

Abstract
Corticotropin-releasing hormone (CRH) is a key regulator of the mammalian stress response, mediating a wide variety of stress-associated behaviors including stress-induced inhibition of reproductive function. To investigate the potential direct action of CRH on pituitary gonadotrope function, we examined CRH receptor expression and second messenger signaling in αT3-1 cells, a murine gonadotrope-like cell line. Reverse transcriptase-polymerase chain reaction (RT-PCR) studies demonstrate that αT3-1 cells express mRNA for the two CRH receptor subtypes, CRH-R1 and CRH-R2, with CRH-R2α as the predominant CRH-R2 isoform. Stimulation of the cells with CRH or urocortin (UCN1) results in rapid, transient increases in the intracellular levels of cAMP that are completely blocked by addition of α-helical CRH 9-41 or astressin, non-selective CRH receptor antagonists. Stimulation of the cells with CRH-R2 specific ligands, urocortin II (UCN2) or urocortin III (UCN3), results in rapid increases in intracellular cAMP levels to 50–60% of the levels observed with UCN1. Treatment with a selective CRH-R2 antagonist, antisauvagine, completely blocks UCN3-mediated increases in cAMP and significantly reduces, but does not completely block UCN1-mediated increases in cAMP, demonstrating that both CRH-R1 and CRH-R2 are functionally active in these gonadotrope-like cells. Finally, UCN treatment significantly increases the transcriptional activity of the glycoprotein hormone α-subunit promoter as assessed by α-luciferase transfection assays. Together, these results demonstrate the functional signaling of CRH receptors in αT3-1 cells, suggesting that CRH may also modulate pituitary gonadotrope function in vivo.
Keywords: CRH, CRF, receptors, gonadotrope, cAMP
Introduction
Corticotropin-releasing hormone (CRH) is a key regulator of the endocrine, behavioral, and autonomic components of the mammalian stress response. Within the endocrine hypothalamic-pituitary-adrenal (HPA) axis, this 41 amino acid peptide is the key mediator of ACTH secretion. Hypothalamic CRH is released in response to stressful stimuli and carried to the anterior pituitary where it activates CRH receptors on anterior pituitary corticotropes, resulting in increased ACTH synthesis and secretion. ACTH acts on the adrenal cortex to increase glucocorticoid release; glucocorticoids then mediate many of the metabolic changes required to respond to the stressor. CRH is also expressed in many other sites in the CNS where it is thought to act as a neurotransmitter to mediate changes in a wide variety of stress-associated behaviors. For example, CRH has been implicated in stress-induced alterations in anxiety-like behavior, feeding behavior, and locomotor activity (Dunn and Berridge 1990; Owens and Nemeroff 1991).
CRH has also been shown to play an important role in stress-induced inhibition of reproductive function. Intracerebroventricular (icv) injection of CRH suppresses the activity of the hypothalamic-pituitary-gonadal (HPG) axis in rats, resulting in decreased gonadotropin- releasing hormone (GnRH) and luteinizing hormone (LH) release (reviewed in (Rivest and Rivier 1995)). Similarly, the repression of pulsatile LH secretion by restraint stress, lipopolysaccharide, insulin-induced hypoglycemia, or fasting can be reversed by icv injection of CRH receptor antagonists (Li, et al. 2006; Li, et al. 2005; Maeda, et al. 1994), suggesting that CRH receptor ligands mediate inhibitory effects of stress on the HPG axis via central pathways (reviewed in (Rivest and Rivier 1995)). While studies suggest that systemic administration of CRH does not alter LH secretion in rats (Rivier and Vale 1984), CRH treatment of primary rat pituitary cultures decreased basal LH secretion (Blank, et al. 1986). Numerous other hypothalamic peptides have also been shown to modulate gonadotropin levels by acting at the pituitary (reviewed in (Evans 1999)). Thus, CRH and other members of the CRH family of peptides, urocortin (UCN1), urocortin II (UCN2) and urocortin III (UCN3), may participate in the stress-mediated regulation of reproduction via multiple mechanisms and sites of action.
CRH and the urocortins mediate their effects via two distinct CRH receptors: CRH-R1 and CRH-R2 (reviewed in (Bale and Vale 2004; Dautzenberg and Hauger 2002; Grammatopoulos and Chrousos 2002; Hillhouse and Grammatopoulos 2006)). The receptors are approximately 70% identical at the amino acid level and are both members of the class B subfamily of seven transmembrane domain G-protein coupled receptors. In most cell types, activation of CRH-R1 or CRH-R2 by CRH or urocortins results in activation of the Gs-adenylyl cyclase pathway and increased cAMP levels; however, recent evidence has demonstrated that in some tissues and cell types both CRH receptors can couple to other G proteins including Gq, Gi, and Go, resulting in activation of various kinases including PKC, MAPK, and Akt (reviewed in (Grammatopoulos and Chrousos 2002; Hillhouse and Grammatopoulos 2006)). CRH-R1 and CRH-R2 differ in their pharmacological properties (reviewed in (Bale and Vale 2004; Dautzenberg and Hauger 2002; Grammatopoulos and Chrousos 2002; Hillhouse and Grammatopoulos 2006)). CRH-R1 has very high affinity for both CRH and UCN1 (Vaughan, et al. 1995). In contrast, CRH-R2 has a significantly higher affinity (20–40 fold) for UCNI than for CRH (Vaughan et al. 1995); UCN2 and UCN3 are relatively selective for CRH-R2, and have been suggested to be CRH-R2 specific ligands (Hsu and Hsueh 2001; Lewis, et al. 2001; Reyes, et al. 2001).
CRH-R1 has been detected in numerous sites in the brain (ie. cerebellum, cerebral cortex, amygdala, various limbic and sensory nuclei), the intermediate lobe of the pituitary, and a subset of corticotrophs in the anterior lobe of the rat pituitary (Potter, et al. 1994; Van Pett, et al. 2000). Recent studies from our laboratory have demonstrated CRH-R1 mRNA not only in a subset of corticotropes, but also in a subset of lactotropes and gonadotropes in murine anterior pituitary (Westphal, et al. 2008). CRH-R2 has been isolated in two alternatively spliced forms in the rodent (α and β; utilize alternative amino terminus) and exhibits an mRNA expression profile that is distinct from that of CRH-R1 (Chalmers, et al. 1995; Lovenberg, et al. 1995b). CRH-R2α is expressed primarily in brain (Chalmers et al. 1995; Lovenberg, et al. 1995a), while CRH-R2β is found largely in the periphery, but is also detected in choroid plexus and cerebral arterioles (Chalmers et al. 1995; Lovenberg et al. 1995a). Within the pituitary, CRH-R2 is expressed predominantly in the posterior lobe; however, a very low but detectable level of CRH-R2 mRNA is also detected in the anterior lobe by in situ hybridization (Kageyama, et al. 2003; Van Pett et al. 2000). Consistent with the potential expression of CRH-R2 in anterior pituitary, Kageyama and colleagues recently demonstrated CRH-R2α mRNA in rat anterior pituitary and CRH-R2 mRNA in rat gonadotropes (Kageyama et al. 2003). These results suggest that both CRH-R1 and CRH-R2 may bind CRH and urocortins not only in the CNS but also in the pituitary to modulate stress-related functions.
