

Abstract
Cysteine and methionine are the two sulfur-containing residues normally found in proteins. Cysteine residues function in the catalytic cycle of many enzymes, and they can form disulfide bonds that contribute to protein structure. In contrast, the specific functions of methionine residues are not known. We propose that methionine residues constitute an important antioxidant defense mechanism. A variety of oxidants react readily with methionine to form methionine sulfoxide, and surface exposed methionine residues create an extremely high concentration of reactant, available as an efficient oxidant scavenger. Reduction back to methionine by methionine sulfoxide reductases would allow the antioxidant system to function catalytically. The effect of hydrogen peroxide exposure upon glutamine synthetase from Escherichia coli was studied as an in vitro model system. Eight of the 16 methionine residues could be oxidized with little effect on catalytic activity of the enzyme. The oxidizable methionine residues were found to be relatively surface exposed, whereas the intact residues were generally buried within the core of the protein. Furthermore, the susceptible residues were physically arranged in an array that guarded the entrance to the active site.
Methionine and cysteine are the two sulfur-containing amino acids that occur in proteins. The structural and catalytic roles of cysteine have been defined for many proteins, but this is not the case for methionine. There are as yet no enzymes for which methionine has been shown to function in the catalytic cycle. Although there are many examples from which methionine has been postulated to play an essential structural role, as yet none have been shown to require the thiol ether, which distinguishes methionine from other residues. For example, in α-antitrypsin, oxidation of Met-358 to methionine sulfoxide destroys the antiproteinase activity, presumably by interfering with complex formation with the target proteinase (1). However, the thiol ether is not required for interaction, since replacement of the methionine with valine gives a fully active antiproteinase (2).
All amino acids are susceptible to oxidation, although their susceptibilities vary greatly (3). Organisms have evolved complex antioxidant defenses to minimize oxidative damage to proteins and other macromolecules. They also possess repair systems for reversing some oxidative modifications and disposal systems for removing modified macromolecules that are not repaired. Oxidative modification of residues within proteins may be mediated by a variety of physiologic and non-physiologic systems, including oxidases, ozone, hydrogen peroxide, superoxide, γ-irradiation, metal-catalyzed oxidation, “leakage” from the electron transport chain, and “auto-oxidation” of flavins or xenobiotics. Methionine residues are remarkable for their high susceptibility to oxidation by most of these systems, with the product generally being methionine sulfoxide. The work of Weissbach, Brot, and colleagues (4) established the widespread existence of methionine sulfoxide reductases, capable of reducing either the free or the protein-bound methionine sulfoxide back to methionine. Thus, a repair mechanism exists for dealing with the product of the relatively facile reaction of oxidants with methionine residues.
This led us to hypothesize that methionine residues could function as a “last chance” antioxidant defense system for proteins. As endogenous components of the protein, their effective concentration is very high, providing effective scavenging of oxidants before they can attack residues that are critical to structure or function. If this hypothesis were correct, one should be able to detect oxidation of solvent-exposed methionine residues before oxidation of other residues. In 1975, Shechter and colleagues reported that only the surface-exposed methionine residues of native proteins were oxidized by chloramine-T or N-chlorosuccinimide (5). More recently, the oxidative inactivation of α-2-macroglobulin has been studied in detail and the results obtained are consistent with the hypothesis (6). This antiproteinase loses activity when exposed to activated neutrophils, or to a model system consisting of chloramine. Consumption of eight equivalents of chloramine caused oxidation of eight methionine residues to methionine sulfoxide. Continued exposure caused oxidation of six additional residues of methionine and of a single tryptophan residue. The fractional loss of the tryptophan residue matched the fractional inactivation of the α-2-macroglobulin. These results are consistent with the suggestion that methionine residues scavenge oxidants that could otherwise attack the tryptophan residue that is essential to function.
The locations of the susceptible methionine residues in the α-2-macroglobulin sequence were not determined, and no crystal structure is available to disclose their three-dimensional location. We therefore chose to study the oxidative modification of bacterial glutamine synthetase by hydrogen peroxide, with the purpose of establishing the susceptibility of methionine residues to oxidation, their location within the protein, and the relation of oxidation to loss of catalytic competence. We found that surface-exposed methionine residues surrounding the entrance to the active site are preferentially oxidized without loss of catalytic activity, consistent with the hypothesis that methionine residues function as an endogenous antioxidant defense system.
