

Abstract
A 16-amino acid residue peptide derived from a consensus motif of natural ferredoxins incorporates a tetranuclear iron sulfur cluster under physiological conditions. Successful assembly of the [4Fe–4S]2+/1+ cluster within a monomeric peptide was demonstrated using size exclusion chromatography, UV-visible, visible CD, and cryogenic EPR spectroscopies. The robustness of [4Fe–4S]2+/1+ formation was tested using peptides with either the ligating cysteine exchanged for alanine or with the intervening amino acids replaced by glycine. The small size of the peptide allows for modular incorporation into more complex protein structures. In one larger structure, we describe a tetra-α-helix bundle that self-assembles both iron–sulfur clusters and hemes, thereby demonstrating feasibility for the general synthesis of maquettes containing multiple, juxtaposed redox cofactors. This is a motif common to the catalytic sites of native oxidoreductases.
The immense complexity inherent in enzyme structure presents a daunting challenge to understanding how proteins fold and assemble cofactors to establish catalytic fitness. The problem of protein folding and secondary structure design has recently been approached with the stratagem of synthesizing scaled-down polypeptides (1–7). Relatively simple design rules have been established for the construction of tetra-α-helix bundles using either a minimalist design approach (3) or a binary patterning strategy (8). We are using designed tetra-α-helix bundles to address the intricacies of protein–cofactor interactions in polypeptides that incorporate biochemical cofactors (9–12). It is our goal to establish the rules for cofactor incorporation and function in these simplified systems to produce minimal, functional, synthetic proteins—i.e., molecular maquettes (9).
We designed and synthesized a tetra-helix bundle, H10H24 (Fig. 1B), that incorporates one to four hemes via bis-His ligation (9). This simplified heme–protein maquette provides, under physiological conditions, for the study of heme–protein interactions, the basis for the variety of function in natural heme proteins. The spectroelectrochemical properties of the hemes in H10H24 are characteristic of those found in b-type cytochromes, as designed, including redox cooperativity due to heme–heme interactions.
Figure 1.
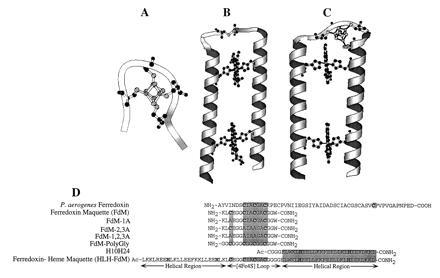
Modular design of the ferredoxin maquette. (A) molscript (13) model of the monomeric hexadecapeptide with bound [4Fe–4S]. (B) Molecular model of the prototype heme–protein maquette H10H24. (C) Computer representation of the modular design helix–loop–helix ferredoxin–heme maquette (HLH–FdM) with a [4Fe–4S] cluster and a pair of bound hemes. (D) Primary sequence alignment illustrating the modular peptide design approach. Given are the sequences for Peptococcus aerogenes Fd (from which the [4Fe–4S]-binding domain was extracted), the natural sequence ferredoxin maquette FdM, versions of the ferredoxin maquette with one, two, and three cysteines to alanine modifications, the glycine-modified ferredoxin maquette, and the HLH–FdM. All synthetic peptides were C-terminally amidated, and the HLH–FdM was N-terminally acetylated.
As widespread and biochemically diverse as the heme proteins, the ferredoxins and related iron sulfur proteins differ from heme proteins in that they display a multitude of cluster nuclearities and coordination geometries (14). The challenge evident in the synthesis of ferredoxin maquettes is to design a peptide that not only self-assembles iron and sulfur ions but also guides the formation of a single cluster architecture. The interaction of iron–sulfur clusters with thiolate ligands in organic solvents has been studied extensively, and, in fact, preassembled clusters have been incorporated into cysteine-containing peptides in dimethyl sulfoxide/water (80:20) (15). However, maquettes offer a tractable system in which to study the assembly of these biological cofactors in aqueous media to determine their delicate interplay with the surrounding heterogeneous chiral protein matrix, a major principle in the acquisition of biological specificity and activity. Additionally, the development of modular protein domains that can be spliced together to form multicofactor peptides offers a systematic approach to the design of more elaborate self-assembling biological structures.
Herein, we describe the design and synthesis of maquettes for ferredoxins containing multinuclear [4Fe–4S] clusters. Using a surprisingly short hexadecapeptide (16) [the creation of which was based on insight gained from inspection of natural proteins (17–22) and inorganic model complexes (23–29)], we have examined the incorporation of a tetra-nuclear iron–sulfur cluster into synthetic peptides under physiological conditions. By substituting the cysteine ligands with noncoordinating alanine residues and exchanging all of the intervening amino acids for glycine, while holding the cysteines in their consensus positions, we have investigated the contributions of each alteration to the assembly of the cluster.
Furthermore, strategies are presented to assemble both [4Fe–4S] and hemes in a tetra-α-helix bundle that demonstrate feasibility for the general synthesis of maquettes containing multiple, juxtaposed, redox cofactors such as hemes, iron–sulfur clusters, flavins, amino acid radicals, and quinones. These motifs are common to the catalytic sites of complex native oxidoreductases—e.g., the active site [4Fe–4S] cluster and siroheme of sulfite reductase (30).
