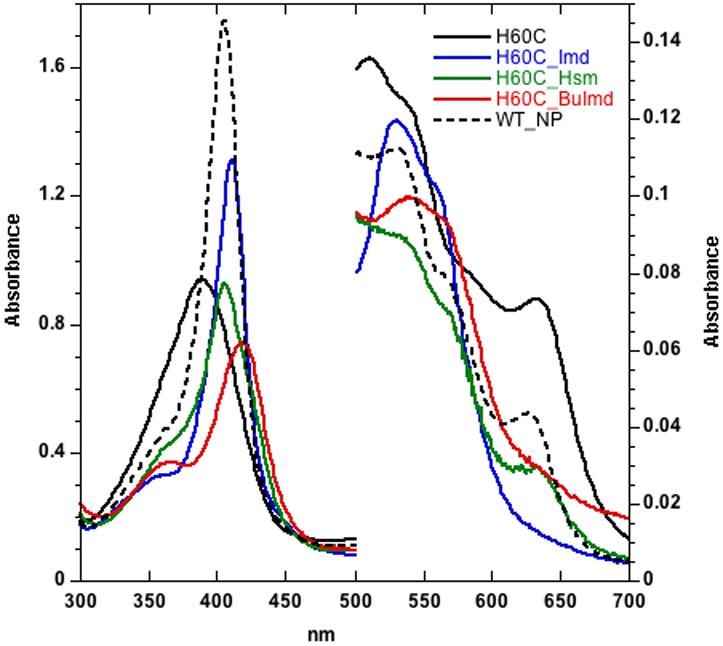
UV/VIS spectra of H60C_NP1 (11 mM, in phosphate buffered saline, pH 7.4) in complex with selected imidazole ligands. NP1 (11 mM, phosphate buffered saline, pH 7.4) is shown for comparison. The Soret maxima are: H60C: 389 nm; H60C/Imd: 411 nm; H60C/Hsm: 405 nm; H60C/nBuIm: 424 nm; NP1: 405 nm.
H60C_NP1 binds to exogenous imidazole ligands more weakly than does NP1, giving a variety of complexes that depend on the ligand. In agreement with previous studies (47), the data of Table 1 shows that wild-type NP1 binds histamine (Kd = 1.7×10-8 M) with a 10 fold higher affinity than imidazole (Kd = 2.5×10-7 M) which has been rationalized based on the X-ray crystal structure showing three hydrogen bonds between the primary amino group of histamine and the protein (36). C- or N-alkyl substitutions of the imidazole ring gave low-spin complexes with similar spectra (data not shown) and were observed to bind to NP1 with affinities intermediate between those of imidazole and histamine (Table 1). The affinity of N-(n-butyl)-imidazole (BuImd) for NP1 (Kd = 5.2×10-8 M) is almost as high as that for histamine, suggesting that the observed hydrogen bonding with histamine is not the only source of the increased affinity, and that the hydrocarbon tail also contributes significantly to the binding affinity. Replacement of the native histidine ligand by cysteine results in significantly reduced binding affinities for all measured imidazole derivatives. The effect is most pronounced for histamine and 4-methylimidazole (Table 1) which show an increase in dissociation constant by more than three orders of magnitude relative to NP1. Nevertheless, UV/Vis titration of H60C_NP1 with imidazole results in difference spectra with sharp isosbestic points (Supporting Information S1). Imidazole and 4-methylimidazole give low-spin complexes with a Soret peak at 411 nm, suggesting a bis-imidazole coordinated heme species. The H60C_NP1/histamine complex shows a predominantly high-spin species with a Soret peak at 405 nm, which is typical of 5 or 6-coordinate imidazole or imidazole/water bound heme and inconsistent with thiolate coordination. These properties suggest that if H60C_NP1 contains a thiolate coordinated heme, this interaction is lost upon binding these ligands. Finally, titration of N-(n-butyl)-imidazole (BuImd) into H60C_NP1 gives two successive complexes. The first species shows a Soret maximum at 424 nm (Figure 1) and is converted to a separate species at 412 nm at high ligand concentrations. The observed Soret band at 424 nm is consistent with a mixed imidazole/thiolate complex as seen in other systems including P450cam, H174C/D235L CcP, and hydroperoxide lyase (27,48,49), while the 412 nm form may result from bis-imidazole coordination.
Table 1.
Dissociation constants at 25°C for ligand complexes of NP1 and H60C_NP
| NP1 | H60C_NP1 | |
|---|---|---|
| Histamine | 17 ± 2 × 10-9 M | 40 ± 10 × 10-6 M |
| Imidazole | 255 ± 5 × 10-9 M | 120 ± 10 × 10-6 M |
| N-(n-Butyl)imidazole | 52 ± 3 × 10-9 M | 7 ± 1 × 10-6 M |
| 4-Methylimidazole | 182 ± 4 ×10-9 M | 560 ± 50 × 10-6 M |
Magnetic circular dichroism
To obtain a more definitive assignment of the axial coordination state of H60C_NP1, magnetic circular dichroism (MCD) spectra for H60C_NP1 were examined. MCD has been used extensively as a fingerprinting technique to identify heme axial coordination states, and has been applied specifically to the identification of thiolate coordination (50-52). As shown in Figure 2, the MCD spectrum of H60C_NP1 is strikingly similar to that of P450cam, particularly in the region of the Soret band where both proteins show a trough at 395 nm with a strong negative ellipticity. In contrast, H175G_CCP, which has similar UV/Vis properties, but has been shown to be coordinated by two waters (50,51), exhibits a dramatically distinct MCD in the Soret region. Also shown in Figure 2 are the MCD spectra of H60C_NP1 in the presence of BuImd and the mixed imidazole/thiolate form of P450cam. The past success of MCD spectroscopy to assign heme coordination in a variety of proteins (52), combined with the similarity of the diagnostic features of the MCD spectra of H60C_NP1 with those of P450cam, provide strong evidence that H60C_NP1 is coordinated by the thiolate of Cys-60 in the ligand-free and in the 424 nm form of the BuImd complex.
Figure 2.

MCD spectra of ligand free H60C_NP1 compared with the 5-coordinate thiolate complex of camphor bound P450cam, and bis-aquo coordinated H175G_CCP (upper panel) suggests that H60C_NP1 is coordinated by Cys-60. In the presence of nBuIm (one equivalent), the MCD of H60_NP1 is converted to a low spin complex (lower panel) that is similar to the mixed imidazole/thiolate complex of P450cam in the presence of imidazole. Samples were prepared in phosphate buffer, pH = 7.4 and 1 mM camphor and MCD spectra run at 4°C.
