

Abstract

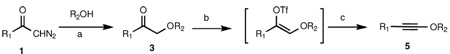
α–Alkoxy ketones 3 can be transformed into 1-alkynyl ethers 5 by a two-step procedure involving formation of the enol triflate or phosphate and base-induced elimination. Performing the same reaction sequence with allylic alcohols (R2OH, R2 = allyl) furnishes instead γ,δ-unsaturated carboxylic acid derivatives 6, derived from [3,3]-sigmatropic rearrangement of the intermediate allyl alkynyl ethers at −78 °C and trapping of the subsequently formed ketene with nucleophiles (Nu-H). Benzyl alkynyl ether 5 (R2 = benzyl) rearranges to indanone 7 upon heating to 60 °C.
Electron-rich alkynes, such as ynamines and ynol ethers, are functional groups that possess significant potential in organic chemistry for the formation of carbon-carbon bonds.1 The synthetic utility of ynamides has been considerably expanded recently, along with the development of new methods for their facile preparation from simple building blocks.2 1-Alkynyl ethers, while possessing many of the reactivity features of ynamides, have been far less investigated due to the relatively few methods currently available for their synthesis.3 In this letter we present a mild and efficient method for the synthesis of diverse 1-alkynyl ethers and demonstrate that a facile sigmatropic rearrangement of allyl and benzyl alkynyl ethers furnishes products containing new carbon-carbon bonds.
We have previously shown that treatment of allyl-1,1-dichlorovinyl ethers with 2.2 equiv of n-butyllithium at −78 °C, followed by quenching of the reaction mixture with excess alcohol, leads to rearranged γ,δ-unsaturated esters in high yield.4 The reaction possesses many of the characteristics of a sigmatropic process, both in terms of its stereospecificity and geometrical requirements. We have proposed that an initially generated allyl alkynyl ether intermediate undergoes a [3,3]-sigmatropic rearrangement either as a neutral or as a negatively charged species.
Due to its reactivity toward a range of functional groups, the use of the nucleophilic base n-BuLi to generate the key intermediate in this reaction may be viewed as a potential limitation. Furthermore, the preparation of the allyl-1,1-dichlorovinyl ether substrate via methylenation of an allylic formate ester requires the use of toxic carbon tetrachloride in combination with triphenylphosphine. Since terminal and internal alkynes have been previously prepared from ketones by treatment of the corresponding enol phosphates or triflates with a non-nucleophilic base,5 we sought to investigate the preparation of 1-alkynyl ethers from α–alkoxy ketones via E2 elimination of the derived enol triflates or phosphates.
Following the procedure of Muthusamy et. al.,6 treatment of a toluene solution of diazoacetophenone 1a and menthol with a catalytic amount (10 mol %) of indium triflate at room temperature furnished α–alkoxy ketone 3a in 91% yield (Scheme 1). Formation of the enol triflate was achieved by stirring 3a with LiHMDS at −78 °C for one hour, followed by quenching with PhNTf2 in DMPU/THF (1:2) and warming to room temperature. The enol triflate obtained (4a) was sufficiently stable to be purified by silica gel chromatography; 1H NMR spectroscopy of the purified material revealed that the triflate was formed as a single Z geometric isomer (the configuration of the double bond was confirmed by NOESY experiments; see supporting information for details). Subsequent treatment with 2 equiv of potassium tert-butoxide at −78 °C in THF smoothly furnished the corresponding alkynyl ether 5a in 75% overall yield from 3a. Alternatively, and more expediently, the crude enol triflate can be directly transformed into 5a in similar overall yield by addition to an excess (3 equiv) of potassium tert-butoxide in THF at −78 °C. The presence of the alkynyl ether moiety in 5a was verified by the chemical shifts of the alkyne carbons in the 13C NMR spectrum (41.8 and 98.5 ppm, respectively), and also by the sharp spike at 2254 cm−1 in the IR spectrum.
Scheme 1.

Preparation of phenylethynyl menthyl ether
To extend this protocol to the preparation of a diverse range of substituted alkynyl ethers, we required access to a variety of α-diazoketones. The procedure of Danheiser et. al.7 allowed the preparation of aryl (1a), alkenyl (Table 1, 1e and 1f), and alkyl (1h, 1i) diazoketones from the corresponding ketones in high yields. However, the preparation of alkynyl diazoketone 1g was problematic due to self-condensation/cycloaddition side reactions.8
Table 1.