MATERIALS AND METHODS
Glutamine Synthetase Preparations.
Unadenylylated glutamine synthetase was purified (7) from an overproducing Escherichia coli, YMC 10/pgln6, and stored at 20 mg/ml in 10 mM imidazole, 100 mM KCl, 1 mM MnCl2 (pH 7.0). Activity was assayed at pH 7.57 with the γ-glutamyl transferase assay (8). Treatment with H2O2 was carried out in 50 mM potassium phosphate (pH 7.5). Both the stock buffer and water were treated with Chelex (Bio-Rad) to minimize metal-catalyzed oxidation of glutamine synthetase (9). One milligram glutamine synthetase in 1 ml total volume was incubated for 2 hr at 37°C in a 12 × 75-mm glass test tube, capped to minimize evaporative losses. H2O2 was varied from 0–160 mM (Fisher, 30%). After incubation, an aliquot was taken for activity determination and the remainder dialyzed with dialysis cassettes (Pierce) to remove H2O2. Samples were dialyzed against five changes of 10 mM potassium phosphate, 100 mM KCl, 10 mM MgCl2 (pH 7.4). Whatever method is used to accomplish this buffer exchange, care must be taken to assure removal of the H2O2. If H2O2 carries over into the formic acid used for CNBr cleavage, it will react to yield performic acid, which readily oxidizes methionine residues.
CNBr Cleavage and Amino Acid Analysis.
CNBr cleaves peptide bonds on the carboxyl side of methionine, yielding homoserine; it does not cleave at methionine sulfoxide (10). CNBr cleavage was carried out on 200 μg of protein dried by vacuum centrifugation (Savant) in 4-ml glass vials (Wheaton 224882) fitted with Teflon-lined caps. CNBr was prepared as a 10 M stock solution in acetonitrile, then diluted to 100 mM with 70% formic acid just before use. One hundred microliters was added to the vial, which was capped and incubated for 1 hr at 70°C in a hood. Five microliters was transferred to another vial for amino acid analysis, and both samples were dried by vacuum centrifugation. Hydrogen chloride hydrolysis and amino acid analysis were carried out on samples with and without CNBr treatment (6).
Proteolytic Susceptibility.
Surface hydrophobicity of the 1-anilinonaphthalene-8-sulfonic acid glutamine synthetases was assessed with the fluorescent probe (Sigma). One hundred micrograms of protein was incubated for 30 min at 37°C with 100 mM 1-anilinonaphthalene-8-sulfonic acid in 50 mM Hepes, 100 mM KCl (pH 7.8) with a total volume of 1 ml as described (11). The 20S proteasome was purified from rat liver essentially as described (11). Proteolytic degradation of glutamine synthetase was assessed with a fluorimetric method adapted to an HPLC detector (12).
Simultaneous Sequencing of Peptide Mixtures.
We wished to quantify the fractional modification of each methionine residue in the oxidatively modified proteins. While this might have been accomplished by reverse-phase HPLC mapping of proteins cleaved by specific proteases, it is a challenging process complicated by varying recoveries and changing retention times caused by oxidation of the methionine residues. We therefore used the technique of simultaneous sequencing of a peptide collection. Simultaneous sequencing by an automated Edman sequencer generates quantitative results from which one can determine the location and extent of covalent modifications, provided that the sequence of the protein is known and the cleavage methods are selected to minimize or eliminate ambiguities. The technique has been used to establish the histidine residues modified by metal-catalyzed oxidation of glutamine synthetase and the cysteine residue modified by glutathiolation in carbonic anhydrase (13, 14).
Treatment of unmodifiied glutamine synthetase with CNBr should yield 17 peptides. Sequencing of this collection of peptides will generate multiple phenylthiohydantoin–amino acids in each cycle of Edman degradation. The peptide collection and resultant phenylthiohydantoin–amino acid patterns will change upon formation of methionine sulfoxides, because CNBr does not cleave these residues. Ambiguities may result from multiple oxidations, but whether this actually occurs can only be determined experimentally for each protein.