Resonance Raman spectroscopy
Resonance Raman (RR) spectroscopy has been used extensively for characterization of the oxidation-, spin-, and coordination-state of heme proteins (53-59) including NP1 (60). The RR spectrum of ferric NP1 (Figure 3A) is consistent with previous observations showing the heme to be predominantly 6-coordinate high-spin (ν3 and ν2 at 1481 and 1559 cm-1) with a small 6-coordinate low-spin contribution (ν3 and ν2 at 1509 and 1578 cm-1). Addition of imidazole (Imd) or N-(n-butyl)-imidazole (BuImd) results in formation of a 6-coordinate low-spin complex, as indicated by the appearance of the ν3 and ν2 bands at 1502 and 1576-1578 cm-1 respectively. In sharp contrast, H60C_NP1 has a strong ν3 band at 1488 cm-1, characteristic of a 5-coordinate, high-spin state (Figure 3B) (56,58), and an unusually weak ν4 band at 1371 cm-1. This unusual ν3/ν4 relative intensity is similar to that for the H25A (proximal His ligand) mutant of human Heme Oxygenase 1 (HO-1) (61), which was suggested to be the result of an unusual proximal ligation of the heme iron. Addition of a 1:1 mole ratio of Imd to H60C_NP1 restores the intensity of the ν4 band, and causes a transition to 6-coordinate low-spin (ν3 at 1504 cm-1), while a similar conversion is only seen after addition of a 5 fold molar excess of BuImd (ν3 at 1500 cm-1) (Figure 3B).
Figure 3.
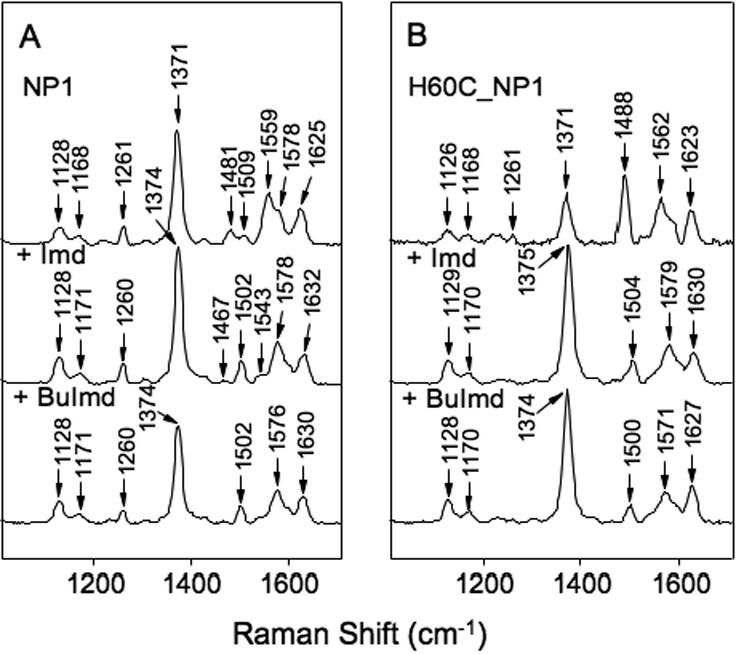
Resonance Raman spectra of ferric heme-iron complexes in the high frequency region. Panel A, NP1. Panel B, H60C_NP1. The top spectrum in each panel was measured without added imidazole ligand whereas +Imd and +BuImd indicate the addition of 5-fold equivalents of imidazole and N-(n-butyl)-imidazole, respectively.
Ferrous deoxy complexes
The high frequency RR spectrum of ferrous deoxy NP1 (ν4 at 1353cm-1, ν3 at 1467cm-1) is typical for a 5-coordinate, high-spin His coordinated heme (Figure 4A) (59) (53,56,58). Addition of Imd or BuImd causes a conversion to a form (ν4 at 1358cm-1, ν3 at 1490cm-1) indicative of 6-coordinate low-spin, as is the appearance of ν2 bands at 1580 (+Imd) and 1577 cm-1 (+BuImd) (Figure 4A) (53,56,58). The RR spectrum of ferrous deoxy H60C_NP1 has a significantly broadened ν4 band at 1357cm-1 (Figure 4B) compared with NP1, and ν3 at 1468 cm-1 indicating a 5-coordinate high-spin heme. However, the axial ligand in this case is unlikely to be a Cys thiolate because the ν4 band for thiolate-ligated ferrous heme typically appears in the 1340-1348 cm-1 range (59,62). Furthermore, the small ν3 peak appearing at 1501 cm-1 (Figure 4B) combined with the position and broadening of the ν4 band suggests a contribution from a 4-coordinate, intermediate-spin state (53,61). If Cys-60 is very weakly coordinated, it could conceivably produce the unusual spectral features noted above for the deoxy and ferric forms of H60C_NP1, including the anomalously high ν4 band position in the deoxy RR spectrum. Taken together, the RR features of ferrous deoxy H60C_NP are consistent with a 5-coordinate heme with either a weakly bound Cys or a water axial ligand. Addition of Imd or BuImd to deoxy H60C_NP1 causes the disappearance of the 1501 cm-1 peak associated with the 4-coordinate intermediate-spin component, leaving only the 5-coordinate high-spin ν3 marker band at 1467-1469 cm-1 (Figure 4B). It is unclear why a 6-coordinate bis-imidazole complex is not seen under these conditions.
Figure 4.
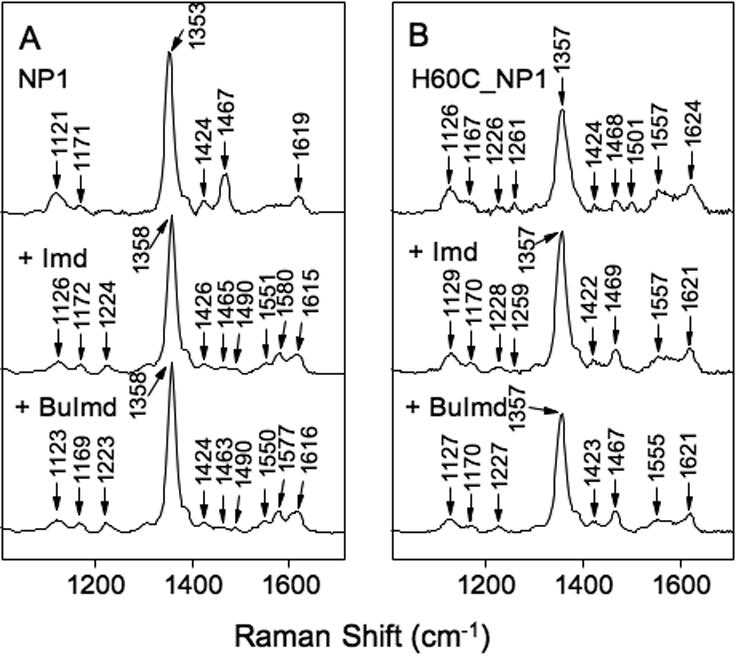
Resonance Raman spectra of deoxy heme-iron complexes in the high frequency region. Panel A, NP1. Panel B, H60C_NP1. The top spectrum in each panel was measured without added imidazole ligand whereas +Imd and +BuImd indicate the addition of 5-fold equivalents of imidazole and N-(n-butyl)-imidazole, respectively.