To investigate the potential action of CRH specifically on pituitary gonadotrope function, we examined CRH receptor expression and functional signaling in αT3-1 cells, a murine gonadotrope precursor cell line. These cells were originally derived by targeted oncogenesis in transgenic mice (Windle, et al. 1990) and they express GnRH receptors and the α-subunit of the glycoprotein hormone, but not LHβ or FSHβ. They exhibit a number of characteristics equivalent to gonadotropes in primary pituitary cultures, demonstrating the utility of these cells as a model system for study of gonadotrope function (Horn, et al. 1991).
Materials and Methods
Cell culture
αT3-1 cells were kindly provided by Dr. Pamela Mellon (University of California, San Diego) (Windle et al. 1990). They were maintained in Dulbecco’s Modified Eagle Medium (Life Technologies) plus 10% fetal calf serum (Hyclone) at 37 degrees in 5% CO2.
cAMP Assays
αT3-1 cells were plated (300,000 cells/well) in 6 well plates (Falcon 3046) and used 2–3 days after plating. Cells were washed 2 times in DMEM and preincubated for 45–60 minutes in DMEM containing 1 mM isobutylmethylxanthine (IBMX, Sigma) to inhibit cAMP phosphodiesterase activity. Preincubation media was removed and DMEM (1 ml) containing 1mM IBMX and various concentrations of human/rat CRF, rat UCN1, mouse UCN2, mouse UCN3, alpha-helical CRH (9–41), astressin (American Peptide Company, Sunnyvale, CA) or antisauvagine (Bachem, Torrance, CA) were added. Cells were incubated at room temperature for the indicated lengths of time before removal of stimulation media. Cells were immediately lysed and intracellular cAMP levels were determined. The Cyclic AMP (3H) Assay System (Amersham Biosciences, GE Healthcare, Piscataway, NJ) was used for the time course, dose response profiles with CRH and UCN1 I, and alpha-helical CRH 9–41 and astressin antagonist studies. In this assay, cells were lysed and incubated 16–24 hours at −20 C with 95% ethanol/20mM HCl (1 ml/well). Cell lysates were then dried under vacuum, resuspended in 300 μl of 0.05 M Tris (pH 7.5), 4 mM EDTA, and cAMP levels were determined in 50 μl samples according to the manufacturer’s protocol using triplicate wells and duplicate assays for each time point or treatment value. Additional dose response studies with UCN1, UCN2, and UCN3 and antisauvagine antagonist studies were performed with the Direct cAMP Enzyme Imunoassay Kit (Assay Designs, Inc. Catalog #901–066, Ann Arbor, MI). After removal of stimulation media, cells were lysed by 20 minute incubation in 1 ml of 0.1 M HCl. Cell lysates were centrifuged at 600 xg at room temperature for 5 minutes and supernatants were used directly in the assay (50 ul/well) or stored at −20C. As above, duplicate assays and duplicate or triplicate wells were used for each treatment condition. Each experiment was performed at least 2 times; dose profiles were repeated 3–5 times.
RNA Isolation and cDNA synthesis
Total RNA was isolated from mouse brain, hypothalamus, heart, and αT3-1 cells using Trizol reagent (Invitrogen). Five micrograms of RNA was used for random hexamer-primed first strand cDNA synthesis (Novagen, Inc). To control for genomic DNA contamination, cDNA synthesis was performed in the presence and absence of SuperScript II Reverse Transcriptase (Life Technologies, Inc.) for each of the RNA samples. The products of cDNA synthesis were purified using QIAquick PCR Purification Kit (Qiagen Inc.).
RT-PCR Reactions
RT-PCR was performed in a final volume of 25 or 50 μl using 1–3 μl of cDNA product, 200 μM deoxyribonucleotides, 1.0 μM primers, Taq buffer with MgCl2 and 1–25 U of Taq DNA Polymerase or Platinum Taq DNA Polymerase (Invitrogen, Inc., CA). Cycle conditions are described below. Based on the mouse CRH-R1 genomic structure (Ensembl ENSMUST00000093925), the primers for mouse CRH-R1 were designed to span at least one intron in the genomic sequence. The primer pair for CRH-R1 (Figure 1A) was as follows: forward primer 5′-GGA-TCA-GCA-GTG-TGA-GAG-CCT-3′ and reverse primer 5′-GTT-CCA-GTG-GAT-GAT-GTT-CCT-3′ (amplifying a 394 bp fragment from nucleotides 109–502 of mouse CRH-R1 cDNA, GenBank accession no. X72305). PCR amplification conditions were 34 cycles of 94 C for 30s, 69 C for 30s, and 72 C for 30s.
The genomic organization and sequence of mouse CRH-R2 was recently published (Chen, et al. 2005). Exons 1 and 2 are spliced to exon 4 to produce CRH-R2β while Exon 3 is spliced to Exon 4 to produce CRH-R2α. Our primers for PCR were designed to span at least one intron. For CRH-R2α, the forward primer was 5′ CTC-CTC-AGC-CTG-CTG-GAG-GCC-AAC-T 3′ and the reverse primer was 5′ GTT-CTC-CAG-GCA-CTC-TCT-GTA-GGC-ATT 3′ (exons 3 to 5, amplifying a 243 bp fragment from nucleotides 158–400 of mouse CRH-R2α cDNA, Genbank accession number AY445512). For CRH-R2β, the forward primer was 5′ GGC-CTA-AGA-GAG-AGG-CCG-GAC-AGA-CCT-CCT-TTG-GA 3′ and the reverse primer was 5′ AGA-ATG-AAG-GTG-GTG-ATG-AGG-TTC-CAG-TGG-ATC-ACA 3′ (Exons 1 to 7, amplifying a 631 bp fragment from nucleotides 40–670 of mouse CRH-R2β cDNA, Genbank accession number NM_009953). Both PCR amplifications included 30 cycles of 92 C for 60s, 66 C for 90s, and 72 C for 90s.