MATERIALS AND METHODS
Chemicals and Solvents.
Iron (III) chloride, sodium sulfide, 2-mercaptoethanol, and trifluoroacetic acid were obtained from Aldrich. Ethanedithiol was purchased from Fluka. Fluorenylmethoxycarbonyl-protected amino acid pentafluorophenyl esters were purchased from PerSeptive Diagnostics (Cambridge, MA) except for fluorenylmethoxycarbonyl-l-Arg(2,2,5,7,8-pentamethyl chromian-6-sulphonyl)–pentafluorophenyl ester, which was obtained from Bachem. All other chemicals and solvents were reagent grade.
Peptide Synthesis.
The peptides were synthesized on a continuous flow MilliGen model 9050 solid phase synthesizer using standard fluorenylmethoxycarbonyl/But protection strategy. The side chain protecting groups used were as follows: histidine (tert-butoxycarbonyl); lysine (tert-butoxycarbonyl); glutamic acid (OBut); cysteine (trityl); arginine (Pmc). The N terminus of the HLH-FdM was acetylated [1:1 (vol/vol) acetic anhydride/pyridine for 30 min]. The peptides were cleaved from the resins and simultaneously deprotected using 90:8:2 trifluoroacetic acid/ethanedithiol/water for 2 h. Crude peptides were precipitated and triturated with cold ether, dissolved in water (0.1% trifluoroacetic acid), lyophilized, and purified to homogeneity by reversed phase C18 HPLC using aqueous–acetonitrile gradients containing 0.1% (vol/vol) trifluoroacetic acid. The peptides were homogeneous and of the correct composition as assayed by analytical HPLC and laser desorption mass spectrometry.
Solution Molecular Mass Determination.
Gel permeation chromatography was performed anaerobically in a Plas-Labs (Lansing, MI) glove box using a Beckman System Gold HPLC with diode array detector fitted with a Superdex 30 column (Pharmacia) eluted at 2 ml/min. The eluting buffer (10 mM KPi/100 mM KCl, pH 8.0) was deoxygenated using successive freeze–pump–thaw cycles. The column was standardized using aprotinin (6.5 kDa), glutathione (0.4 kDa), apo-FdM-1,2,3A (1.4 kDa), and the homodimer of FdM-1,2,3A (2.8 kDa).
[4Fe–4S]2+/1+ Incorporation.
The [4Fe–4S] cluster was incorporated into apopeptide using modified literature methodologies (31–32). Under strictly anaerobic conditions, a solution of 2% (vol/vol) 2-mercaptoethanol in 50 mM Hepes buffer (pH 8) containing 20 μM peptide was equilibrated for 2 h to reduce any adventitious disulfides. A 50-mM solution of FeCl3 was slowly added to a final concentration of 120 μM before addition of a 50-mM solution of Na2S to a total concentration of 120 μM. The order of addition of ferric chloride and sodium sulfide was inconsequential to the yield of the reaction.
UV-Visible (UV-Vis) Spectroscopy.
UV-vis spectra were recorded on a Perkin–Elmer Lambda 2 spectrophotometer using quartz cells of 0.2- and 1.0-cm path lengths. Peptide concentrations were between 3 and 5 μM as determined spectrophotometrically using ɛ280 = 5600 M−1·cm−1 for Trp.
CD Spectropolarimetry.
CD spectra were recorded on an Aviv Associates (Lakewood, NJ) model 62DS spectropolarimeter using rectangular quartz cells of 0.2- and 1.0-cm path lengths. Thermal control was maintained by a thermoelectric module with a Neslab Instruments (Portsmouth, NH) CFT-33 refrigerated recirculating water bath as a heat sink. Peptide concentrations for UV-CD experiments were between 5 and 10 μM as determined spectrophotometrically using ɛ280 = 5600 M−1·cm−1 for Trp. The hexadecapeptide was a random coil, and the FdM was helical (60%) as determined using CD spectrometry.
EPR Spectroscopy.
EPR spectroscopy was performed using a Bruker (Billerica, MA) model ESP300E spectrometer. Temperature control was maintained by an Oxford ESR model 900 continuous flow cryostat interfaced with an Oxford model ITC4 temperature controller. Frequency was measured by a Hewlett–Packard model 5350B frequency counter. Typical EPR parameters included: sample temperature, 10K; microwave frequency, 9.449 GHz; microwave power, 1 mW; modulation frequency, 100 kHz; modulation amplitude, 20.0 G; and time constant, 164 ms. Unless otherwise noted, protein concentrations were 20 μM.
Redox Potentiometry.
Chemical redox titrations were performed in a cuvette within an inert atmosphere glove box with platinum working and calomel reference electrodes (33). Ambient redox potentials (measured against the standard hydrogen electrode) were adjusted by addition of aliquots (<1 μl) of sodium dithionite or potassium ferricyanide. Titrations were performed in 50 mM Hepes (pH 8.0) and 100 mM KCl. Electrode-solution mediation was facilitated by the following mediators at a 10-μM concentration: neutral red, 2-hydroxy-1,4-naphthoquinone, safranine T, and phenosafranine. After equilibration at each potential, a 300-μl sample was removed and frozen in liquid nitrogen, and the EPR spectrum was recorded. Each spectrum is presented as the difference between an oxidized reference spectrum (−200 mV) and the reduced spectrum. The doubly integrated spectral area was plotted against potential, and the data were fit to the Nernst equation with n = 1.0 (fixed), as shown in the Inset to Fig. 5C for HLH–FdM.