Ferrous-CO Complexes
Assignment of the Fe-CO and C-O stretching modes of the Fe2+-CO complexes is useful because they are influenced by the type of proximal ligand and the polarity of the distal pocket environment (63,64). The RR spectrum of the Fe2+-CO complex of NP1 in the high frequency region displays a characteristic 6-coordinate low-spin pattern, with peaks at 1371 cm-1 (ν4), 1495 cm-1 (ν3) and 1578 cm-1 (ν2) (Figure 5B). Addition of Imd or BuImd results in only small changes, indicating that these ligands do not directly displace His-60 or CO (Supporting Information S2). The Fe2+-CO complex of H60C-NP1 displays similar 6-coordinate features with ν3 = 1497 cm-1 and ν2 = 1576 cm-1 (Figure 5B). The νFe-CO band for NP1 and H60C_NP1 was seen at 493 cm-1 and 529 cm-1 respectively (Figure 5A) (56,59,63,65), while the νC-O band for NP1 and H60C_NP were observed at 1965 and 1962 cm-1. Confirmation of these assignments is provided by the observed shifts to lower frequencies in the presence of 13C18O (Figure 6A). Additionally, νC-O for NP1 and H60C_NP1 was seen at ~1965 cm-1 and 1962 cm-1 in the 12C16O - 13C18O difference spectrum (Figure 6B).
Figure 5.
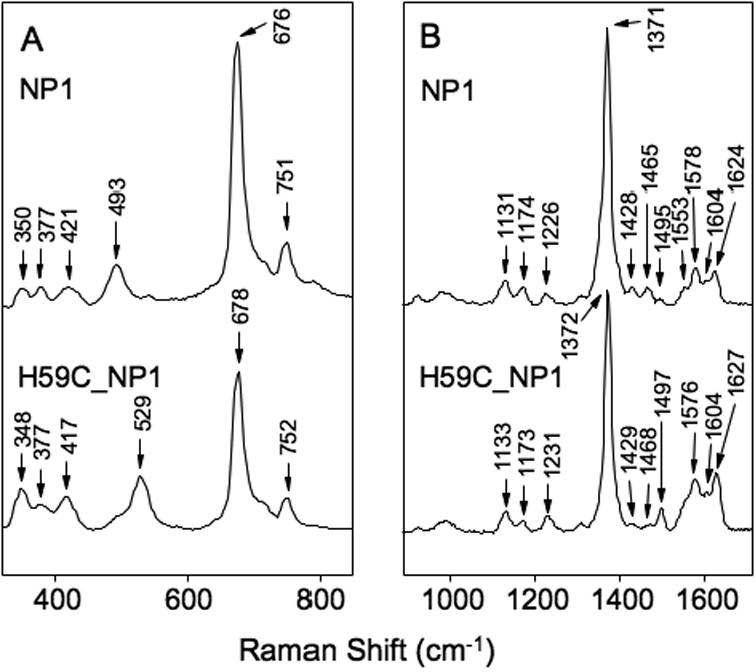
Resonance Raman spectra of NP1-CO and H60C_NP1-CO complexes. Panel A, low frequency region. Panel B, high frequency region. The spectra were without added imidazole ligand.
Figure 6.
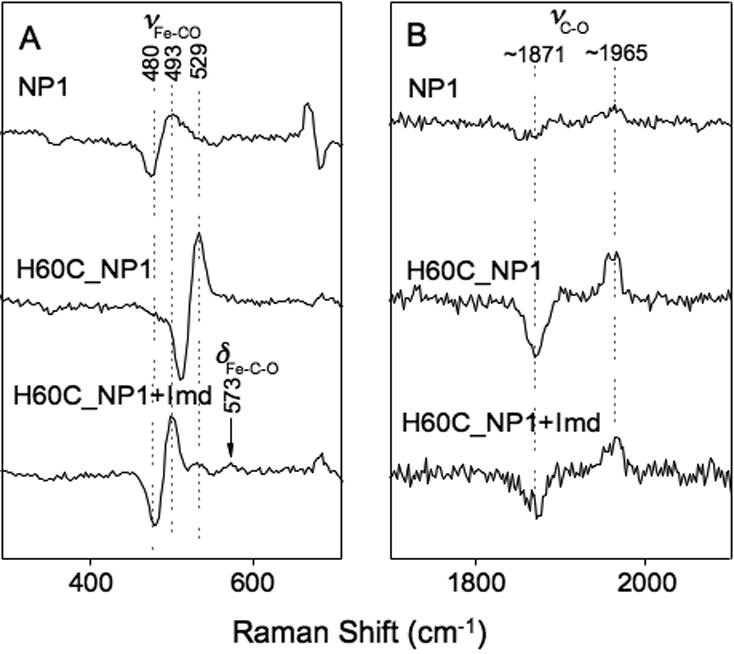
Fe2+·CO - Fe2+·13C18O resonance Raman difference spectra for NP1 and H60C_NP1. Panel A, low frequency region showing the Fe-CO stretching vibrational band. Panel B, high frequency region showing the internal C-O stretching vibrational band. H60C_NP1 +Imd represents the difference spectrum measured with 5-fold molar excess of added imidazole.
Plots of νFe-CO versus νC-O for a wide variety of CO-heme complexes show distinctive linear correlations where the proximal ligand identity and polarity of the distal environment can be inferred (56,63,65-67). The observed 493 and 1965 cm-1 frequencies for the CO complex of WT-NP1 are typical for a heme protein with a neutral histidine proximal ligand and a hydrophobic distal environment (63), while the 529/1962 cm-1 pairing for H60C_NP1 places it unambiguously in the region corresponding to 5-coordinate CO-heme complexes (Figure 7) (63,65,66). This suggests that weak axial coordination by Cys-60 is lost upon CO binding, a result that is consistent with other examples of engineered cysteine coordination (14,27).
Figure 7.