The CNBr peptide collection of each of the 16 glutamine synthetase preparations was loaded onto the sequencing column of a Hewlett–Packard G1005A automated sequencer equipped with a model 1040 diode array spectrophotometer. Five cycles were run on each protein, providing an overdetermination useful for confirming the status of each methionine. In addition to cleavage at each methionine by CNBr, the 70% formic acid also cleaved each of the three Asp–Pro sequences (15). These additional peptides were useful for assessing recovery of peptides from each sample. For example, cleavage at Asp-103–Pro-104 places an arginine in cycle 2, the only one that occurs in that cycle. Examination of the yield of arginine established that recovery was reproducible from sample to sample. Table 1 shows the expected effect of oxidation of each methionine in the first five cycles.
Table 1.
Susceptibility of methionine residues in glutamine synthetase to oxidation and the expected decrease in residue yields during simultaneous peptide sequencing
| Methionine no. | Oxidized by H2O2 | Cycle 1 | Cycle 2 | Cycle 3 | Cycle 4 | Cycle 5 |
|---|---|---|---|---|---|---|
| 8 | Yes | Leu | Asn | Glu | His | Glu |
| 48 | Yes | Phe | Asp | Gly | Ser | Ser |
| 65 | Yes | Val | Leu | Met | ||
| 68 | Yes | Pro | Asp | Ala | Ser | Thr |
| 195 | Yes | Cys | Leu | Val | Met | |
| 199 | No | Glu | Gln | Gln | ||
| 202 | No | Gly | Leu | Val | Val | Glu |
| 228 | No | Thr | Lys | Lys | Ala | Asp |
| 256 | ? | Pro | Lys | Pro | Met | |
| 260 | No | Phe | Gly | Asp | Asn | Gly |
| 268 | No | His | Cys | His | Met | |
| 272 | No | Ser | Leu | Ser | Lys | Asn |
| 331 | Yes | Leu | Ala | Tyr | Ser | Ala |
| 376 | No | Ala | Gly | Leu | Asp | Gly |
| 392 | Yes | Asp | Lys | Asn | Leu | Tyr |
| 455 | Yes | Thr | Pro | His | Pro | Val |
Oxidation of a methionine residue prevents cleavage by CNBr, thus causing a decrease in yield of residues in the following peptide. Short, hydrophilic peptides may exhibit poor recovery, and that was the case for P257KPM so that the status of Met-256 could not be determined. Oxidation of the carboxyl-terminal methionine of such peptides causes an increase in recovery of the preceeding residues because the resultant peptide is longer. This phenomenon was useful in monitoring the status of Met-68, whose oxidation caused an increase in valine in cycle 1 because of improved retention of Val-66. Also, methionine sulfoxide was reduced back to methionine under conditions of Edman sequencing so that yields of methionine during sequencing increased as residues were oxidized. For example, no methionine was detected in cycle 3 of the control glutamine synthetase, but yields increased as Met-68 was oxidized. The second column refers to oxidation by exposure to hydrogen peroxide.
RESULTS
Methionine/Methionine Sulfoxide by Amino Acid Analysis.
Exposure of glutamine synthetase to increasing concentrations of hydrogen peroxide for a fixed time (2 hr) generated a series of modified proteins with increasing methionine sulfoxide content, approaching a plateau of eight residues at 160 mM peroxide (Fig. 1). Extrapolation to infinite peroxide concentration suggests that a maximum of 10 of the 16 methionine residues per subunit could be oxidized (Fig. 1 Inset). The γ-glutamyl transferase activity of the enzyme was almost unchanged, decreasing only 15% in the sample exposed to 160 mM peroxide. Thus, oxidation of methionine residues provides a route for scavenging hydrogen peroxide without loss of catalytic activity.
Figure 1.