Figure 5.
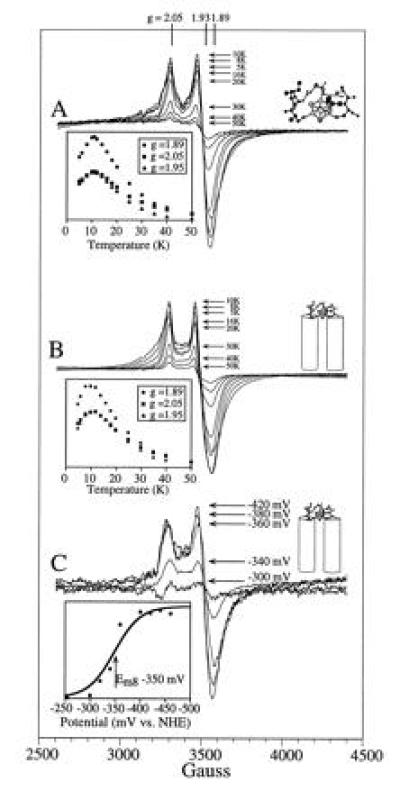
EPR characterization of the ferredoxin maquette. (A) Temperature dependence of the hexadecapeptide [4Fe–4S] EPR signal. Under the conditions used—i.e., 1 mW of power—the absolute value of the signal intensities at the signal extrema (g = 2.05, 1.95, and 1.89) are as shown in the Inset. (B) Temperature dependence of the [4Fe–4S] EPR signal from the ferredoxin–heme maquette. Signal intensities are as shown in the inset. The observed behavior of the [4Fe–4S] in the HLH–FdM is identical to that of the hexadecapeptide shown in A. (C) Redox potentiometry of the HLH–FdM. EPR redox titration of the [4Fe–4S]2+/1+ couple is shown. The determined reduction potential of −350 mV vs. normal hydrogen electrode (Inset) is typical of native [4Fe–4S] ferredoxins.
RESULTS
Design of FdM.
Insight into the design of ferredoxin maquettes was gained from structures and consensus motifs of natural proteins and from model compounds drawn from coordination chemistry (17–29). P. aerogenes ferredoxin (17) offered two crystallographically characterized [4Fe–4S]2+,1+ binding domains, and a set of closely related variants in other species provided the primary consensus sequence -Cys-(Xaa)2-Cys-(Xaa)2-Cys-(Xaa)10–50-Cys. We used the natural sequence provided by P. aerogenes, -Cys-Ile-Ala-Cys-Gly-Ala-Cys-, for three of the cysteines, as shown in Fig. 1D. The N-terminal sequence that includes the fourth cysteine was designed for future accommodation into a HLH–FdM. In contrast, this fourth cysteine in natural ferredoxins is normally located toward the C terminus and is followed by a proline (14). A C-terminal tryptophan placed at the end of a glycine linker served as a spectroscopic tag. The resulting sequence for the FdM was NH2-KLCEGGCIACGACGGW-CONH2.
Design of HLH–FdM.
As indicated above, the natural sequence FdM was designed to lend itself to integration into larger structures as a basis for the construction of more sophisticated multicofactor maquettes. Using helices consistent with the prototype heme–peptide maquette H10H24, a tetra-helix bundle that binds one to four hemes (9), we designed a [4Fe–4S]–heme maquette, HLH–FdM. The peptide HLH–FdM, Ac-L KKLREEH LKLLEEF KKLLEEH LKLCE GGCIACGACGGGG ELWKL HEELLKK FEELLKL HEERLKK L-CONH2, is designed to contain two helical regions of ≈27 amino acids, each joined by the FdM sequence that serves to terminate the α helices and to ligate a [4Fe–4S] cluster in a rudimentary turn. The helices are comprised of minor variations of four coiled-coil heptad repeats of [Leua-Glub-Gluc-Leud-(Leu)e-Lysf-(Lys/Leu)g]4 (3). By analogy to H10H24, His were placed in four heptad a positions (positions 8, 22, 46, and 60) in HLH–FdM for the purposes of binding hemes. Phenylalanine residues were placed at the center of each helical region (a positions 15 and 53) to separate the two heme-binding sites, and a pair of Arg were placed at d positions 5 and 63 to modulate the redox potential of the heme in the H8,H60-binding site. Additionally, a single tryptophan was located at the end of the C-terminal helix, position 43, as a convenient spectroscopic marker. The N-terminal Cys–Cys linkage of the earlier H10H24 design, expected to interfere with [4Fe–4S] incorporation, was replaced by the ferredoxin–maquette sequence, resulting in the 67-amino acid HLH monomer HLH–FdM. Thus, the resulting HLH–FdM peptide contains a hydrophobic core analogous to that of H10H24 with a [4Fe–4S]-binding domain between the helices.
UV-Vis Spectroscopy.