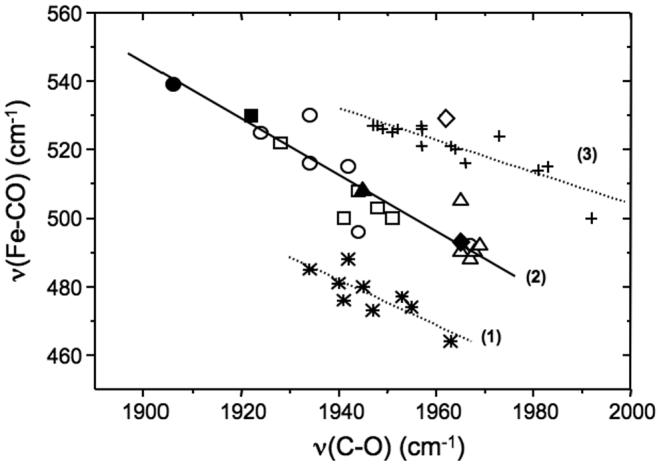
Correlation of Fe-CO and C-O stretching frequencies for different heme proteins and porphyrin derivatives. Lines (1) and (2) are for complexes in which the proximal ligand is thiolate and histidine, respectively. Line (3) is for five-coordinate CO adducts. The open circles, open squares and open triangles correspond to data obtained for mutant and pH-variant forms of horse radish peroxidase, cytochrome c peroxidase and myoglobin, respectively. The corresponding filled symbols represent data obtained from the wild-type proteins. The filled and open diamonds correspond to wild-type NP1 and the mutant H59C_NP1 data, respectively, measured in this work. Figure adapted from refs. (63,65,66).
Crystal structures of H60C_NP1 ligand complexes
Crystals of H60C_NP1 in complex with histamine and imidazole were grown using high concentrations of ammonium phosphate as the primary precipitant. Under such conditions, nitrophorin crystallizes in space group P21 with two protein molecules in the asymmetric unit. Attempts to crystallize H60C_NP1 in the absence of exogenously added imidazole ligands have not yet been successful. This, on its own, may suggest conformational differences between ligand free H60C_NP1 and H60C_NP1 in complex with coordinating ligands. Data collection and refinement statistics for both H60C_NP1 structures are summarized in Table 2. Refinement of the structures was done without applying non-crystallographic symmetry (NCS) averaging. The quality of the refinement compares favorably with two previously published structures of NP1 and the NP1/histamine complex (36). A stereo view of the heme binding cavity and representative electron density for the 1.7 Å structure of the imidazole complex is shown in Figure 8.
Table 2.
Data collection and refinement statistics
| Complex | H60C_NP1/imidazole | H60C_NP1/histamine |
|---|---|---|
| PDB code | 1U17 | 1 U18 |
| Data Collection | ||
| Resolution (Å)a | 30-1.7 (1.70-1.81) | 27-1.96 (2.06-1.96) |
| Space group | P1 21 1 | P1 21 1 |
| Cell a,b,c (Å), β (°) | 39.01, 73.99, 65.22; 99.16 | 38.820, 73.85, 65.38; |
| Unique reflections | 40,082 | 26,735 |
| Completenessa (%) | 99.5 (90.7) | 92.5 (70.4) |
| Multiplicity | 3.9 (3.7) | 2.1 (1.4) |
| IΣI | 8.1 (1.0) | 11.2 (3.4) |
| Rsymb | 0.074 (0.37) | 0.038 (0.125) |
| Refinement | ||
| Rcrysc | 0.182 | 0.195 |
| Rfreed (5%) | 0.237 | 0.226 |
| R.m.s.d. bonds (Å) | 0.021 | 0.032 |
| R.m.s.d. angles (Å) | 1.96 | 2.00 |
| ESUe (Å) | 0.094 | 0.130 |
| Atoms: protein (chain A/B) | 1447 / 1447 | 1439 / 1439 |
| Atoms: heme, ligand, (chain | 43, 5, 5 / 43, 5, 5 | 43, 8 / 43, 8 |
| No. waters | 310 | 210 |
| <B> protein (Å2) | 23 / 25 | 35 / 41 |
| <B> heme (Å2) | 22 /25 | 33 / 41 |
| <B> ligand (Å2) | 18 /19 | 32 / 37 |
| <B> water (Å2) | 31 | 38 |
Values in parentheses are for the highest resolution shell
Rsym = (Σh|Ih-<I>|) / (Σh|Ih), where <I> is the mean intensity of all symmetry related reflections Ih.
Rcrys = (Σ|Fobs-Fcalc|) / (ΣFobs).
Rfree as for Rcrys using a random subset of the data not included in the refinement
ESU, Estimated overall coordinate error based on maximum likelihood
Figure 8.

Stereoview showing residues surrounding the heme group in the H60C_NP1/Imd complex. The 2Fo-Fc electron density map is contoured at 2s and representative for the entire structure. Residue Cys-60 points away from the heme towards a hydrophobic pocket formed in part by Tyr-41 and Phe-69.
The structure of the H60C_NP1/histamine complex has been refined to 1.96 Å for residues 2-185. Figure 9 shows an overlay near the heme center for NP1/Hsm and H60C_NP1/Hsm. The side chain of Cys-60, as well as the heme and the histamine ligand are clearly defined in electron density maps. The distance between the iron and the Nε of histamine is 2.29 Å, approximately 0.2 Å longer than the distance observed in the bisimidazole heme complex discussed below. The distal histamine ligand binds to H60C_NP1 at essentially the identical position as observed in the NP1/Hsm complex (36). Cys-60 is not coordinated to the heme in H60C_NP/Hsm but is instead directed towards a hydrophobic pocket formed by reorienting the side chains of Tyr-41 and Phe-69 (Figures 8 and 9). Proximal heme coordination is provided by a solvent molecule, either a water molecule or ammonia acquired from the crystallization solution. However, this solvent appears to be weakly coordinated, with distances of 2.3 Å and 2.5 Å for the two molecules in the asymmetric unit. The distance between the Cys-60 sulfur atom and the iron differs slightly between the two molecules in the asymmetric unit (chain A 5.45 Å and chain B 5.11 Å). Without movement of the heme group or protein backbone, this distance could only be reduced to 4.1 Å by rotation about the Cα-Cβ bond, which is larger than that needed for heme-thiolate coordination (~2.3 Å for P450cam). This implies that movements of the heme and/or protein backbone must accompany coordination of Cys-60 in solution.
Figure 9.

Stereoview of the H60C_NP1/Hsm complex (dark) overlaid with the structure of the NP1/Hsm complex (light colored). Structures were superimposed using residues Tyr-59 to Tyr-61. The movement of residue Tyr-41 is the most significant structural change.