All PCR products were electrophoresed and visualized on an ethidium bromide-stained 1.0% agarose gel. The 1 Kb Plus DNA ladder (Figure 1A) or 1Kb DNA ladder (Figure 1B and 1C) (Invitrogen) was used for DNA size standards.
Transient transfections and reporter constructs
Cultures of αT3-1 cells were plated on 6-well plates (400,000 cells/well) in DMEM + 10% FCS + gentamicin (50μg/ml). Cells were transfected 12–24 hours later using Fugene (see manufacturer’s recommendations, Roche, Indianapolis, IN) with 1.75 μg luciferase reporter DNA and 0.25 μg RSV-βgal DNA per well. The α-subunit-luciferase construct was kindly provided by Dr. Sally Camper (University of Michigan) and contains 480 bp of the mouse glycoprotein hormone α-subunit promoter (−480 to +43 bp) fused to the luciferase reporter gene (Brinkmeier, et al. 1998). RSV-βgal was use to normalize for transfection efficiency. For all studies, 10 μM forskolin (Calbiochem, San Diego, CA) or various concentrations of UCN peptides were applied at 40 hours post-transfection. Cells were harvested 4 hours later (44 hours post-transfection) in cold 1X PBS, pelleted and lysed in 100μl lysis buffer (0.25 M Tris (pH=8.0), 0.1M EDTA, 15mM MgSO4, 1mM DTT, and 1% Triton X-100) and incubated on ice for 10 min. Lysates were centrifuged at 10,000rpm for 10 min at 4C and 5μl of each supernatant was added to 100 μl of luciferase assay buffer (Cortright, et al. 1997) and assayed 30 seconds in a Turner 20/20 luminometer; β-galactosidase activity was assayed as previously described (Cortright, et al. 1997). Data are represented as fold induction over untreated vehicle control (untreated controls = 1.0). Experiments were performed in duplicate and each experiment was repeated at least three independent times. Fold inductions from 3 independent experiments were combined for the α-luciferase transfection data presented here.
Data analysis and statistical methods
All data are presented as mean +/− standard error of the mean (SEM). Statistical significance of the cAMP responses over time and in the presence of CRH receptor antagonists was determined by ANOVA with post-hoc analysis using StatView (Abacus Concepts). Duplicate or triplicate wells were used for all cAMP assays and at least 2–5 independent assays were performed for each study. The EC50 values for CRH, UCN1, UCN2, and UCN3 were determined by non-linear regression analyses using GraphPad PRISM (version 3.0) software (GraphPad). Statistical analysis for the transient transfection studies was performed using a one-way ANOVA followed by multiple comparison post-hoc analysis, where all selected groups were analyzed simultaneously. Analysis was performed using Statview and p values that reached 95% confidence levels are included in each figure legend.
Results
Expression of CRH receptor mRNA in αT3-1 cells
RT-PCR was utilized to characterize the expression of CRH-R1 and CRH-R2 mRNA in αT3-1 cells. cDNA synthesis reactions were performed in the presence (+) and absence (−) of reverse transcriptase to provide controls for genomic DNA contamination of the cDNA. CRH-R1 and CRH-R2 PCR primer pairs were also specifically designed to span at least one intron. Mouse hypothalamus (Hy) and mouse total brain (Br) cDNA were used as positive controls for CRH-R1 and CRH-R2 respectively. As can be seen in Figure 1A, the primer pair for CRH-R1 demonstrates the presence of an appropriately sized PCR fragment in cDNA from αT3-1 cells (αT+) and mouse hypothalamus (Hy+) (394 bp in Figure 1A). No bands were seen in the minus reverse transcriptase lanes (−), suggesting the absence of genomic DNA contamination. RT-PCR with two additional primer pairs confirmed the presence of CRH-R1 in αT3-1 cells (data not shown; (Westphal et al. 2008)).
Figure 1.
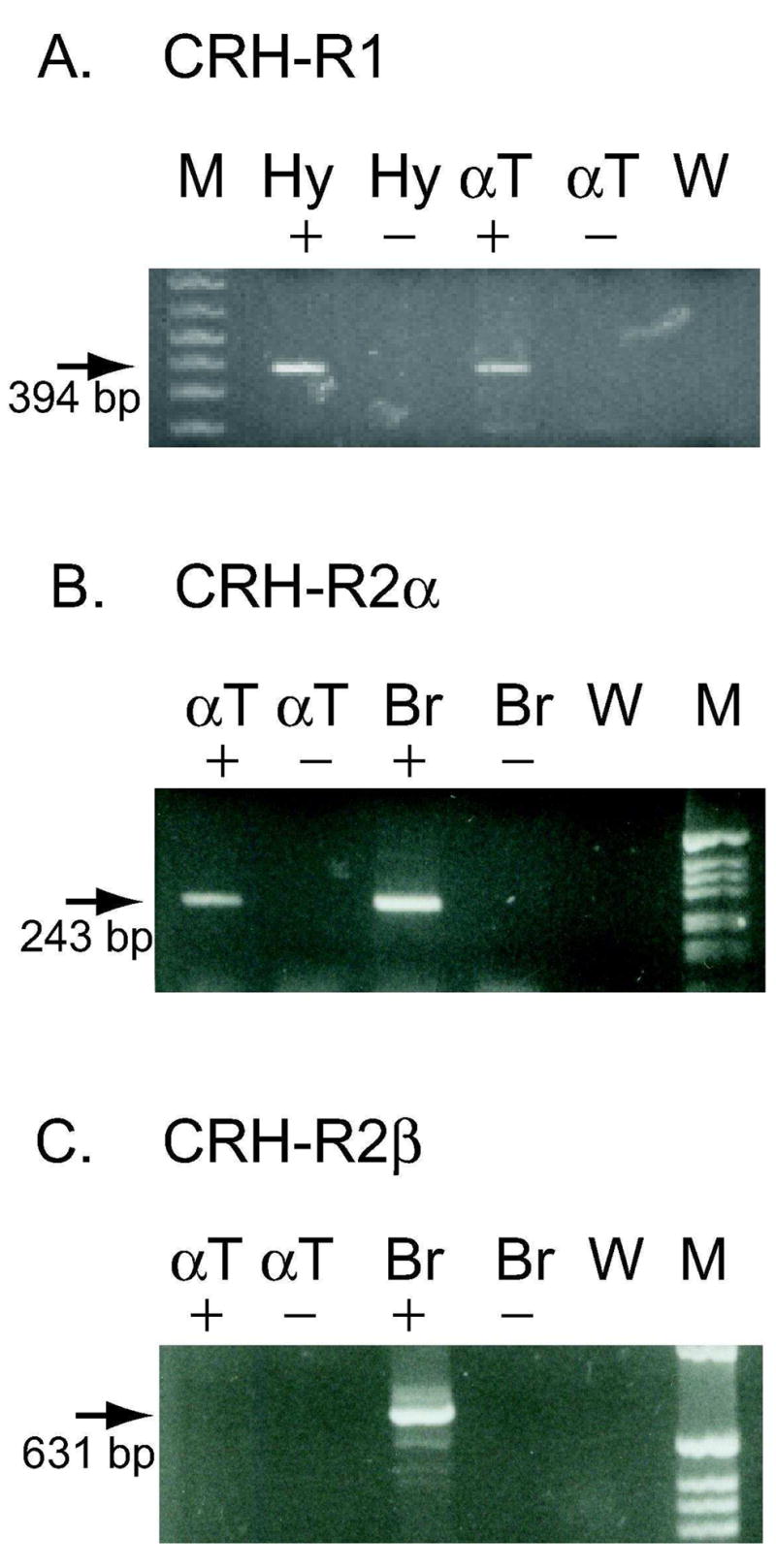