In aqueous buffer, the UV-vis spectra of the oxidized FdM and HLH–FdM have broad maxima at 385 and 392 nm, respectively, as shown in Fig. 2A. These spectra are similar to those observed for P. aerogenes ferredoxin (34) and are consistent with incorporation of a [4Fe–4S] cluster. Reduction with dithionite yielded virtually featureless visible spectra, shown in Fig. 2A, for both FdM and HLH–FdM.
Figure 2.
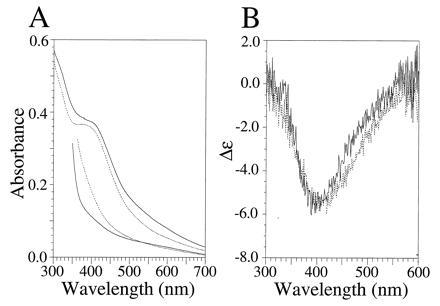
UV-vis and visible CD characterization of the [4Fe–4S] ferredoxin maquettes FdM and HLH–FdM. (A) UV-vis spectra of oxidized (upper traces) and dithionite-reduced (lower traces) [4Fe–4S] ferredoxin maquette (broken line) and HLH [4Fe–4S] ferredoxin maquette (solid line). Oxidized and reduced spectra were obtained from samples with equivalent peptide concentrations. The rising absorbance below 400 nm is due to dithionite. (B) Visible CD spectrum of the oxidized FdM (broken line) and HLH–FdM (solid line). All spectra contained 33 μM peptide in an anaerobic cuvette of 1.0-cm path length (10 mM KPi and 100 mM KCl, pH 8.0).
CD Studies.
In aqueous buffer, the natural sequence ferredoxin–maquette FdM has a UV-CD spectrum typical of a random coil peptide with no discernible secondary structure. In contrast, the HLH–FdM possesses a CD spectrum characteristic of highly α-helical peptides, with minima at 208 and 222 nm clearly demonstrating that addition of the ferredoxin loop results in no significant alteration in the secondary structure of the helices. In degassed aqueous buffer (10 mM KPi/100 mM KCl, pH 8.0), the Θ222/Θ208 ratio is ≥1.0, which is diagnostic of coiled coil structure (35, 36). After incorporation of the [4Fe–4S] cluster, the visible CD spectrum of each peptide was taken to aid in evaluation of the nuclearity of the incorporated cluster. As shown in Fig. 2B, both FdM and HLH–FdM display visible CD spectra with negative Cotton effects at 410 and 405 nm, respectively, illustrating that the cluster resides in a chiral environment. The lack of a positive Cotton effect in this spectral region is consistent with a [4Fe–4S] cluster and is quite distinct from the Cotton effect of the vis-CD spectra typically observed for [2Fe–2S] clusters (37).
Gel Permeation Chromatography.
The aggregation state of FdM and HLH–FdM were evaluated using gel permeation chromatography. The ferredoxin–maquette FdM eluted as a singular species with an apparent molecular mass of 1.6 kDa, as shown in Fig. 3, trace A. Size exclusion chromatography demonstrated that the reconstituted peptide was monomeric and provided an optical spectrum consistent with [4Fe–4S] incorporation. The peptide containing two cysteines, FdM-2,3A, eluted as two components (Fig. 3, trace B) with apparent molecular masses consistent with a monomer (1.8 kDa) and a dimer (3.5 kDa), indicating formation of a [4Fe–4S] cluster from two peptides in low yield. The HLH–FdM eluted with an apparent molecular mass of 19.6 kDa based on a column standardized with globular proteins. This peptide coeluted with H10H24, a known tetra-helix bundle (9), indicating that the related proteins have similar hydrodynamic radii.
Figure 3.
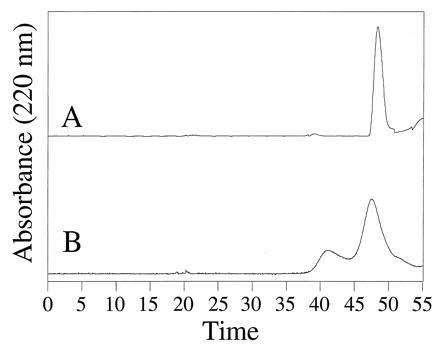
Size exclusion characterization of the ferredoxin–maquette aggregation state. The chromatograms of FdM (trace A) and FdM-2,3A (trace B) illustrate the incorporation of [4Fe–4S] into monomeric peptide for FdM and dimeric FdM-2,3A.
EPR of FdM.