The structure of the H60C_NP1/imidazole complex at 1.7 Å is compared with NP1/histamine in Figure 10. The position of the imidazole rings are shifted and rotated compared to the positions occupied by the corresponding histamine and histidine ligands. The orientation of the distal imidazole ligand is similar to the orientation observed for the histamine ligand, potentially as a result of interactions with the sidechains of Leu-124 and Leu-134 in the distal cavity. However, the proximal imidazole ligand is rotated by approximately 90° compared to the proximal histidine of NP1 such that the proximal and distal imidazole rings of H60C_NP1/imidazole are oriented at roughly 90° to each other. In addition, the smaller imidazole rings exhibit small differences in tilt with respect to the plane of the heme compared with histamine and histidine coordinated forms. On the other hand, the iron-imidazole distances for the two molecules in the asymmetric unit (1.91 Å - 2.12 Å) are very similar to those seen in the NP1/histamine complex (1.96 Å - 2.10 Å).
Figure 10.

Stereoview of the H60C_NP1/Imd complex (dark) overlaid with the structure of the NP1/Hsm complex (light colored). Structures were superimposed using residues Tyr-59 to Tyr-61. Significant differences can be found in the movement of residue Tyr-41, the orientation of the proximal imidazole ligands and in the distal side the flipping of residue Gly-132.
The most significant difference in protein structure between H60C_NP1/histamine and H60C_NP1/imidazole appears to be related to hydrogen bonding interactions between the aminoethyl side chain of histamine and the protein. These involve residues Asp-31 and Glu-33 near the flexible A-B loop, as well as Leu-131 in β-strand H, near the G-H β-turn (residues 127-129). As a consequence of the loss of these interactions in the imidazole complex, the surface loop between Leu-124 and Leu-134 undergoes a conformational change as shown in Figure 11. Residues Lys-126 to Asp-133 are displaced by an average RMSD of 1.9 Å between the imidazole and histamine complexes. Asp-133 exhibits a 70° difference in ϕ, allowing Leu-131 and Gly-132 to reorient toward the protein surface. These changes are associated with a 170° ϕ rotation at Gly-132 and a 120° ψ rotation of Leu-131. In addition, the carbonyl of Gly-132, which forms a hydrogen bond via a bridging water molecule to the Nδ of histamine, instead makes a hydrogen bond with the amide of Leu-124 in the imidazole complex, and contributes to the β-sheet hydrogen bonding network between strands G and H.
Figure 11.

Stereoview of the GH loop region. Structures H60C_NP1/Hsm (dark) and H60C_NP1/Imd (light colored) were superimposed. The Hsm ligand is visible on the left. Significant changes occur between residue Cys-123 and Leu134, see text for details.
During refinement, small conformational differences were seen between the two protein chains within the asymmetric unit. To analyze these differences, “all-alanine” models of both chains were superimposed and the mean deviation for each residue was calculated. Truncating amino acid side chains eliminates deviations resulting from side chain orientation, allowing the analysis to report on crystal packing induced changes in the protein backbone. A plot of the root mean square deviations (rmsd) obtained for the superposition of the two H60C_NP1 structures showed the most significant deviations near the A-B loop between residues 35-39 (rmsd >2 Å) and the region of the B-C loop between residues 49-53 (rmsd 1.4 Å) (Supporting Information S3). An additional small difference between chains A and B is seen only for the imidazole complex between residues 166-170, which in chain A folds into a single type I β-turn (residue 167-170), while chain B forms an additional type IV β-turn (residues 166-169). It is of interest that similar changes in the A-B loop have been observed in NP4 in response to ligand binding (68,69).
Discussion
Replacement of the axial histidine heme ligand of NP1 by a cysteine has afforded a number of insights into the ability of this atypical heme binding protein architecture to accommodate changes at the active site. The introduction of a fifth cysteine residue into the NP1 protein sequence does not interfere with recombinant expression in E.coli, in vitro refolding, heme reconstitution or purification of the protein. The two native disulfide bonds are formed properly and no oxidation of the free Cys-60 residue is observed. UV/Vis, MCD, and RR spectroscopy provide strong evidence that in the absence of exogenous ligands, H60C_NP1 contains a thiolate coordinated heme, which is lost upon reduction or addition of exogenous ligands. For small imidazole ligands, bisimidazole complexes are formed with the displacement of the proximal cysteine ligand. Variation of the exogenous ligand allowed formation of two additional complexes which were assigned to mono-imidazole (histamine) or mixed imidazole/thiolate (N-(n-butyl)imidazole) forms. These substituted imidazoles are presumably too large to readily occupy the proximal heme cavity, preventing bis-imidazole coordination.
While crystals of the thiolate coordinated forms of H60C_NP have not been obtained to date, the structures of the imidazole and histamine complexes are consistent with the spectroscopic properties of the protein in solution. The differences in the propensity of thiolate and imidazole coordinated forms to crystallize suggests a conformational change in the structure associated with Cys-60 coordination. Indeed, as suggested by the structures of the imidazole and histamine complexes, the Cys-60 side chain cannot be moved to a coordinating distance without additional adjustment of the protein backbone or heme position. Model building suggests that the most likely scenario involves the movement of the heme slightly towards the barrel opening and towards β-strand C combined with reorientation of the cysteine side chain toward the iron. To accommodate this shift, several side chains, including Tyr-41, adjacent to the flexible AB surface loop, would have to adjust their positions. Alternatively, movements of β-strand C, containing Cys-60, toward the heme may allow thiolate coordination, but the potential disruption of the extended β-sheet will probably prevent large changes of this nature. The proposed shift in heme position would interrupt a number of favorable hydrophobic interactions between the heme and cavity floor, and this penalty may be one of the factors resulting in weak coordination by Cys-60. Histidine to cysteine mutants in a number of predominantly α-helical heme proteins have also allowed introduction of engineered thiolate coordination (25-27). While these studies initially suggested that the thiolate ligand was lost on reduction or ligand binding, recent results have shown that for many of these cases, the cysteine remains bound as a neutral protonated thiol ligand (30). In contrast, the H60C_NP1 mutant clearly undergoes dissociation of the bound thiolate upon ligand binding. This difference may be due in part to the unusual β-barrel architecture of the nitrophorins such that the protein environment immediately surrounding the heme is less able to adapt to the shorter cysteine ligand. The introduction of a weak thiolate coordination into a β-barrel heme pocket may provide a useful model for comparing the structural and ligand induced responses in other thiolate heme proteins.