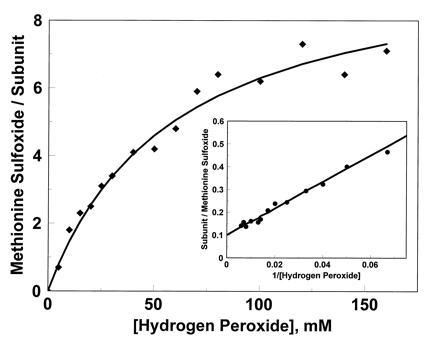
Oxidation of methionine residues in glutamine synthetase by hydrogen peroxide, determined by amino acid analysis. A double reciprocal plot of the same data is shown (Inset), demonstrating that extrapolation to infinite peroxide concentration would cause oxidation of 10 of the 16 methionine residues in each subunit.
Effect of Oxidation on Susceptibility to Proteolysis.
Oxidation of glutamine synthetase by a metal-catalyzed system was previously shown to cause loss of catalytic activity and to render the protein susceptible to degradation by either the 20S proteasome (multicatalytic proteinase) or a specific bacterial protease (9, 16). The modifications “sensed” by the degrading proteases have not been firmly established, but an increase in surface hydrophobicity did correlate well with susceptibility to proteolysis (17). We found that the proteolytic susceptibility of the hydrogen peroxide-treated proteins also correlated with increased surface hydrophobicity (Fig. 2), consistent with the hypothesis that exposure of hydrophobic patches renders proteins susceptible to proteolytic degradation. No increase in proteolytic susceptibility nor in hydrophobicity occurred until ≈6 residues of methionine were oxidized. If this relationship also holds in vivo, then there exists a window of opportunity for reduction of the oxidized residues by methionine sulfoxide reductases without competition from proteases.
Figure 2.
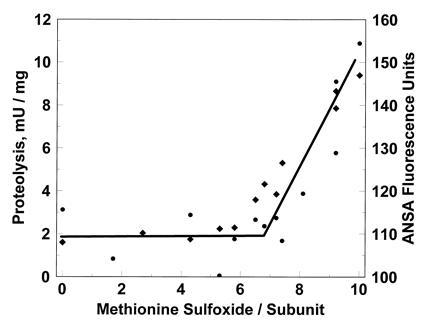
The susceptibility of oxidatively modified glutamine synthetases to proteolysis by the proteosome (♦) correlated with increased surface hydrophobicity (•).
Identification of the Oxidized Methionine Residues.
The technique of simultaneous sequencing of the CNBr peptide collection allowed determination of the status of each methionine except Met-256 (Table 1, Fig. 3). Even at the highest concentration of peroxide, seven methionine residues remain intact, whereas eight are oxidized. We could not detect differences in susceptibility among the eight oxidized residues.
Figure 3.
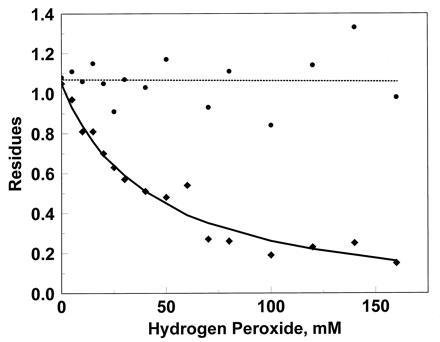
Examples of results from simultaneous sequence analysis of CNBr peptide collections of glutamine synthetases exposed to hydrogen peroxide. (♦—-♦), aparagine in cycle 2, which monitors Met-8; (•- - - -•), lysine in cycle 4, which monitors Met-272.
The three-dimensional structure of the 12-subunit glutamine synthetase has been determined by Eisenberg and colleagues (18), and the locations of the methionine residues were mapped onto their structure (Fig. 4). The oxidized residues are relatively exposed, whereas the intact residues are generally buried within the core of the protein. Furthermore, the susceptible residues are physically arranged in an intriguing formation. The active site is formed by two subunits that create a bay through which substrates enter and products leave. The eight oxidizable methionine residues are arrayed along the border of the bay, forming a phalanx that guards the active site.
Figure 4.