Expression of CRH-R1 and CRH-R2α RNA in αT3-1 cells. RT-PCR was utilized to test for the expression of CRH receptor RNA in αT3-1 cells. Figure 1A utilizes primers specific for CRH-R1 and Figures 1B and 1C utilize primers specific for CRH-R2α and CRH-R2β respectively. The presence or absence of RT during cDNA synthesis is indicated by + or − signs. A) Panel A shows an amplified product of 394 bp in hypothalamus and αT3-1 cDNA lanes (see arrow). Hy + (hypothalamus, with RT), Hy − (hypothalamus, without RT), αT + (αT3-1, with RT), αT − (αT3-1, without RT), W, water control. B) Panel B shows an amplified product of 243 bp in αT3-1 (αT +) and brain (Br +) cDNA lanes (see arrow). These bands are consistent with the expression of CRH-R2α in αT3-1 and total brain RNA samples. C) Panel C shows amplified products of 631 bp only in the brain (Br +) lane (see arrow). The absence of amplified product in the αT + lane indicates that CRH-R2β is not detectable in αT3-1 RNA. M represents 1 kb Plus (Invitrogen) DNA ladder in Panel A and 1 kb (Invitrogen) DNA ladder in Panels B and C.
RT-PCR was also performed using multiple sets of primer pairs for CRH-R2. CRH-R2 is expressed in two alternatively spliced forms in the rodent, CRH-R2α and CRH-R2β (14, 19). These alternatively spliced transcripts differ in the 5′untranslated region and the NH2-terminus of the encoded protein. Primer pairs were selected that contained the 5′ forward primer within the alternatively spliced region of the transcript, providing specificity for the CRH-R2α or CRH-R2β mRNAs. Results shown in Figure 1B demonstrate the presence of an appropriately sized fragment for CRH-R2α (243 bp) in αT3-1 (αT+) and mouse total brain (Br+) cDNA. The 243 bp fragment from αT3-1 cDNA was subcloned; DNA sequence analysis confirmed that the fragment contained the CRH-R2α cDNA sequence. Additional primer pairs also confirmed the presence of CRH-R2α in αT3-1 cells (data not shown). The cDNA samples used for the PCR in Figure 1B were also tested with several different CRH-R2β primer pairs. While the 631 bp CRH-R2β–specific PCR product is readily detected in total brain (Br+) (Figure 1C) or mouse heart (not shown), CRH-R2β is not detected in the same αT3-1 cDNA samples (αT+) that readily detected CRH-R2α. Additional primer pairs also failed to demonstrate detectable levels of CRH-R2β in the αT3-1 samples. Together, the data in Figure 1A-C clearly demonstrate the presence of CRH-R1 and CRH-R2α mRNA in αT3-1 cells, with non-detectable levels of CRH-R2β transcripts.
Stimulation of cAMP production in αT3-1 cells by CRH and UCN1
CRH receptors have been shown to couple to Gs in many tissues and cell lines, resulting in increased intracellular cAMP levels. To test the intracellular signaling of the CRH receptors in αT3-1 cells, cells were treated with 200 nM CRH or UCN1 in the presence of 1 mM IBMX for various lengths of time and cAMP production was examined. UCN1 and CRH bind to both CRH-R1 and CRH-R2, although CRH binds to CRH-R2 with a 14 fold lower affinity than CRH-R1 (Ki=13 nM and 0.95 nM, respectively), while UCN1 binds to both CRH-R1 and CRH-R2 with very high affinity (Ki<1.0 nM) (Donaldson, et al. 1996). As shown in Figure 2, treatment of αT3-1 cells with CRH or UCNI resulted in rapid increases in intracellular cAMP levels. The CRH- and UCN1-mediated increases in cAMP were maximal at 2–5 minutes and were significantly increased over levels in vehicle-treated cells (dotted line). The increased cAMP levels were usually 2–4 fold over basal levels, and maximal levels of cAMP did not significantly differ between CRH and UCN1 treatments within experiments. Interestingly, intracellular cAMP levels decreased rapidly from peak values even in the presence of 1 mM IBMX.
Figure 2.
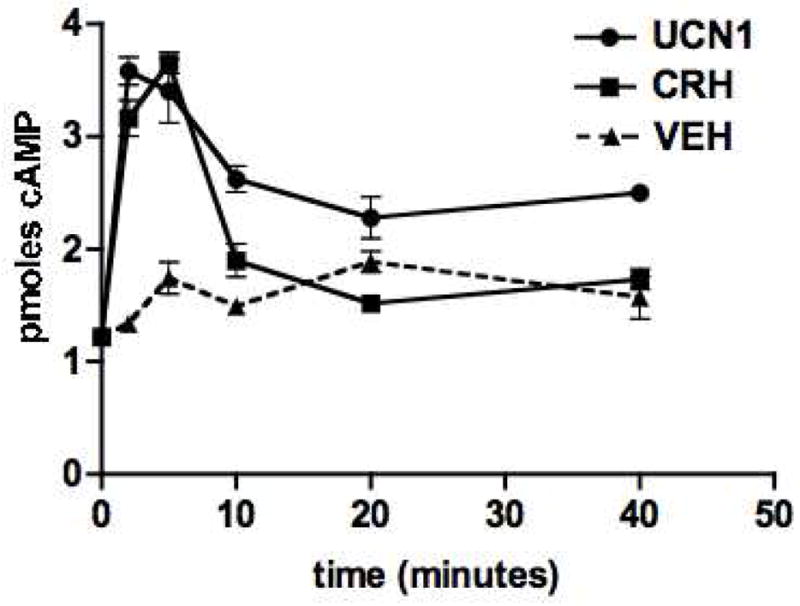
Time-dependent increases in intracellular cAMP levels in αT3-1 cells after treatment with CRH or UCN1. Cells were treated with 200 nM CRH or UCN for 2, 5, 10, 20 and 40 minutes in the presence of 1 mM IBMX. cAMP assays were performed on cell lysates. The addition of CRH (□) or UCN1 (O) resulted in a rapid increase in intracellular cAMP levels as compared to vehicle treatment (dotted line). Points represent the average (+/− SEM) for triplicate wells from a representative experiment.