Superposition of the two imidazole coordinated H60C_NP1 structures with NP1 reveals only minor changes in the position of the heme within the relatively open β-barrel cavity of NP1. In addition there is no significant electron density near the heme methyl groups that would suggest heme orientational disorder, as seen previously for NP1 and NP4 by NMR (70) and crystallography (71). The fact that little shift or disorder is observed in H60C_NP1 indicates that the heme position within NP1 has been co-optimized for axial coordination geometry, hydrogen bonding to the heme propionates, and shape complementarity. Indeed, of the eleven residues which are observed to be in Van der Waals contact with the heme of NP1, six of these interactions (Tyr-29, Tyr-41, Leu-58, Phe-87, Tyr-106 and Leu-124) are seen in both monomers of all three structures discussed here. On the other hand, differences are observed in the hydrogen bonding networks with heme propionates. While hydrogen bonding to Asp-71 Oδ is universally observed, Lys-126 Nζ interacts directly with both heme propionates only in chains A of the two histamine complexes. Additionally, Asp-35 Oδ hydrogen bonds to a heme propionate only in NP1/Hsm. Thus, despite the rather open heme binding cavity of nitrophorins, it appears that in the absence of protein-linked axial coordination, the predominant native heme position remains as the lowest energy conformation due to hydrophobic Van der Waals interactions with the proximal and peripheral heme pocket. It would be of interest to examine the solution equilibrium thermodynamics of heme orientational disorder in H60C_NP1 as a probe of how axial coordination contributes to the disorder that has been seen in these systems.
Finally, significant differences in protein loop conformation are seen in the H60C_NP1 mutants between the two molecules of the asymmetric unit. In particular, the flexibility of the AB loop is interesting, as this loop has been shown to undergo a ligand induced movement towards the protein cavity in a closely related isozyme, NP4. This AB loop movement in response to NO binding was proposed to be functionally significant as a means to trap NO in the binding cavity (68,72). The same movement was later observed upon imidazole binding to NP4 (71) and has been invoked as the reason for multiphasic kinetics of NO release from NP4 (69,73). In our structures of H60C_NP1, we observed a less pronounced but similar conformational change in the AB loop region for the two molecules within the asymmetric unit. These differences must solely be the result of crystal packing effects rather than ligand binding.
Conclusion
We have shown in this study that replacement of the native histidine heme ligand in nitrophorin 1 with cysteine results in cysteine coordination to the ferric heme iron. However, this coordination may be weak, as it is lost upon reduction or the introduction of most, but not all exogenous imidazole ligands. Structures of the complexes indicate that the unusual b-barrel architecture of the protein may prevent movements of the heme and/or protein backbone that would allow the introduced cysteine from providing a strong coordination geometry. These factors may have important implications for future efforts to introduce unusual coordination states in these systems.
Supplementary Material
Acknowledgements
The plasmid for the NP1 gene was provided by Prof. F. A. Walker, and we thank Prof. Walker for a number of helpful discussions. We thank Dr. C. D. Stout for advice and help during X-ray data acquisition and structure refinement. We also thank David Ginsberg for assistance during protein expression and purification. This work was supported by National Institute of Health Grants GM41049 (to DBG) and GM26730 (to JHD).
Abbreviations
- BuImd
N-(n-butyl)imidazole
- CcP
cytochrome c Peroxidase
- H60C_NP1
His-60 to Cys-60 mutant of nitrophorin 1
- Hsm
histamine
- Imd
imidazole
- Mb
myoglobin
- MCD
magnetic circular dichroism
- NP1
wild-type nitrophorin 1 from Rhodnius prolixus
- RR
resonance Raman
- UV/Vis
Ultraviolet/Visible
Contributor Information
Stefan W. Vetter, Department of Molecular Biology, The Scripps Research Institute, 10550 North Torrey Pines Road, La Jolla, CA 92037, current address: Department of Chemistry and Biochemistry, Florida Atlantic University, 777 Glades Road, Boca Raton, FL 33431
Andrew C. Terentis, Department of Chemistry and Biochemistry, Florida Atlantic University, 777 Glades Road, Boca Raton, FL 33431
John H. Dawson, Department of Chemistry and Biochemistry, University of South Carolina, Columbia, SC 29208
David B. Goodin, Department of Molecular Biology, The Scripps Research Institute, 10550 North Torrey Pines Road, La Jolla, CA 92037
References
- 1.Armstrong FA, Bond AM, Buchi FN, Hamnett A, Hill HA, Lannon AM, Lettington OC, Zoski CG. Analyst. 1993;118(8):973–978. doi: 10.1039/an9931800973. [DOI] [PubMed] [Google Scholar]
- 2.Iffland A, Tafelmeyer P, Saudan C, Johnsson K. Biochemistry. 2000;39(35):10790–10798. doi: 10.1021/bi001121e. [DOI] [PubMed] [Google Scholar]
- 3.Musah RA, Goodin DB. Biochemistry. 1997;36(39):11665–11674. doi: 10.1021/bi9708038. [DOI] [PubMed] [Google Scholar]