Scope of alkynyl ether sythesis via elimination of enol triflatesa
 | |||||
|---|---|---|---|---|---|
| entry | R1 | R2 | 3 (% yield) | 5 | 5 (% yield) |
| 1 | Ph | menthyl | 91 | a | 75 |
| 2 | Ph | CH2CH(CH2)3 | 89 | b | 82 |
| 3 | Ph | t-Bu | 67 | c | 68 |
| 4 | Ph | Ph | 78 | d | 70 |
| 5 | 1-cyclohexenyl | menthyl | 84 | e | 90 |
| 6 | (CH3)2CCH | CH2CH(CH2)3 | 81 | f | 68 |
| 7 | PhCC | menthyl | 69b | g | 64 |
| 8 | t-Bu | CH2CH(CH2)3 | 87 | h | --c |
| 9 | C6H13 | menthyl | 90 | i | --c |
Reaction conditions: a. R2OH (1.5 equiv), In(OTf)3 (10 mol %), toluene, rt; b. LiHMDS, THF, −78 °C, then PhNTf2, DMPU, −78 °C-rt; c. KOt-Bu, THF, −78 °C.
Yield of 3g prepared in three steps from menthol via an alternative route described in the text.
No alkynyl ethers were detected in the reaction mixture; see text for details.
The three-step protocol for alkynyl ether synthesis (diazoketone-alcohol coupling, enol triflate formation, and elimination) was carried out starting with a number of different diazoketones and alcohols (Table 1). Primary, secondary, tertiary alcohols and phenols all work equally well, furnishing the expected alkynyl ethers (entries 1–4) in good yields. Aromatic (R1 = Ph entries 1–4), α,β-unsaturated (R1 = 1-cyclohexenyl, (CH3)2CCH, entries 5 and 6) and alkynyl (R = CCPh, entry 7) ketones 3 were all effective precursors of alkynyl ethers. Due to the problems encountered in preparing alkynyl diazoketones (vide supra), an alternative protocol for the synthesis of α-alkoxy ketone 3g was developed, involving reaction of the sodium alkoxide of menthol with chloroacetic acid,9 formation of the Weinreb amide,10 and addition to phenylethynyl lithium (2 equiv) at −78 °C. Enol triflate formation then proceeded uneventfully, furnishing a ~1:1 mixture of E and Z geometrical isomers. Subsequent treatment with KOt-Bu under standard conditions then allowed the preparation of compound 5g, a 1,3-diynyl menthyl ether.
Unlike results for aromatic, alkenyl, and alkynyl ketone substrates, aliphatic α-alkoxy ketones failed to generate the corresponding enol triflates upon treatment with LiHMDS/PhNTf2 (Table 1, entries 8 and 9). A survey of different reaction conditions and triflating agents (Tf2O, Comins’ triflimide) yielded no improvement, with high Rf decomposition products obtained in all cases. We suspect that the enol triflates derived from aliphatic ketones are unstable, perhaps decomposing to allenic compounds that undergo side reactions.
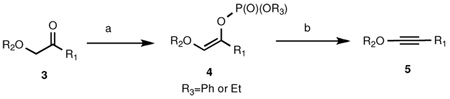
To address this important limitation, we investigated the preparation of putatively more stable enol phosphates from α-alkoxy ketones.11 Indium-triflate catalyzed coupling of tert-butyl diazoacetate with 4-penten-1-ol provided ketone 3h in 89% yield. Addition of a toluene solution of KHMDS to a mixture of 3h and diethylphosphoryl chloride in THF at −78 °C furnished enol phosphate 4h (again as a single Z geometric isomer; see NOESY data in the supporting information) in 81% yield (Scheme 2). Disappointingly, no reaction was observed upon treatment of the enol phosphate with KOt-Bu in THF at −78 °C; even after warming the reaction to room temperature and then to reflux, only starting material was isolated. Use of the stronger bases LDA or LiTMP also led to no reaction. Finally, it was found that addition of 2–3 equiv Schlosser’s base (n-BuLi-KOt-Bu) to the enol phosphate at −78 °C led to the rapid formation of the alkynyl ether in 60% yield.