Direct evidence for the successful assembly of a [4Fe–4S]2+/1+ cluster within monomeric peptide, FdM, was obtained by cryogenic EPR spectroscopy (Fig. 4, trace A). As isolated, the material displays a weak g = 2 EPR signal that is sometimes observed in bacterial ferredoxin preparations and is perhaps indicative of a minor component of an oxidized [3Fe–4S]1+ cluster. Reduction of the sample with sodium dithionite yielded a rhombic EPR spectrum (Fig. 4, trace A) with resonances at g = 2.05, 1.93, and 1.89, signifying a paramagnetic (S = 1/2) [4Fe–4S]1+ cluster (29). EPR signals characteristic of a S = 3/2 [4Fe–4S]1+ cluster were not observed under various conditions of peptide concentration, microwave power, and temperature. Detailed examination of the (S = 1/2) [4Fe–4S]1+ cluster signal revealed a microwave power half-saturation of 20 mW at 10 K, a value typical of those observed for the exchange-coupled multinuclear clusters in natural [4Fe–4S] ferredoxins. Variable temperature studies at 1-mW microwave power provided the signal heights shown in the Inset to Fig. 5A, demonstrating saturation of the signal below 11 K. Spin quantitation of the reduced state indicated >60% [4Fe–4S] incorporation into the peptide. Additionally, EPR spectroscopy under various oxidation-reduction conditions provided no evidence for incorporation of mononuclear, binuclear [2Fe–2S], or high potential iron protein-type [4Fe–4S]3+/2+ clusters and only a minor amount of the trinuclear [3Fe–4S] cluster. Therefore, these data indicate high specificity for the [4Fe–4S]2+/1+ cluster. Clearly, the free energy of formation of the tetra-nuclear cluster from iron and sulfur ions is sufficient to drive the assembly assisted by a single, surprisingly short peptide. Thus, assembly and establishment of spectral and electrochemical properties of the [4Fe–4S] ferredoxin in an aqueous environment requires, to a first approximation, 16 residues or less.
Figure 4.
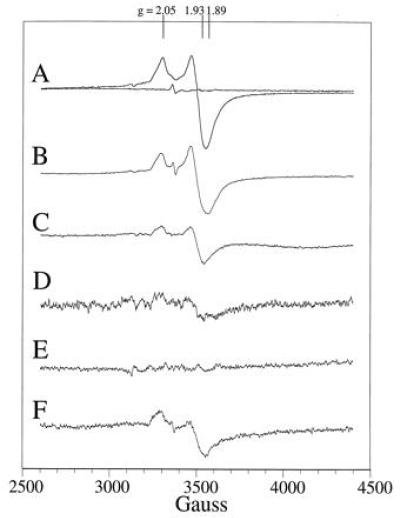
EPR characterization of the ferredoxin maquettes. (Trace A) EPR spectrum of the oxidized and reduced ferredoxin maquette at 10 K. (Trace B) EPR spectrum-modified ferredoxin maquette with one cysteine substituted for an alanine (FdM-1A) after [4Fe–4S] incorporation and dithionite reduction. (Trace C) EPR spectrum of the dithionite-reduced [4Fe–4S] ferredoxin maquette (FdM-2,3A) with two cysteine residues replaced by alanine at 10 K. (Trace D) EPR spectrum at 10-fold amplification of the modified ferredoxin maquette with three cysteines substituted for alanine (FdM-1,2,3A) after [4Fe–4S] incorporation and dithionite reduction. (Trace E) EPR spectrum of l-cysteine at 10 K after attempted [4Fe–4S] incorporation and dithionite reduction. (Trace F) Dithionite-reduced EPR spectrum of 80 μM glycine-containing ferredoxin maquette at 10 K at 5-fold magnification.
EPR of FdM Variants.
Some contributions from the cysteines and the intervening amino acids to the assembly of this ferredoxin maquette were obtained from simple comparisons of the relative yield of the [4Fe–4S] assembly in FdM variants (Fig. 1D). Replacement of a single cysteine with an alanine (FdM-1A in Figs. 1 D and 4, trace B) results in cluster formation at only slightly lower yield. Hydroxide ion or 2-mercaptoethanol, both present in the reaction media, are possible exogenous ligands for this cluster. Exchange of two cysteines ligands for alanine to force assembly from two separate hexadecapeptides lowers the relative yield to 35% (FdM-2,3A in Figs. 1D and 4, trace C), roughly the amount of dimer peptide observed in the size exclusion experiment. Removal of three of the cysteines to force assembly from three or four peptides results in almost total loss of yield (Fig. 4, trace D) as does the use of l-cysteine alone (Fig. 4, trace E). Likewise, exchanging all of the amino acids for glycine while holding the cysteines in their consensus positions leads to a less than 10% yield (FdM-PolyGly in Figs. 1D and 4, trace F), underscoring the importance of the intervening amino acids on cluster assembly.
EPR of HLH–FdM.
Fig. 5B shows that the ferredoxin maquette FdM can be constrained to the tetra-α-helix bundle and still assemble the [4Fe–4S] cluster without change in EPR spectral characteristics. The EPR spectral properties (line shape and position) of the [4Fe–4S]1+ bound to HLH–FdM, shown in Fig. 5B, are indistinguishable from those of the hexadecapeptide FdM. Fig. 5C demonstrates that the measured redox potential (Em8 = −350 mV) is comparable to that of native ferredoxins (14, 34). Straightforward addition of hemin [Fe(protoporphyrin IX)Cl] to HLH–FdM that includes the [4Fe–4S] cluster leads to rapid and complete incorporation of axially ligated bis-His heme. Additionally, heme binding results in no significant alteration of the EPR properties and hence disruption of the [4Fe–4S] cluster in the loop region. Of interest, addition of hemin before [4Fe–4S] incorporation initially results in absorption spectra consistent with thiolate ligation to the ferric hemes (λmax at 358, 418, 539, and 565 nm) (38), which slowly convert into those typical of the bis-His heme (λmax at 413 and 535 nm). These data demonstrate the incorporation of hemes into the bis-His binding sites within the [4Fe–4S] peptide and confirm the incorporation of the iron–sulfur cluster.