- 4.Musah RA, Jensen GM, Bunte SW, Rosenfeld RJ, Goodin DB. J. Mol. Biol. 2002;315:845–857. doi: 10.1006/jmbi.2001.5287. [DOI] [PubMed] [Google Scholar]
- 5.Hirst J, Goodin DB. J. Biol. Chem. 2000;275:8582–8591. doi: 10.1074/jbc.275.12.8582. [DOI] [PubMed] [Google Scholar]
- 6.Erman JE, Vitello LB, Miller MA, Kraut J. J. Am. Chem. Soc. 1992;114(16):6592–6593. [Google Scholar]
- 7.Joo H, Lin Z, Arnold FH. Nature. 1999;399(17):670–673. doi: 10.1038/21395. [DOI] [PubMed] [Google Scholar]
- 8.Cirino PC, Arnold FH. Current Opinion Chem. Biol. 2002;6(2):130–135. doi: 10.1016/s1367-5931(02)00305-8. [DOI] [PubMed] [Google Scholar]
- 9.Glieder A, Farinas ET, Arnold FH. Nature Biotechnology. 2002;20(11):1135–1139. doi: 10.1038/nbt744. [DOI] [PubMed] [Google Scholar]
- 10.May O, Voigt CA, Arnold FH. Enzyme Catalysis in Organic Synthesis. 2nd Edition Vol. 1. 2002. pp. 95–138. [Google Scholar]
- 11.Cirino PC, Arnold FH. Angew. Chem. Int. Ed. 2003;42(28):3299–3301. doi: 10.1002/anie.200351434. [DOI] [PubMed] [Google Scholar]
- 12.Peters MW, Meinhold P, Glieder A, Arnold FH. J. Am. Chem. Soc. 2003;125:13442–13450. doi: 10.1021/ja0303790. [DOI] [PubMed] [Google Scholar]
- 13.Farinas ET, Alcalde M, Arnold FH. Tetrahedron. 2004;60:525–528. [Google Scholar]
- 14.Ozaki S.-i., Roach MP, Matsui T, Watanabe Acc. Chem. Res. 2001;34:818–825. doi: 10.1021/ar9502590. [DOI] [PubMed] [Google Scholar]
- 15.Sigman JA, Kwok BC, Lu Y. J. Am. Chem. Soc. 2000;122(34):8192–8196. [Google Scholar]
- 16.Allocatelli CT, Cutruzzola F, Brancaccio A, Vallone B, Brunori M. FEBS Letters. 1994;356(1):151. doi: 10.1016/0014-5793(94)00918-x. [DOI] [PubMed] [Google Scholar]
- 17.Ikeda-Saito M, Hori H, Andersson LA, Prince RC, Pickering IJ, George GN, Sanders C. d., Lutz RS, McKelvey EJ, Mattera R. J. Biol. Chem. 1992;267(32):22843–22852. [PubMed] [Google Scholar]
- 18.Sono M, Roach MP, Coulter ED, Dawson JH. Chem. Rev. 1996;96:2841–2887. doi: 10.1021/cr9500500. [DOI] [PubMed] [Google Scholar]
- 19.Atkins WM, Sligar SG. J. Am. Chem. Soc. 1989;111:2715–2717. [Google Scholar]
- 20.Poulos TL. J. Biol. Inorg. Chem. 1996;1:356–359. [Google Scholar]
- 21.Goodin DB, McRee DE. Biochem. 1993;32:3313–3324. [PubMed] [Google Scholar]
- 22.Watanabe Y, Groves JT. Enzymes. 3rd Ed. Vol. 20. 1992. pp. 405–452. [Google Scholar]
- 23.Dawson JH. Science. 1988;240(4851):433–439. doi: 10.1126/science.3358128. [DOI] [PubMed] [Google Scholar]
- 24.Green MT, Dawson JH, Gray HB. Science. 2004;304:1653–1656. doi: 10.1126/science.1096897. [DOI] [PubMed] [Google Scholar]
- 25.Adachi S, Nagano S, Ishimori K, Watanabe Y, Morishima I, Egawa T, Kitagawa T, Makino R. Biochemistry. 1993;32(1):241–252. doi: 10.1021/bi00052a031. [DOI] [PubMed] [Google Scholar]
- 26.Liu Y, Moeenne-Loccoz P, Hildebrand DP, Wilks A, Loehr TM, Mauk AG, Ortiz de Montellano PR. Biochemistry. 1999;38(12):3733–3743. doi: 10.1021/bi982707s. [DOI] [PubMed] [Google Scholar]
- 27.Sigman JA, Pond AE, Dawson JH, Lu Y. Biochemistry. 1999;38(34):11122–11129. doi: 10.1021/bi990815o. [DOI] [PubMed] [Google Scholar]
- 28.Hildebrand DP, Ferrer JC, Tang H-L, Smith M, Mauk AG. Biochemistry. 1995;34:11598–11605. doi: 10.1021/bi00036a036. [DOI] [PubMed] [Google Scholar]
- 29.Wang W-H, Lu J-X, Yao P, Xie Y, Huang Z-X. Prot. Engineering. 2003;16(12):1047–1054. doi: 10.1093/protein/gzg134. [DOI] [PubMed] [Google Scholar]
- 30.Perera R, Sono M, Sigman JA, Pfister TD, Lu Y, Dawson JH. Proc. Natl. Acad. Sci. 2003;100(7):3641–3646. doi: 10.1073/pnas.0737142100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ribeiro JMC, Hazzard JMH, Nussenzveig RH, Champagne DE, Walker AF. Science. 1993;260(23 April):539–541. doi: 10.1126/science.8386393. [DOI] [PubMed] [Google Scholar]
- 32.Oliveira PL, Kawooya JK, Ribeiro JMC, Meyer T, Poorman R, Alves EW, Walker FA, Machado EA, Nussenzveig RH, Padovan GJ, Masuda H. J. Biol. Chem. 1995;270(18):10897–10891. doi: 10.1074/jbc.270.18.10897. [DOI] [PubMed] [Google Scholar]
- 33.Montfort WR, Weichsel A, Andersen JF. Biochim. Biophys. Acta. 2000;1482:110–118. doi: 10.1016/s0167-4838(00)00165-5. [DOI] [PubMed] [Google Scholar]
- 34.Flower DR, North AC, Attwood TK. Protein Sci. 1993;2:753–761. doi: 10.1002/pro.5560020507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Flower DR, North ACT, Sansom CE. Biochim. Biophys. Acta. 2000;1482:9–24. doi: 10.1016/s0167-4838(00)00148-5. [DOI] [PubMed] [Google Scholar]
- 36.Weichsel A, Andersen JF, Champagne DE, Walker FA, Montfort WR. Nat. Struct. Biol. 1998;5(4):304–309. doi: 10.1038/nsb0498-304. [DOI] [PubMed] [Google Scholar]
- 37.Andersen JF, Champagne DE, Weichsel A, Ribeiro JMC, Bulfour CA, Dress V, Montfort WR. Biochemistry. 1997;36:4423–4428. doi: 10.1021/bi9628883. [DOI] [PubMed] [Google Scholar]
- 38.Akoyunoglou J-HA, Olcott HS, Brown WD. Biochemistry. 1963;2(5):1033–1041. doi: 10.1021/bi00905a021. [DOI] [PubMed] [Google Scholar]
- 39.Kriss GA. In: Fitting models of UV and optical spectral data: using SPECFIT in IRAF. Crabtree DR, Hanisch RJ, Barnes J, editors. Astronomical data analysis software & systems III; San Fransisco: 1994. [Google Scholar]
- 40.Huff AM, Chang CK, Cooper DK, Smith KM, Dawson JH. Inorg. Chem. 1993;32:1460–1466. [Google Scholar]