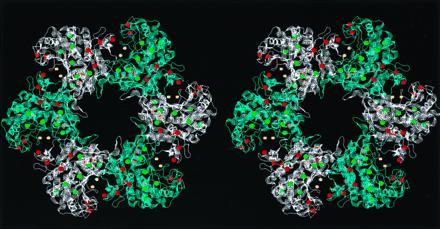
Location of oxidized and intact methionine residues in glutamine synthetase. This stereo figure was created by the program rasmol (19) using the coordinates determined by Almassy et al. (18), deposited in the Brookhaven Data Base (reference 2GLS). For clarity, only one of the two hexamers is shown, and subunits are alternately colored blue and white. The sulfur groups of methionine residues are shown as balls, with intact residues in green and the oxidized residues in red. The active sites are formed by two adjacent subunits, and the Mn2+ in the core of the active sites is shown in yellow. The oxidizable methionine residues appear to form an array about the entrance to the active site bay.
DISCUSSION
Methionine residues have long been known to undergo oxidation to methionine sulfoxide, often with a concomitant effect on an overall biological function, and the review by Vogt (20) provides a useful compilation of many examples. Recently, several investigators have noted that methionine residues within a protein exhibit variability in their susceptibility to oxidation. Susceptibility generally correlates with the surface exposure of the residue, although residues near the methionine can modulate its susceptibility (21). Hsu and colleagues (7) studied oxidation by hydrogen peroxide of recombinant human stem cell factor, which contains five methionine residues. The two surface-exposed residues, Met-1 and Met-159, were readily oxidized, but with negligible effects on biological activity. Met-27 was oxidized at about one-third the rate of the rapidly oxidized residues, again with little effect on activity. The remaining two residues, Met-36 and Met-48, were much less susceptible to oxidation and modification of either residue was accompanied by a substantial loss of biological activity. Similarly, Gitlin and colleagues (22) established that oxidation of Met-111 in interferon α-2b did not alter its biological activity. Nabuchi et al. (23) reported studies on hydrogen peroxide-mediated oxidation of the two methionine residues present in human parathyroid hormone. Oxidation of Met-8 slightly reduced biological activity, whereas oxidation of Met-18 substantially reduced activity. Keck showed that two surface-exposed methionine residues of interferon or three methionine residues of tissue plasminogen activation could be oxidized without loss of biological activity (24). A similar result with keratinocyte growth factor has been summarized recently (25). As noted in the introduction, α-2-macroglobulin will likely provide another example, although the location of the initially oxidized eight methionine residues is not yet known. Yao et al. (26) reported the rates of oxidation of methionine residues in calmodulin exposed to hydrogen peroxide. They were not able to obtain rates for each methionine residue, but the available results were consistent with the conclusion that susceptibility to oxidation was proportional to the surface exposure of the residue. These studies of various biologically active proteins support the hypothesis that surface-exposed methionine residues effectively scavenge oxidizing agents, while generally preserving the biological function of the molecule. In these cases, no other specific role of methionine has been ascertained.
While solvent-exposed methionine residues are likely to protect from environmentally proximate oxidizing agents, residues in or near active sites may protect enzymes from “auto-oxidation” by substrates or cofactors. For example, oxidation of a single methionine in rabbit 15-lipoxygenase was known to be mediated by substrates or products, and appearance of the methionine sulfoxide had been correlated with loss of catalytic activity. However, the studies of Gan and colleagues (27) established that replacement of the methionine residue by leucine did not prevent inactivation by substrates, demonstrating that formation of methionine sulfoxide was not the cause of inactivation. We suggest that oxidation of the active site methionine may actually retard the inactivation of the lipoxygenase. Whether the resulting methionine sulfoxide can be reduced back to methionine in vivo is unknown.
Methionine is readily oxidized by a variety of agents, as noted above. Indeed, a common laboratory method of scavenging chloramines, hypochlorous acid, or CNBr is simply to add a stoichiometric excess of methionine. In addition to their facile oxidation, surface-exposed methionine residues of proteins provide an enormously high concentration of antioxidant at the protein surface, an ideal combination for defense against oxidation of key residues within the protein or even for protecting other molecules against oxidation, for example at sites of inflammation. For simplicity, consider a spherical protein of diameter 60 Å and assume that eight methionine residues are distributed within that volume. Then the concentration of methionine is ≈100 mM, and if one considered the residues restricted to a shell around the surface of the protein, then the effective concentration would be even higher.