Scheme 2.

Preparation of alkyl-substituted alkynyl ethers
A variety of alkyl-substituted alkynyl ethers may be prepared efficiently from the corresponding enol phosphates (Table 2, entries 1–3). Either the diphenyl phosphate or the diethylphosphate derivative may be employed for the elimination step, and the order of addition of base and substrate appears to have no effect on the reaction yield (addition of n-BuLi to a mixture of enol phosphate and KOt-Bu vs. addition of the enol phosphate to pre-formed BuK). Performing the two-step sequence on hexyl and isopropyl ketones 3i and 3j furnished enol phosphates 4i and 4j and alkynyl ethers 5i and 5j, respectively, in good yields. This method is also an effective means of synthesizing aryl and alkenyl-substituted alkynyl ethers (Table 2, entries 4 and 5).
Table 2.
Scope of alkynyl ether synthesis via elimination of enol phosphatesa
 | ||||||
|---|---|---|---|---|---|---|
| entry | R1 | R2 | 4 | % yield 4 | 5 | % yield 5 |
| 1 | t-Bu | CH2CH(CH2)3 | hb | 81 | h | 60 |
| 2 | C6H13 | menthyl | ic | 89 | i | 72 |
| 3 | i-Pr | menthyl | jb | 78 | j | 85 |
| 4 | 1-cyclohexenyl | t-Bu | kb | 72 | k | 70 |
| 5 | Ph | t-Bu | lb | 82 | c | 56 |
Reaction conditions: a. KHMDS, ClPO(OR3)2, THF, 78 °C; b. n-BuLi, KOt-Bu, THF, −78 °C.
The diethylphosphate derivative was synthesized.
The diphenylphosphate derivative was synthesized.
Interestingly, when methyl ketone 3m (Scheme 3) was treated with KHMDS and ClPO(OEt)2, a 3:1 mixture of enol phosphates 4n and 4m was obtained in 65% yield. Subjection of this mixture to n-BuLi/KOt-Bu at −78 °C cleanly gave rise to allenyl ether 8 in 85% yield.
Scheme 3.

Formation of allenyl ether 8 from methyl ketone 3m
Finally, we wished to examine the special case of alkynyl ethers derived from allylic and benzylic alcohols. Early investigations by Arens12 and Schmid13 revealed that substituted and unsubstituted benzyl alkynyl ethers undergo [3,3]-sigmatropic rearrangement followed by intramolecular ketene trapping to form indanones in high yield. Interestingly, these processes took place in refluxing CCl4, at temperatures well below those typical for uncatalyzed aromatic Claisen rearrangements.14 In(OTf)3-catalyzed coupling of benzyl alcohol and diazoacetophenone 1a furnished α-benzyloxy acetophenone 3o in 87% yield (Scheme 4). Enol triflate formation proceeded smoothly to afford 4o, which when treated with KOt-Bu in THF at −78 °C gave rise to benzyl alkynyl ether 5o in 75% yield. This compound proved to be thermally labile, partially rearranging to indanone 7a upon heating above room temperature for rotary evaporation; it also readily decomposed on silica gel chromatography. A solution of 5o in toluene was thus heated to 60 °C for 60 minutes to effect conversion to indanone 7a in quantitative yield.
Scheme 4.