Dose-dependent stimulation of cAMP production in αT3-1 cells by CRH and UCN1
Dose response studies were performed using various concentrations of CRH or UCN1 in the presence of 1 mM IBMX. As time course experiments had shown maximal intracellular cAMP levels at 2–5 minutes after addition of ligand, all treatments were for 3 minutes. As seen in Figure 3, cAMP levels increased in αT3-1 cells in a dose-dependent fashion in response to CRH or UCN1 treatment. CRH and UCN1 were similar in potency and showed equivalent maximum cAMP levels across multiple experiments. The half-maximal stimulatory (EC50) concentrations of CRH and UCN1 were not significantly different with values of 14.5 nM +/− 5.9 (n=3) and 21.2 nM +/− 5.1 (n=7), respectively.
Figure 3.
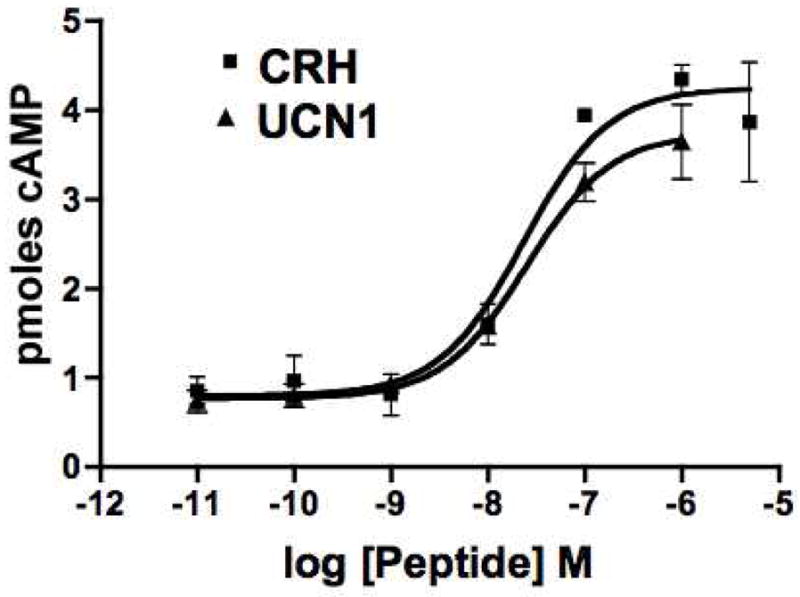
Dose-dependent increases in cAMP levels in αT3-1 cells following treatment by CRH (□) or UCN1 (Δ). Intracellular levels of cAMP are shown after treatment of αT3-1 cells with various concentrations of CRH or UCN. Treatments were for 3 minutes. Points are the average (+/− SEM) from duplicate assays on triplicate wells from a representative experiment. The maximal cAMP levels are not significantly different following CRH or UCN1 treatment and the EC50 values are also not significantly different.
Inhibition of CRH- and UCN1-mediated increases in cAMP with non-selective CRH receptor antagonists
We used CRH receptor antagonists (α-helical CRH 9–41 and astressin) that block both CRH-R1 and CRH-R2 (reviewed in (Hauger, et al. 2006)) to determine whether the CRH- and UCNI-mediated increases in intracellular cAMP were specifically mediated by CRH receptors. Cells were pretreated for 1 hour with 1 mM IBMX and fresh media was added that contained 100 nM CRH or UCN1 in the presence or absence of 10 μM α-helical CRH 9–41 (9–41, Figure 4A) or 1 uM astressin (AST, Figure 4B). Cells were also treated with vehicle or receptor antagonist alone. All treatments were for 3 minutes. α-helical CRH 9–41 treatment alone showed a small but significant increase in intracellular cAMP levels (Figure 4A). CRH or UCN1 treatment alone also resulted in a significant increase in cAMP, while co-administration with α-helical CRH 9–41 reduced cAMP values to vehicle-treated levels. The α-helical CRH 9-41 results shown here are consistent with previous studies that suggest that it is not a pure antagonist, demonstrating weak intrinsic agonist activity (Baldwin, et al. 1991; Hauger et al. 2006). A second non-selective CRH receptor antagonist, astressin (Figure 4B), also showed complete suppression of the CRH- and UCN1-mediated increases in cAMP, with no significant effect of astressin alone. Together these studies clearly demonstrate that CRH and UCN1 mediate significant increases in intracellular cAMP levels that can be effectively inhibited by CRH receptor antagonists that block both CRH-R1 and CRH-R2 signaling.
Figure 4.