It is notable that a significant number of methionine residues in glutamine synthetase may be oxidized without an increase in surface hydrophobicity or proteolytic susceptibility. Proteins within this window may be repaired by reduction of the methionine sulfoxide to methionine. The net effect is the catalytic scavenging of reactive species as shown in these schematic equations:
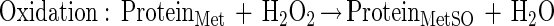 |
1 |
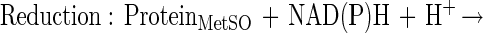 |
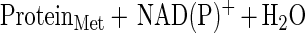 |
2 |
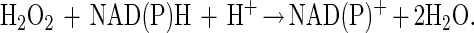 |
This example points out that cyclic oxidation and reduction of methionine residues both scavenges H2O2 and drives an NAD(P)H oxidation reaction. Other reactive species may be similarly scavenged, including superoxide, ozone, hypochlorous acid, and chloramines. Other proteins such as thioredoxin and thioredoxin reductase would likely be important in these catalytic cycles.
Other investigators have pointed out that it may sometimes be desirable to inactivate molecules exposed to oxidizing conditions. For example, activated neutrophils release reactive oxygen species and proteases at sites of inflammation. The well-established oxidative inactivation of α-1-antitrypsin prevents this antiprotease from interfering with the action of elastase at the site. We would therefore expect that the oxidizable Met-358 would be surface exposed, whereas the other methionine residues would be relatively inaccessible, so that they do not react with the oxidizing agents before Met-358. The available structures are consistent with this expectation (28).
Returning to the general hypothesis that methionine residues function as endogenous antioxidants, it follows that mutants enriched at known positions in surface-exposed methionine residues should be more resistant to oxidative inactivation. These modified proteins may also have longer half-lives in vivo, especially if a methionine sulfoxide reductase is functional in the cell. Conversely, proteins engineered to decrease their exposed methionine residues would be more susceptible to oxidative inactivation. Increased oxidative inactivation would also be a consequence of impaired methionine sulfoxide reductase activity.
In summary, methionine residues may act as endogenous antioxidants. Surface-exposed residues react readily with oxidizing agents at physiological pH, and their effective concentration at the protein surface is very high. Other residues within the critical regions of the protein are thus protected, and the existence of a repair mechanism means that each methionine may scavenge many oxidizing molecules.
References
- 1.Johnson D, Travis J. J Biol Chem. 1979;254:4022–4026. [PubMed] [Google Scholar]
- 2.Rosenberg S, Barr P J, Najarian R C, Hallewell R A. Nature (London) 1994;312:77–80. doi: 10.1038/312077a0. [DOI] [PubMed] [Google Scholar]
- 3.Stadtman E R. Annu Rev Biochem. 1993;62:797–821. doi: 10.1146/annurev.bi.62.070193.004053. [DOI] [PubMed] [Google Scholar]
- 4.Brot N, Weissbach H. Arch Biochem Biophys. 1983;223:271–281. doi: 10.1016/0003-9861(83)90592-1. [DOI] [PubMed] [Google Scholar]
- 5.Shechter Y, Burstein Y, Patchornik A. Biochemistry. 1975;14:4497–4503. doi: 10.1021/bi00691a025. [DOI] [PubMed] [Google Scholar]