Synthesis and thermal rearrangement of phenylethynyl benzyl ether
The previous studies of Katzenellenbogen,15 as well as our own investigations,4 have indicated that allyl-1-alkynyl ethers undergo rapid sigmatropic rearrangement at low temperatures (−33 °C, −78 °C) once formed. Therefore, we wished to assess if sigmatropic rearrangement would also take place upon t-BuOK treatment of α-allyloxy ketone-derived vinyl trifates at −78 °C. Reaction of prenol or allyl alcohol with α–diazoacetophenone in the presence of indium triflate furnished α-alkoxyketones 3q and 3r in 88% and 82% yields, respectively (Scheme 5). Enol triflate 4q, derived from 3q (LHMDS, Tf2NPh, DMPU, −78 °C–rt), was treated with 3 equiv of potassium tert-butoxide in THF at −78 °C; after warming to room temperature, tert-butyl ester 6a was obtained in 78% yield. Performing the same reaction with 3 equiv each of potassium tert-butoxide and morpholine at −78 °C furnished amide 6b in 69% yield. Enol triflate 4r, derived from 3r (LHMDS, Tf2NPh, DMPU, −78 °C–rt), was similarly transformed into menthyl ester 6c (54%, as a 1:1 mixture of diastereomers) by treatment with 2.5 equiv of the potassium alkoxide of menthol in THF at −78 °C. Surprisingly, however, attempts to prepare the simple methyl ester 6d met with little initial success. Addition of 4r to a solution of excess potassium methoxide in THF at −78 °C led to the exclusive production of 3r, presumably arising from nucleophilic attack of methoxide at the triflate sulfur atom. Addition of KOtBu to a solution of 4r and methanol at −78 °C also gave rise to 3r as major product, as well as minor amounts of rearranged methyl and tert-butyl ester products; inclusion of a tertiary amine catalyst (such as triethylamine)16 led to no improvement in the yields of rearranged products obtained. Finally, it was found that rapid, sequential addition of 2.5 equiv KOtBu and 2.5 equiv MeOH to a THF solution of 4r at −85 °C led to the clean production of 6d in 95% yield.17 Repetition of this reaction with geraniol instead of methanol gave rise to γ,δ-unsaturated geranyl ester 6e in 77% yield.
Scheme 5.

Synthesis of γ,δ-unsaturated esters and amides
A logical mechanistic pathway (Scheme 6) for this process may involve hindered alkoxide-induced E2 elimination of triflate ion from A to produce the corresponding allyl alkynyl ether B, which undergoes [3,3]-sigmatropic rearrangement to furnish allyl ketene intermediate C. Nucleophilic trap of the ketene by the excess alkoxide base or other nucleophile present leads to enolate D, which is protonated by the conjugate acid of the base (t-BuOH, menthol, etc.) to furnish the γ,δ-unsaturated carboxylic acid derivative.
Scheme 6.

Mechanistic proposal for the formation of γ,δ-unsaturated carboxylic acid derivatives
In summary, we have developed an efficient procedure for the synthesis of a diverse range of alkynyl ethers from α-diazoketones and alcohols. We have also shown that allyl and benzyl alkynyl ethers undergo sigmatropic rearrangement and nucleophilic trapping to produce α-substituted ketones, esters, and amides. Further studies on this useful low-temperature sigmatropic rearrangement are in progress and will be reported in due course.
Supplementary Material
Detailed experimental procedures, spectroscopic data, and 1H and 13C NMR spectra for all compounds in Table 1 and Table 2 and Scheme 3–Scheme 5, as well as NOESY spectra for compounds 4a and 4h. This material is available free of charge via the Internet at http://pubs.acs.org
Acknowledgment
We thank the National Institutes of Health (SC2 GM081064-01), the ACS Petroleum Research Fund (No. PRF 45277-B1) and the Henry-Dreyfus Teacher-Scholar Award for their generous support of our research program. We also thank Mr. Shayan Rab (USC), Mr. Nick Vidar (CSUN), and Ms. Yen-Nhi Do Nguyen (CSUN) for preparing 5b, 5c, and 7a, respectively.