DISCUSSION
Using inspiration gleaned from structural biology and inorganic coordination chemistry, we have investigated the feasibility of [4Fe–4S] cluster incorporation into relatively simple polypeptides containing cysteine under physiological conditions. As designed, the 16-amino acid natural ferredoxin maquette FdM self-assembles a [4Fe–4S]2+/1+ cluster into a monomeric peptide at micromolar concentrations under physiological conditions to provide a synthetic ferredoxin. When this modular [4Fe–4S]-binding peptide was incorporated into a 67-amino acid HLH peptide, a tetra-helix bundle was formed that bound both heme and the [4Fe–4S] cluster. This provides the first example of incorporation of two distinct redox cofactors into a single designed protein, a requisite step in the fabrication of functional complex synthetic oxidoreductases.
Biochemical self-assembly reactions of ferric chloride and sodium sulfide in the presence of 2-mercaptoethanol were used to incorporate the [4Fe–4S] clusters into the peptides studied in aqueous solution under fastidiously anaerobic conditions. The identity of the cluster incorporated into the natural sequence ferredoxin maquette FdM was determined by a combination of EPR, UV-visible and visible-CD spectroscopies. Although the observed g values (gav. < 2 [gav. = gx + gy + gz)3 = average g value] of the EPR resonances from the dithionite-reduced FdM cannot be used alone to discriminate between a (S = 1/2) [4Fe–4S]1+ and a (S = 1/2) [2Fe–2S]1+ cluster, the relaxation properties are most consistent with a tetra-nuclear cluster. The absorption spectra of the oxidized clusters are also consistent with a tetra-nuclear cluster and are similar to those observed for P. aerogenes ferredoxin (34). The lack of a positive Cotton effect in the visible-CD further indicates that the bound cluster is tetra-nuclear and resides in a chiral environment. Furthermore, UV-CD spectropolarimetry illustrates that the hexadecapeptide FdM contains no discernible structure in the absence of bound [4Fe–4S], implying that the robust nature of [4Fe–4S] self-assembly drives the peptide folding.
The simplicity of the natural sequence ferredoxin maquette FdM allows for investigations into the roles that each ligand and the intervening amino acids play in biochemical [4Fe–4S] cluster assembly under physiological conditions. A peptide containing only three cysteine ligands (FdM-1A) is still capable of assembling the [4Fe–4S] cluster, only in slightly lower yield, as illustrated in Fig. 4, trace B. In this case, the identity of the exogenous ligand could be 2-mercaptoethanol or hydroxide ion [which is sometimes observed in natural [4Fe–4S] proteins (39)] or perhaps could be another peptide. When the number of cysteine residues per peptide is lowered to two, assembly of the [4Fe–4S] cluster from a pair of peptides results in reduced yield, as shown in Figs. 3, trace B, and 4, trace C. Replacement of three of the cysteines (Fig. 4, trace D) or use of l-cysteine alone (Fig. 4, trace E) (which forces assembly from four separate ligands) results in almost total loss of cluster formation. The intervening amino acids play a significant role in cluster assembly, as indicated in Fig. 4, trace F, where all of the amino acids were exchanged for glycine while holding the cysteines in their consensus positions, leading to a <10% yield. Similar glycine-containing peptides incorporate prefabricated iron–sulfur clusters in organic solvents in high yield (15), but, in aqueous solution, the [4Fe–4S] cluster assembly by FdM-PolyGly is very poor. The observed differences between aqueous and organic solvents illustrate one of the advantages of using maquettes to probe cofactor and protein assembly.
The small size and robustness of the natural sequence ferredoxin maquette FdM lend it to integration into larger structures as a basis for the construction of more sophisticated multicofactor maquettes. The one suggested in Fig. 1 C and D has the hexadecapeptide constrained as a loop in a HLH structure established to assemble into a tetra-α-helix bundle. Additionally, the helices in this architecture are equipped with His to ligate hemes. Fig. 5 B and C show that the hexadecapeptide can be constrained on the tetra-α-helix bundle and still assemble the [4Fe–4S] cluster without change in EPR spectral properties with a redox potential (Em8 = −350 mV) comparable to native [4Fe–4S] ferredoxins (14). The similarities between the spectroelectrochemical properties of the [4Fe–4S] cluster bound to FdM and the larger HLH–FdM indicate that they reside in similar environments. In particular, the redox potential of each suggests a high degree of solvent exposure. This is consistent with the observation that natural [4Fe–4S] clusters with low potentials are often sequestered from solvent (less than −500 mV in PsaC of photosystem I; see ref. 31).
The modular design of multicofactor maquettes is illustrated by straightforward addition of hemin (not shown) to HLH–FdM (including the [4Fe–4S] cluster), which leads to rapid and complete incorporation of axially ligated bis-His heme [Fe (III) protoporphyrin IX]. The lack of alteration of the EPR properties of the [4Fe–4S] cluster in the loop indicates that the incorporation of the hemes into the helical regions does not adversely affect the [4Fe–4S] cluster. In the absence of bound [4Fe–4S], the kinetically driven formation of a thiolate-ligated heme gradually exchanges into the thermodynamically favored bis-His-ligated product. The ligation of a [4Fe–4S] cluster with cysteine and the incorporation of hemes into the bis-His-binding sites in a single peptide provides the initial example of using differential ligation motifs to incorporate distinct cofactors in a designed protein.