- 6.Reddy V Y, Desrochers P E, Pizzo S V, Gonias S L, Sahakian J A, Levine R L, Weiss S J. J Biol Chem. 1994;269:4683–4691. [PubMed] [Google Scholar]
- 7.Hsu Y R, Narhi L O, Spahr C, Langley K E, Lu H S. Protein Sci. 1996;5:1165–1173. doi: 10.1002/pro.5560050619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stadtman E R, Smyrniotis P Z, Davis J N, Wittenberger M E. Anal Biochem. 1979;95:275–285. doi: 10.1016/0003-2697(79)90217-3. [DOI] [PubMed] [Google Scholar]
- 9.Rivett A J, Levine R L. Arch Biochem Biophys. 1990;278:26–34. doi: 10.1016/0003-9861(90)90226-o. [DOI] [PubMed] [Google Scholar]
- 10.Fliss H, Weissbach H, Brot N. Proc Natl Acad Sci USA. 1983;80:7160–7164. doi: 10.1073/pnas.80.23.7160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Friguet B, Szweda L I, Stadtman E R. Arch Biochem Biophys. 1994;311:168–173. doi: 10.1006/abbi.1994.1222. [DOI] [PubMed] [Google Scholar]
- 12.Sahakian J A, Szweda L I, Friguet B, Kitani K, Levine R L. Arch Biochem Biophys. 1995;318:411–417. doi: 10.1006/abbi.1995.1248. [DOI] [PubMed] [Google Scholar]
- 13.Apffel A, Sahakian J, Levine R L. Protein Sci. 1994;3:99. (abstr.). [Google Scholar]
- 14.Cabiscol E, Levine R L. Proc Natl Acad Sci USA. 1996;93:4170–4174. doi: 10.1073/pnas.93.9.4170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Marcus F. Int J Pept Protein Res. 1985;25:542–546. doi: 10.1111/j.1399-3011.1985.tb02208.x. [DOI] [PubMed] [Google Scholar]
- 16.Roseman J E, Levine R L. J Biol Chem. 1987;262:2101–2110. [PubMed] [Google Scholar]
- 17.Cervera J, Levine R L. FASEB J. 1988;2:2591–2595. doi: 10.1096/fasebj.2.10.2898411. [DOI] [PubMed] [Google Scholar]
- 18.Almassy R J, Janson C A, Hamlin R, Xuong N H, Eisenberg D. Nature (London) 1986;323:304–309. doi: 10.1038/323304a0. [DOI] [PubMed] [Google Scholar]
- 19.Sayle R A, Milner-White E J. Trends Biochem Sci. 1995;20:374. doi: 10.1016/s0968-0004(00)89080-5. [DOI] [PubMed] [Google Scholar]
- 20.Vogt W. Free Rad Biol Med. 1995;18:93–105. doi: 10.1016/0891-5849(94)00158-g. [DOI] [PubMed] [Google Scholar]
- 21.von Eckardstein A, Walter M, Holz H, Benninghoven A, Assmann G. J Lipid Res. 1991;32:1465–1476. [PubMed] [Google Scholar]
- 22.Gitlin G, Tsarbopoulos A, Patel S T, Sydor W, Pramanik B N, Jacobs S, Westreich L, Mittelman S, Bausch J N. Pharm Res. 1996;13:762–769. doi: 10.1023/a:1016059902645. [DOI] [PubMed] [Google Scholar]
- 23.Nabuchi Y, Fujiwara E, Ueno K, Kuboniwa H, Asoh Y, Ushio H. Pharm Res. 1995;12:2049–2052. doi: 10.1023/a:1016281031373. [DOI] [PubMed] [Google Scholar]
- 24.Keck R G. Anal Biochem. 1996;236:56–62. doi: 10.1006/abio.1996.0131. [DOI] [PubMed] [Google Scholar]
- 25.Spahr, C. S., Narhi, L., Speakman, J., Lu, H. S. & Hsu, Y. R. (1996) Protein Sci. 5, Suppl. 1, 119. [DOI] [PMC free article] [PubMed]
- 26.Yao Y, Yin D, Jas G S, Kuczer K, Williams T D, Schoneich C, Squier T C. Biochemistry. 1996;35:2767–2787. doi: 10.1021/bi951712i. [DOI] [PubMed] [Google Scholar]
- 27.Gan Q F, Witkop G L, Sloane D L, Straub K M, Sigal E. Biochemistry. 1995;34:7069–7079. doi: 10.1021/bi00021a019. [DOI] [PubMed] [Google Scholar]
- 28.Loebermann H, Tokuoka R, Deisenhofer J, Huber R. J Mol Biol. 1984;177:531–557. [PubMed] [Google Scholar]