References
- 1.(a) Shindo M. Tetrahedron. 2007;63:10. doi: 10.1016/j.tet.2007.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Brandsma L, Bos HJ, Arens JF. In: The Chemistry of Acetylenes. Viehe HG, editor. New York: Marcel Dekker; 1969. p. 751. [Google Scholar]; (b) Stang PJ, Zhdankin VV. In: The Chemistry of Triple-Bonded Functional Groups. Patai S, editor. New York: John Wiley & Sons; 1994. chapter 19. [Google Scholar]
- 2.(a) For reviews of the chemistry of ynamines and ynamides, see:Zificsak CA, Mulder JA, Hsung RP, Rameshkumar C, Wei LL. Tetrahedron. 2001;57:7575.Mulder JA, Kurtz KCM, Hsung RP. Synlett. 2003;10:1379.Katritzky AR, Jiang R, Singh SK. Heterocycles. 2004;63:1455.(d) Tetra-hedron-Symposium-In-Print: Chemistry of Electron-Deficient Ynamines and YnamidesTetrahedron. 2006;62(Issue No 16)
- 3.(a) Bruckner D. Synlett. 2000;10:1402. [Google Scholar]; (b) Moyano A, Charbonnier F, Greene AE. J. Org. Chem. 1987;52:2919. [Google Scholar]; (c) Pericas MA, Serratosa F, Valenti E. Tetrahedron. 1987:2311. [Google Scholar]; (d) Smithers RH. Synthesis. 1985:556. [Google Scholar]; (e) Himbert G, Loffler A. Synthesis. 1992:495. [Google Scholar]
- 4.Christopher A, Brandes D, Kelly S, Minehan TG. Org. Lett. 2006;8:451. doi: 10.1021/ol052685j. [DOI] [PubMed] [Google Scholar]
- 5.(a) Brummond KM, Gesenberg KD, Kent JL, Kerekes AD. Tetrahedron Lett. 1998;39:8613. [Google Scholar]; (b) Negishi E, King AO, Tour JM. Org. Syn. 1990;Coll. Vol. 7:63. [Google Scholar]
- 6.Muthusamy S, Babu SA, Gunanathan C. Tetrahedron Lett. 2002;43:3133. [Google Scholar]
- 7.Danheiser RL, Miller RF, Brisbois RG, Park SZ. J. Org. Chem. 1990;55:1959. [Google Scholar]
- 8.This problem has been previously noted:Boyer JH, Selvarajan R. J. Org. Chem. 1971;36:1679.
- 9.Gibson HW, Berg MA, Clifton Dickson J, Lecavalier PR, Wang H, Merola JS. J. Org. Chem. 2007;72:5759. doi: 10.1021/jo070786k. [DOI] [PubMed] [Google Scholar]
- 10.Nahm S, Weinreb SM. Tetrahedron Lett. 1981;22:3815. [Google Scholar]
- 11.1-Ethynyl ethers have been previously prepared by base-induced elimination of enol phosphates derived from acetate esters:Cabezas JA, Oehlschlager AC. J. Org. Chem. 1994;59:7523.
- 12.Olsman H, Graveland A, Arens JF. Rec. Trav. Chim. Pays-Bas. 1964;83:301. [Google Scholar]
- 13.Wunderli A, Zsindely J, Hansen HJ, Schmid H. Chimia. 1972;26:643. [Google Scholar]
- 14.Typical reaction temperatures for sigmatropic rearrangement of allyl phenyl ether range from 170°(neat) to 220°C (in diphenyl ether). For mechanistic investigations of the thermal aromatic Claisen rearrangement, see:Meyer MP, DelMonte AJ, Singleton DA. J. Am. Chem. Soc. 1999;121:10865.Kupczyk-Subotkowska L, Subotkowski W, Saunders WH, Shine HJ. J. Am. Chem. Soc. 1992;114:3441.Gozzo FC, Fernandes SA, Rodrigues DC, Eberlin MN, Marsaioli AJ. J. Org. Chem. 2003;68:5493. doi: 10.1021/jo026385g.
- 15.Katzenellenbogen JA, Utawanit T. Tetrahedron Lett. 1975;16:3275. [Google Scholar]
- 16.Tertiary amines have been used extensively to catalyze the addition of alcohols to ketenes: seeLarsen RD, Corley EG, Davis P, Reider PJ, Grabowski EJJ. J. Am. Chem. Soc. 1989;111:7650.Cannizaro CE, Houk KN. J. Am. Chem. Soc. 2004;126:10992. doi: 10.1021/ja035899+.Pracejus H, Kohl G. Justus Liebigs Ann. Chem. 1969;722Hodous BL, Ruble JC, Fu GC. J. Am. Chem. Soc. 1999;121:2637.
- 17.At −85 °C, deprotonation of the enol triflate by KOt-Bu is rapid but nucleophilic addition of KOt-Bu to the intermediate ketene is slow. Thus, when methanol is added rapidly after the addition of KOt-Bu, the remaining alkoxide base (~1.5 equiv) rapidly deprotonates methanol to form potassium methoxide, which then adds to the ketene intermediate to form therresponding methyl ester enolate; upon protonation, product 6d results.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Detailed experimental procedures, spectroscopic data, and 1H and 13C NMR spectra for all compounds in Table 1 and Table 2 and Scheme 3–Scheme 5, as well as NOESY spectra for compounds 4a and 4h. This material is available free of charge via the Internet at http://pubs.acs.org