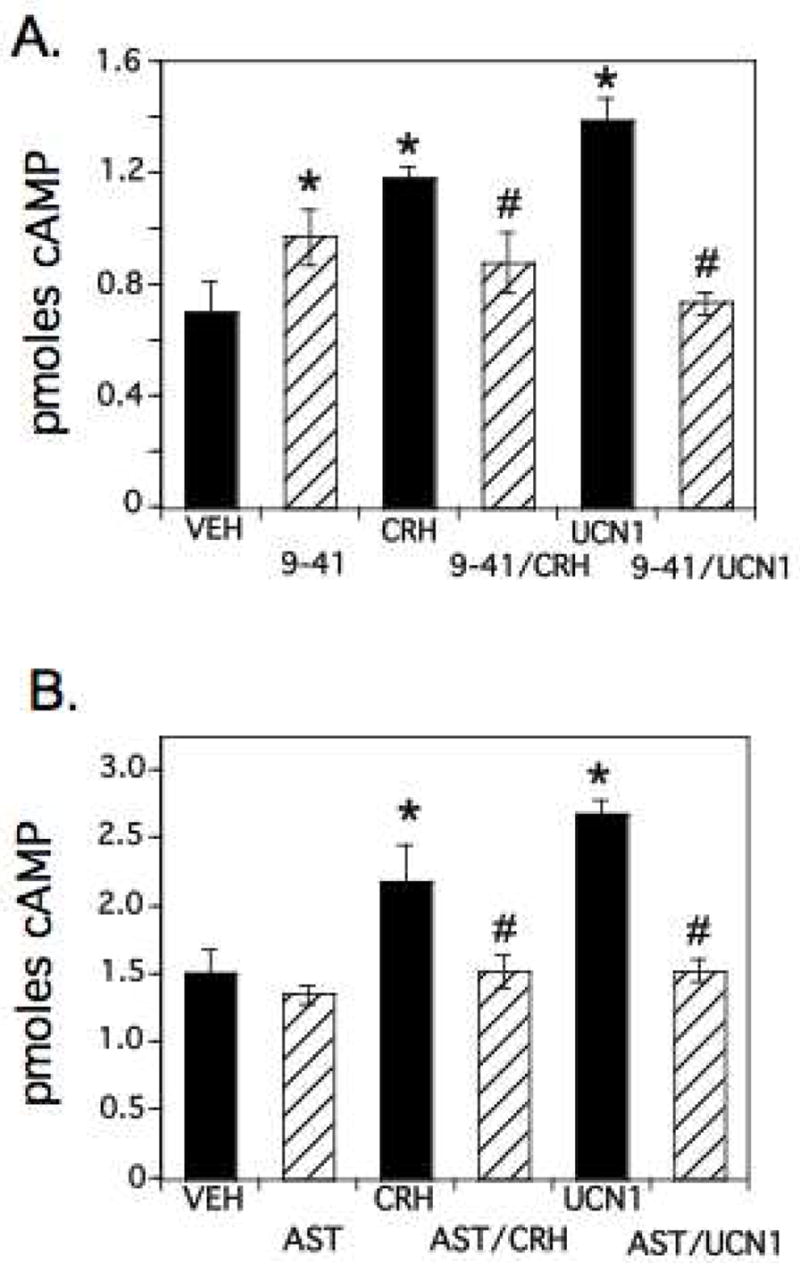
Inhibition of CRH- or UCN1-mediated stimulation of cAMP levels by CRH receptor antagonists α-helical CRH 9–41 or astressin. A) αT3-1 cells were treated with DMEM/IBMX containing vehicle, 10μM α-helical CRH 9–41 (9–41), 100 nM CRH, 100 nM UCN1, or 9–41 plus CRH or UCN1 for 3 minutes. Intracellular cAMP levels after 9–41, CRH or UCN1 treatments were significantly increased from vehicle-treated levels (VEH; significance from VEH indicated by *, p<0.05). In addition, treatments with 9–41 in the presence of CRH or UCN1 blocked the CRH- and UCN1-mediated increases in intracellular cAMP (#, significantly different from CRH or UCN1 treatments alone, p<0.05). B) αT3-1 cells were treated as above except that astressin (AST, 1 μM) was used alone or in combination with 100nM CRH or UCN1. These results also show significant inductions in cAMP levels with CRH and UCN1 and significant repressions of CRH- or UCN-mediated increases in cAMP by the addition of astressin. * and # are used as in Panel A.
Contributions of CRH-R2 to the cAMP signaling in αT3-1 cells
To determine the contribution of CRH-R2 to the overall cAMP increases in αT3-1 cells in response to CRH ligands, we stimulated cells with various doses of UCN2 and UCN3, CRH-R2-specific ligands (Jahn, et al. 2004). The EC50 values for cAMP stimulation for UCN2 and UCN3 were 20.3 nM +/− 9.7 (n=3) and 32.5 nM +/− 8.2 (n=3), not significantly different from each other or the EC50 value of UCN1 (21.2+/− 5.2 nM, n=7). At maximal doses of UCN2 and UCN3 (100nM), we observed increases in cAMP levels that reached 60.0 +/−7.6 % (n=6) and 54.3 +/− 6.0% (n=13) of the levels obtained with UCN1 or CRH (90.9 +/− 11.0 % relative to UCN1) (Figure 5A), demonstrating that both CRH-R1 and CRH-R2 can contribute to the cAMP increases in αT3-1 cells.
Figure 5.
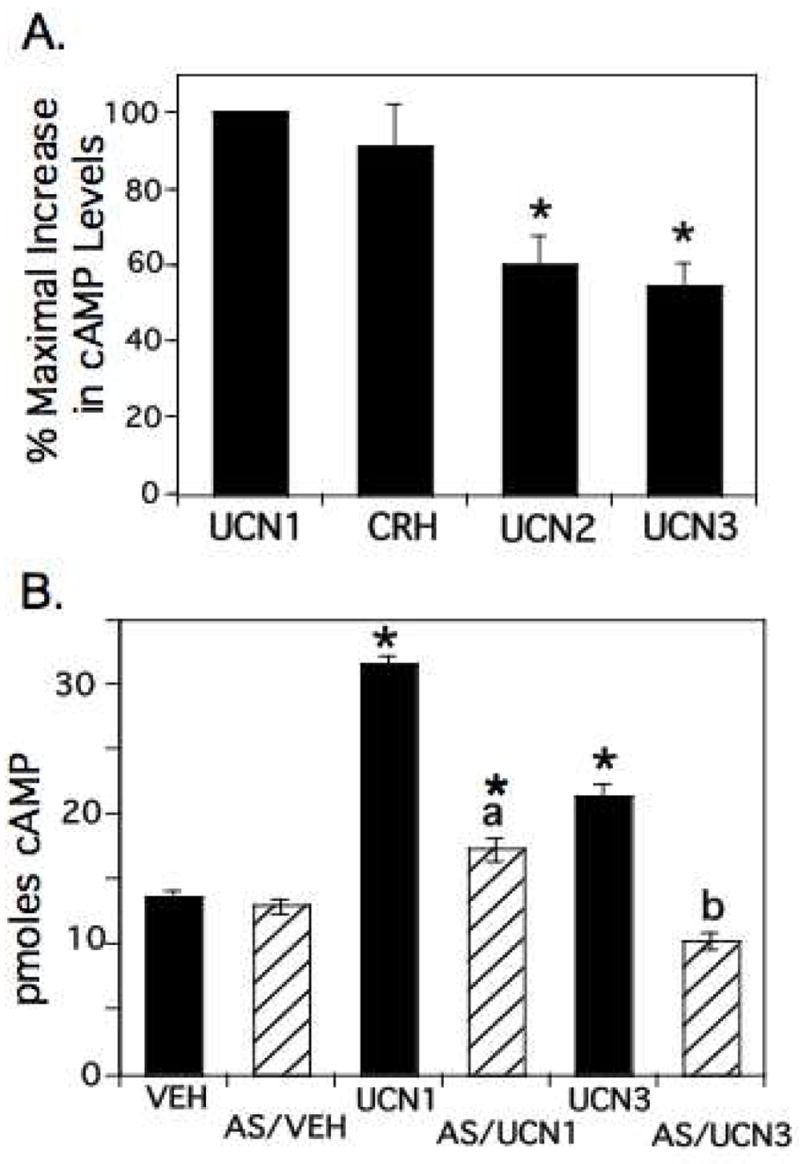
Functional role of CRH-R2 in cAMP signaling in αT3-1 cells. A) αT3-1 cells were treated with vehicle, CRH, UCN1, UCN2, or UCN3 (100nM) for 3 minutes. Intracellular cAMP levels were determined and the increase in cAMP levels under each treatment condition (from VEH) was normalized to that observed with UCN1 (set to 100%). UCN2 and UCN3 showed significantly lower elevations in cAMP, showing only 50–60% of the increases obtained with UCN1 or CRH (*, significantly different from UCN1, p<0.05), suggesting that CRH-R2 contributes approximately 50% to the cAMP increases observed after stimulation with UCN1. B) αT3-1 cells were pretreated with antisauvagine (AS, 1uM) or vehicle for 15 minutes followed by a 3 minute treatment with UCN1 or UCN3 (100 nM) in the presence or absence or antisauvagine. Intracellular cAMP levels after UCN treatments were significantly increased from vehicle-treated levels (VEH; significantly different from VEH indicated by *, p<0.05), with UCN1-mediated increases being significantly greater than UCN3-mediated increases. There was no significant difference in intracellular cAMP levels between VEH and AS treatments. Treatment with AS in the presence of UCN3 completely blocked the UCN3-mediated increases (b, significantly different from UCN3, p<0.05), resulting in cAMP levels not significantly different from VEH or AS. Treatment with AS in the presence of UCN1 significantly reduced cAMP levels from UCN1 (a, significantly different from UCN1, p<0.05), but cAMP levels remained significantly greater than VEH, AS/VEH or AS/UCN3 levels, suggesting that activated CRH-R1 was responsible for the remaining cAMP increase.