- 41.Collaborative Computational Project, N Acta Crystallogr. 1994;D 50:760–763. [Google Scholar]
- 42.Pflugrath JW. Acta Crystallogr. 1999;D 55:1718–1725. doi: 10.1107/s090744499900935x. [DOI] [PubMed] [Google Scholar]
- 43.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Acta Crystallogr. 1998;D 54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 44.Bailey S. Acta Cryst. 1994;D 50:760–763. [Google Scholar]
- 45.McLachlan AD. Acta Crystallogr. 1982;A 38:871–873. [Google Scholar]
- 46.Hutchinson EG, Thornton JM. Prot. Sci. 1996;5:212–220. doi: 10.1002/pro.5560050204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ribeiro JMC, Walker FA. J. Exp. Med. 1994;180(December):2251–2257. doi: 10.1084/jem.180.6.2251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sono M, Andersson LA, Dawson JH. J. Biol. Chem. 1982;257(14):8308–8320. [PubMed] [Google Scholar]
- 49.Psylinakis E, Davoras EM, Ioannidis N, Trikeriotis M, Petrouleas V, Ghanotakis DF. Biochim. Biophys. Acta. 2001;1533:119–127. doi: 10.1016/s1388-1981(01)00150-0. [DOI] [PubMed] [Google Scholar]
- 50.Hirst J, Wilcox SK, Ai J, Moënne-Loccoz P, Loehr TM, Goodin DB. Biochemistry. 2001;40(5):1274–1283. doi: 10.1021/bi002090q. [DOI] [PubMed] [Google Scholar]
- 51.Hirst J, Wilcox SK, Williams PA, Blankenship J, McRee DE, Goodin DB. Biochemistry. 2001;40(5):1265–1273. doi: 10.1021/bi002089r. [DOI] [PubMed] [Google Scholar]
- 52.Sono M, Stuehr DJ, Ikeda-Saito M, Dawson JH. J. Biol. Chem. 1995;270(34):1993–19948. doi: 10.1074/jbc.270.34.19943. [DOI] [PubMed] [Google Scholar]
- 53.Andersson L, Mylrajan M, Sullivan E, Jr, Strauss S. J. Biol. Chem. 1989;164:19099–19102. [PubMed] [Google Scholar]
- 54.Hu S, Morris IK, Singh JP, Smith KM, Spiro TG. J. Am. Chem. Soc. 1993;115:12446–12458. [Google Scholar]
- 55.Hu S, Smith KM, Spiro TG. J. Am. Chem. Soc. 1996;118:12638–12646. [Google Scholar]
- 56.Lou BS, Snyder JK, Marshall P, Wang JS, Wu G, Kulmacz RJ, Tsai AL, Wang J. Biochemistry. 2000;39(40):12424–12434. doi: 10.1021/bi001257c. [DOI] [PubMed] [Google Scholar]
- 57.Spiro TG, Li XY, editors. Biological Applications of Raman Spectroscopy. John Wiley & Sons; New York: 1987. [Google Scholar]
- 58.Spiro TG, Strong JD, Stein P. J. Am. Chem. Soc. 1979;101:2648–2655. [Google Scholar]
- 59.Wang J, Caughey WS, Rousseau DL. Resonance Raman Scattering: a Probe of Heme Protein-bound Nitric Oxide. In: Feelisch M, Stamler J, editors. Methods in Nitric Oxide Research. J. Wiley; Chichester ; New York: 1996. [Google Scholar]
- 60.Maes EM, Walker FA, Montfort WR, Czernuszewicz RS. J. Am. Chem. Soc. 2001;123(47):11664–11672. doi: 10.1021/ja0031927. [DOI] [PubMed] [Google Scholar]
- 61.Sun J, Loehr TM, Wilks A, Ortiz de Montellano PR. Biochemistry. 1994;33(46):13734–13740. doi: 10.1021/bi00250a026. [DOI] [PubMed] [Google Scholar]
- 62.Anzenbacher P, Evangelista-Kirkup R, Schenkman J, Spiro TG. Inorg. Chem. 1989;28(25):4491–4495. [Google Scholar]
- 63.Vogel KM, Kozlowski PM, Zgierski MZ, Spiro TG. Inorg. Chim. Acta. 2000;297(12):11–17. [Google Scholar]
- 64.Spiro TG, Kozlowski P. Acc. Chem. Res. 2001;34:137–144. doi: 10.1021/ar000108j. [DOI] [PubMed] [Google Scholar]
- 65.Li T, Quillin ML, Phillips GN, Olson JS. Biochemistry. 1994;33:1433–1446. doi: 10.1021/bi00172a021. [DOI] [PubMed] [Google Scholar]
- 66.Feis A, Rodriguez-Lopez JN, Thorneley RNF, Smulevich G. Biochemistry. 1998;37:13575–13581. doi: 10.1021/bi981399v. [DOI] [PubMed] [Google Scholar]
- 67.Terentis AC, Thomas SR, Takikawa O, Littlejohn TK, Truscott RJW, Armstrong RS, Yeh S-R, Stocker R. J. Biol. Chem. 2002;277:15788–15794. doi: 10.1074/jbc.M200457200. [DOI] [PubMed] [Google Scholar]
- 68.Weichsel A, Andersen JF, Roberts SA, Montfort WR. Nat. Struct. Biol. 2000;7(7):551–554. doi: 10.1038/76769. [DOI] [PubMed] [Google Scholar]
- 69.Maes EM, Weichsel A, Anderson JF, Shepley D, Montfort WR. Biochemistry. 2004;43:6679–6690. doi: 10.1021/bi049748a. [DOI] [PubMed] [Google Scholar]
- 70.Shokhireva TK, Shokhirev NV, Walker FA. Biochemistry. 2003;42:679–693. doi: 10.1021/bi026765w. [DOI] [PubMed] [Google Scholar]
- 71.Roberts SA, Weichsel A, Qiu Y, Shelnutt JA, Walker FA, Montfort WR. J. Biol. Chem. 2001;40(38):11327–11337. doi: 10.1021/bi0109257. [DOI] [PubMed] [Google Scholar]
- 72.Andersen JF, Ding XD, Balfour C, Shokhireva TK, Champagne DE, Walker AF, Montfort WR. Biochemistry. 2000;39:10118–10131. doi: 10.1021/bi000766b. [DOI] [PubMed] [Google Scholar]
- 73.Nienhaus K, Maes EM, Weichsel A, Montfort WR, Nienhaus GU. J. Biol. Chem. 2004;279(38):39401–39407. doi: 10.1074/jbc.M406178200. [DOI] [PubMed] [Google Scholar]
- 74.Skerra A. Curr. Opin. Mol. Ther. 2007;9(4):336–344. [PubMed] [Google Scholar]
- 75.Hohlbaum AM, Skerra A. Expert Rev. Clin. Immunol. 2007;3(4):491–501. doi: 10.1586/1744666X.3.4.491. [DOI] [PubMed] [Google Scholar]
- 76.Schlehuber S, Skerra A. Expert Opin. Biol. Ther. 2005;5(11):1453–1462. doi: 10.1517/14712598.5.11.1453. [DOI] [PubMed] [Google Scholar]
- 77.Skerra A. Methods Princ. Med. Chem. 2003;19:187–211. Protein-Ligand Interactions. [Google Scholar]
- 78.Skerra A. Rev. Mol. Biotechnol. 2001;74(4):257–275. doi: 10.1016/s1389-0352(01)00020-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.