Although the detailed overall structure of the [4Fe–4S]–heme maquette remains to be established (see suggestions in Fig. 6), we can be certain that remarkably straightforward tactics are available for the synthesis of multicofactor proteins. This assertion is based on the combination of both high specificity and favorable free energy derived from the intrinsic coordination chemistry of the two distinct iron cofactors within the selected polypeptides. Under physiological conditions, the enthalpy of metal ligand interactions clearly overwhelms the opposing entropic demands attendant with cluster assembly into minimal polypeptides at micromolar concentrations. Indeed, the cusps of assembly failure are only approached after substantial enhancement of entropy by either dividing the cysteine between two hexadecapeptides or exchanging the consensus sequence for glycine. The robustness of [4Fe–4S] cluster self-assembly suggests that it plays structural roles not only in protein assembly but also in driving the formation of catalytically fit sites in proteins. These roles may go back to the early, anoxygenic Earth, where an abundance of iron and sulfur is thought to have interacted with cysteine in short peptides (40).
Figure 6.
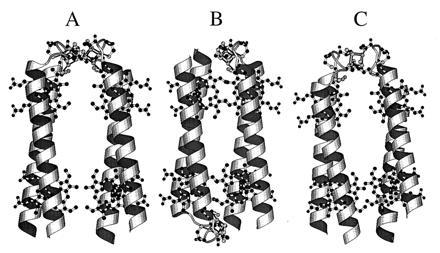
Proposed models for the HLH–FdM. (A) molscript model of a possible structure for the HLH–FdM with a pair of [4Fe–4S] clusters bound between parallel HLH. (B) Molecular model of a possible structure for the HLH–FdM with a pair of [4Fe–4S] clusters bound between a pair of antiparallel-oriented, HLH peptides. The distance between the two [4Fe–4S] clusters is in the range of 7–15 Å. (C) Computer model of a possible structure for the HLH–FdM with a single [4Fe–4S] cluster bound between a pair of parallel HLH–FdM peptides. The absence of paramagnetic interaction between the iron–sulfur clusters in HLH–FdM suggests that the model in A in unlikely.
Acknowledgments
We thank Dr. Kim Sharp for aid in preparing Figs. 1 and 6. This work was supported by grants from the National Institutes of Health (GM 27309 and GM 41048). B.R.G. and F.R. acknowledge receipt of postdoctoral fellowships from the National Institutes of Health and the European Molecular Biology Organization, respectively. Mass spectroscopic analyses were performed by the Protein Chemistry Laboratory of the University of Pennsylvania.
Footnotes
Abbreviations: HLH–FdM, helix–loop–helix ferredoxin–heme maquette; UV-vis, UV-visible.
References
- 1.DeGrado W F, Wasserman Z R, Lear J D. Science. 1989;243:622–628. doi: 10.1126/science.2464850. [DOI] [PubMed] [Google Scholar]
- 2.Handel T M, Williams S A, DeGrado W F. Science. 1990;261:879–885. doi: 10.1126/science.8346440. [DOI] [PubMed] [Google Scholar]
- 3.Bryson J W, Betz S F, Lu H S, Suich D J, Zhou H X, O’Neil K T, DeGrado W F. Science. 1995;270:935–941. doi: 10.1126/science.270.5238.935. [DOI] [PubMed] [Google Scholar]
- 4.Ghadiri M R, Granja J R, Buehler L K. Nature (London) 1994;369:301–304. doi: 10.1038/369301a0. [DOI] [PubMed] [Google Scholar]
- 5.Ghadiri M R, Choi C. J Am Chem Soc. 1990;112:1630–1632. [Google Scholar]
- 6.Struthers M D, Cheng R P, Imperiali B. Science. 1996;271:342–344. doi: 10.1126/science.271.5247.342. [DOI] [PubMed] [Google Scholar]
- 7.Hecht M H, Richardson J S, Richardson D C. Science. 1990;261:884–891. doi: 10.1126/science.2392678. [DOI] [PubMed] [Google Scholar]
- 8.Kamtekar S, Schiffer J M, Xiong H, Babik J M, Hecht M H. Science. 1993;262:1680. doi: 10.1126/science.8259512. [DOI] [PubMed] [Google Scholar]
- 9.Robertson D E, Farid R S, Moser C C, Urbauer J L, Mulholland S E, Pidikiti R, Lear J D, Wand A J, DeGrado W F, Dutton P L. Nature (London) 1994;368:425–32. doi: 10.1038/368425a0. [DOI] [PubMed] [Google Scholar]
- 10.Choma C T, Lear J D, Nelson M J, Dutton P L, Robertson D E, DeGrado W F. J Am Chem Soc. 1994;116:856–865. [Google Scholar]