To further confirm the contribution of CRH-R2 to the UCN1-mediated increases in cAMP, we utilized a selective CRH-R2 antagonist, antisauvagine (Brauns, et al. 2002; Ruhmann, et al. 1998). This peptide has a 100–400 fold greater selectivity for CRH-R2 than CRH-R1. Cells were pretreated with antisauvagine for 15 minutes before addition of UCN1 or the CRH-R2 specific agonist, UCN 3. As shown in Figure 5B, cAMP levels after antisauvagine treatment alone did not significantly differ from vehicle. However, antisauvagine completely blocked the UCN3-mediated increase in cAMP, while only partially blocking (75% repression) the UCN1-mediated cAMP increases, consistent with both CRH-R1 and CRH-R2 contributing to the UCN1-mediated cAMP increases.
UCN1 increases α-luciferase transcriptional activity
While LHβ and FSHβ are not produced in αT3-1 cells, the glycoprotein hormone α-subunit is actively transcribed and translated in these cells and its expression is highly regulated by GnRH, gonadal steroids, and a variety of peptides including pituitary adenylyl cyclase activating polypeptide (PACAP) and endothelin 1, working through numerous intracellular signaling pathways (reviewed in (Evans 1999; McArdle and Counis 1996)). PACAP has been shown to increase intracellular cAMP levels in αT3-1 cells, resulting in increased transcriptional activity and steady state levels of α-subunit mRNA (Attardi and Winters 1998; Burrin, et al. 1998; Schomerus, et al. 1994; Tsujii, et al. 1995). As cAMP levels are also rapidly increased by CRH or UCN in these cells, we utilized a mouse α-subunit promoter-luciferase reporter construct (α-luciferase) in transfection studies to assess the increased transcriptional activity of the α-subunit promoter in αT3-1 cells after treatment with UCN1. As seen in Figure 6, UCN1 significantly increased α-luciferase activity in a dose-dependent manner from 30–300 nM, reaching levels that were not significantly different from those induced by forskolin, a direct activator of adenylyl cyclase. As the mouse α-subunit promoter is known to contain cAMP response elements and has previously been shown to be activated by increased cAMP/PKA activation (Attardi and Winters 1998; Schoderbek, et al. 1992), these results are consistent with the CRH-mediated elevations in cAMP causing an increase in α-subunit promoter-directed transcriptional activity in αT3-1 cells.
Figure 6.
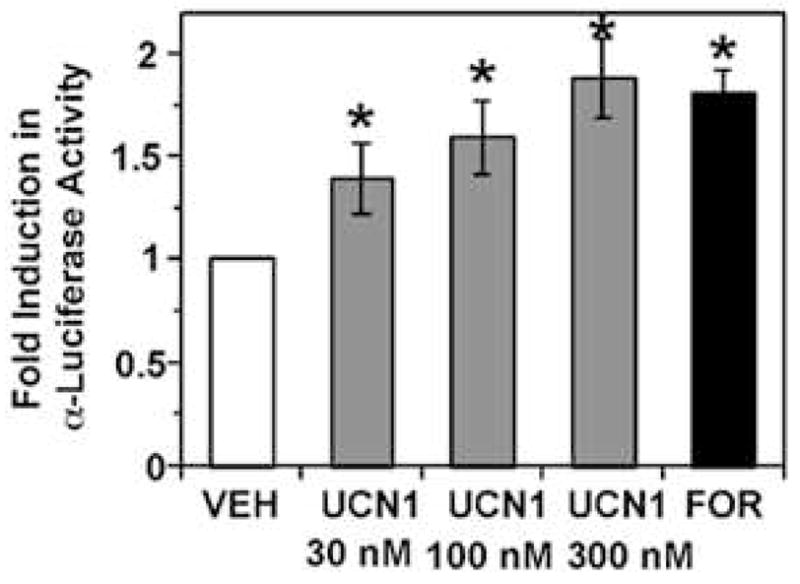
Dose-dependent increases in α-luciferase activity in transiently transfected αT3-1 cells following treatment with UCN1. Treatment of α-luciferase-transfected αT3-1 cells with UCN1 (30 nM, 100nM, or 300nM, 4h) or forskolin (10 μM, 4h) significantly induced α-luciferase promoter activity. The relative promoter activity is represented as fold induction over control. Values represent the mean ± SEM (n=3). *, P < 0.05, as compared to vehicle control.