- 11.Rabanal F, DeGrado W F, Dutton P L. J Am Chem Soc. 1996;118:473–474. [Google Scholar]
- 12.Kalsbeck W A, Robertson D E, Pandey R K, Smith K M, Dutton P L, Bocian D F. Biochemistry. 1996;35:3429–3438. doi: 10.1021/bi952662k. [DOI] [PubMed] [Google Scholar]
- 13.Kraulis P. J Appl Crystallogr. 1991;24:946–950. [Google Scholar]
- 14.Cammack R. In: Advances in Inorganic Chemistry. Sykes G, editor. San Diego: Academic; 1992. pp. 1–322. [Google Scholar]
- 15.Que L, Jr, Anglin J R, Bobrik M A, Davison A, Holm R H. J Am Chem Soc. 1974;96:6042–6048. doi: 10.1021/ja00826a014. [DOI] [PubMed] [Google Scholar]
- 16.Fraústo da Silva J J R, Williams R J P. The Biological Chemistry of the Elements: The Inorganic Chemistry of Life. New York: Oxford Univ. Press; 1991. [Google Scholar]
- 17.Adman E T, Sieker L C, Jensen L H. J Biol Chem. 1973;248:3987–3996. [PubMed] [Google Scholar]
- 18.Beinert H. FASEB J. 1990;4:2483–2491. doi: 10.1096/fasebj.4.8.2185975. [DOI] [PubMed] [Google Scholar]
- 19.Johnson M K. In: Encyclopedia of Inorganic Chemistry. King R B, editor. Vol. 4. Chichester, U.K.: Wiley; 1994. pp. 1896–1915. [Google Scholar]
- 20.Finnegan M G, Conover R C, Park J-B, Zhou Z H, Adams M W W, Johnson M K. Inorg Chem. 1995;34:5358–5369. [Google Scholar]
- 21.Houseman A L, Oh B H, Kennedy M C, Fan C, Werst M M, Beinert H, Markley J L, Hoffman B M. Biochemistry. 1992;31:2073–80. doi: 10.1021/bi00122a026. [DOI] [PubMed] [Google Scholar]
- 22.Lauble H, Kennedy M C, Beinert H, Stout C D. J Mol Biol. 1994;237:437–51. doi: 10.1006/jmbi.1994.1246. [DOI] [PubMed] [Google Scholar]
- 23.Ibers J A, Holm R H. Science. 1980;209:223–235. doi: 10.1126/science.7384796. [DOI] [PubMed] [Google Scholar]
- 24.Holm R H, Ciurli S, Weigel J A. Prog Inorg Chem. 1990;38:1–74. [Google Scholar]
- 25.Holm R H. Adv Inorg Chem. 1992;38:1–71. [Google Scholar]
- 26.Laskowski E J, Frankel R B, Gillum W O, Papaefthymiou G C, Renaud J, Ibers J A, Holm R H. J Am Chem Soc. 1978;100:5322–5337. [Google Scholar]
- 27.Coucouvanis D. Acc Chem Res. 1991;24:1–8. [Google Scholar]
- 28.Kanatzidis M G, Hagen W R, Dunham W R, Lester R K, Coucouvanis D. J Am Chem Soc. 1985;107:953–961. [Google Scholar]
- 29.Bertini I, Ciurli S, Luchinat C. Struct Bonding (Berlin) 1995;30:1–53. [Google Scholar]
- 30.Crane B R, Siegel L M, Getzoff E D. Science. 1995;270:59–65. doi: 10.1126/science.270.5233.59. [DOI] [PubMed] [Google Scholar]
- 31.Zhao J, Li N, Warren P V, Golbeck J H, Bryant D A. Biochemistry. 1992;31:5093–5099. doi: 10.1021/bi00137a001. [DOI] [PubMed] [Google Scholar]
- 32.Yu L, Zhao J, Lu W, Bryant D A, Golbeck J H. Biochemistry. 1993;32:8251–8258. doi: 10.1021/bi00083a028. [DOI] [PubMed] [Google Scholar]
- 33.Dutton P L. Methods Enzymol. 1978;54:411–435. doi: 10.1016/s0076-6879(78)54026-3. [DOI] [PubMed] [Google Scholar]
- 34.Stombaugh N A, Sundquist J E, Burris R H, Orme-Johnson W H. Biochemistry. 1976;15:2633–2638. doi: 10.1021/bi00657a024. [DOI] [PubMed] [Google Scholar]
- 35.Zhou N E, Kay C M, Hodges R S. J Biol Chem. 1992;267:2664–2670. [PubMed] [Google Scholar]
- 36.Zhou N E, Kay C M, Sykes B D, Hodges R S. Biochemistry. 1993;32:6190–6197. doi: 10.1021/bi00075a011. [DOI] [PubMed] [Google Scholar]
- 37.Stephens P J, Thomson A J, Dunn J B R, Keiderling T A, Rawlins J, Rao K K, Hall D O. Biochemistry. 1978;17:4770–4778. doi: 10.1021/bi00615a026. [DOI] [PubMed] [Google Scholar]
- 38.Dawson J H, Sono M. Chem Rev. 1987;87:1255–1276. [Google Scholar]
- 39.Robbins A H, Stout C D. Proc Natl Acad Sci USA. 1989;86:3639–3643. doi: 10.1073/pnas.86.10.3639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Blöchl E, Keller M, Wächtershäuser G, Stetter K O. Proc Natl Acad Sci USA. 1992;89:8117–8120. doi: 10.1073/pnas.89.17.8117. [DOI] [PMC free article] [PubMed] [Google Scholar]