Discussion
We have demonstrated via RT-PCR that CRH-R1 and CRH-R2α mRNAs are expressed in αT3-1 cells. These G-protein coupled receptors are functional and couple through Gs, as treatment of these cells with CRH or UCN1 results in significant increases in intracellular cAMP levels that can be blocked completely by the presence of non-selective CRH receptor antagonists. Our results also demonstrate that both CRH-R1 and CRH-R2α are functional in these cells as CRH-R2-specific ligands increase cAMP levels to only 50–60% of the levels observed with non-selective agonists (CRH and UCN1), and CRH-R2-selective antagonists do not completely abolish the UCN1 mediated increases in cAMP. In addition, we have shown that UCN1 treatment of αT3-1 cells results in increased transcriptional activity of the glycoprotein hormone α-subunit promoter, a promoter known to be positively regulated by elevated cAMP levels. These results are the first to demonstrate both the expression and functional signaling of CRH-R1 and CRH-R2α in murine gonadotrope-like cells, and they suggest a potential direct role for CRH or the other CRH-like ligands, UCN1, UCN2, and UCN3, in modulation of gonadotropin transcription and secretion at the level of the anterior pituitary gonadotrope. This proposed regulation at the pituitary could complement or modulate the CRH-mediated regulation of hypothalamic GnRH production and secretion.
In the rodent, CRH-R1 mRNA has been localized primarily in the brain and intermediate and anterior lobes of the pituitary (Potter et al. 1994; Van Pett et al. 2000). While CRH-R1 mRNA had previously been detected in a subset of rat corticotropes (Potter et al. 1994), recent studies from our laboratory have also demonstrated CRH-R1 mRNA in a subset of cells expressing ACTH, prolactin, or LHβ transcripts in the mouse anterior pituitary (Westphal et al. 2008), consistent with the expression of CRH-R1 in the mouse gonadotrope-like αT3-1 cell line. CRH-R2 expression in the rodent is quite distinct from CRH-R1, with a more restricted pattern in brain but numerous sites of expression in the periphery (Chalmers, et al. 1996; Van Pett et al. 2000). While the two rodent isoforms of CRH-R2, CRH-R2α and CRH-R2β, show comparable pharmacologic properties (Hauger et al. 2006), they differ significantly in their tissue-specific expression. In situ hybridization studies using probes from the common region of CRH-R2 have demonstrated CRH-R2 mRNA expression in the posterior pituitary, with a weak but clearly detectable signal in the anterior pituitary (Van Pett et al. 2000), most likely in gonadotropes (Kageyama et al. 2003). CRH-R2α mRNA has also been detected in anterior pituitary by ribonuclease protection assay (Kageyama et al. 2003). Consistent with these results, we have detected CRH-R2α mRNA, but not CRH-R2β, in αT3-1 cells by RT-PCR. Together these results suggest that a population of gonadotropes may express both CRH-R1 and CRH-R2α at certain times in development or in response to specific hormonal signals.
The expression of CRH-R1 and R2α on αT3-1 cells that also express GnRH receptors is quite interesting in light of recent studies demonstrating the significant population of anterior pituitary cells in rat and mouse that exhibit multiple hypothalamic releasing hormone (HRH) receptors (multiresponsive cells) (Nunez, et al. 2003; Villalobos, et al. 2004). Recent studies suggest that 38% of female mouse anterior pituitary cells express multiple HRH receptors, with almost 50% of LH-positive cells being multiresponsive (Nunez et al. 2003). Our results are consistent with these studies and suggest that anterior pituitary cells are more multifunctional than previously thought, with great potential for cross-talk between hypothalamic releasing factors and pituitary hormonal release. Interestingly, CRH has recently been shown to induce thyrotropin release from the amphibian pituitary gland via CRH-R2 signaling (Okada, et al. 2007).
In response to treatment with CRH or UCN1, the αT3-1 cells show a very rapid increase in intracellular cAMP levels, demonstrating the positive coupling of the CRH receptors to Gs and adenylyl cyclase after ligand stimulation. The rapid rise in intracellular cAMP levels in the αT3-1 cells in response to CRH or UCN with a peak at 2–5 minutes followed by a rapid decline is similar to results seen with murine atrial cardiomyocyte tumor cells, AT-1 cells, which express endogenous CRH-R2β mRNA (Heldwein, et al. 1996). The rapid decline in cAMP levels even in the presence of IBMX is quite interesting, and it may suggest that one or both types of CRH receptors on αT3-1 cells is/are desensitized quickly following exposure to ligand. This theory is supported by reports of rapid agonist-induced CRH-R1 desensitization and internalization (Roseboom, et al. 2001; Teli, et al. 2005). Alternatively, the dose of IBMX used in these studies may not be sufficient to inhibit all phosphodiesterase activity in these cells.
The responses to CRH and the urocortins in the αT3-1 cells were dose-dependent and specific for CRH receptors, as the presence of non-selective CRH receptor antagonists, α-helical CRH 9-41 or astressin, completely inhibited the intracellular cAMP increases. Similar peak cAMP values were observed with CRH or UCN1, ligands that activate both CRH-R1 and CRH-R2α. In contrast, peak cAMP values with UCN2 and UCN3 stimulation reached levels only 50–60% of UCN1, consistent with their CRH-R2 selectivity. Similarly, the significant, but incomplete inhibition of UCN1-mediated increases in cAMP production in αT3-1 cells with antisauvagine, the CRH-R2-specific antagonist (100–400 fold greater binding to CRH-R2), was consistent with the UCN1-mediated activation of both CRH-R1 and CRH-R2α. The EC50 values for cAMP production in αT3-1 cells were not significantly different between CRH, UCN1, UCN2 or UCN3, and ranged from 14 to 32 nM. Studies in cell lines that have been stably or transiently transfected with CRH receptors have shown EC50 values for CRH-R1 of 1 to 3 nM for both CRH and UCN1 and EC50 values for CRH-R2 of 0.2 to 20 nM (Vaughan et al. 1995). Primary cultures or cultured cell lines expressing endogenous CRH receptors have shown higher EC50 values (Heldwein et al. 1996).
While stress-induced suppression of GnRH and LH release is clearly dependent on central actions of CRH (Li et al. 2006; Li et al. 2005), the finding that αT3-1 cells, a gonadotrope-like cell line, express CRH receptors suggests an additional and potential direct role for CRH or urocortins on gonadotrope function, consistent with recent studies which have identified CRH-R1 or CRH-R2 mRNA in rat or mouse gonadotropes (Kageyama et al. 2003; Westphal et al. 2008). Our studies here extend previous results to demonstrate that these CRH receptors are functional, increasing intracellular cAMP levels resulting in transcriptional activation of cAMP-responsive genes including the glycoprotein hormone α-subunit. PACAP has also been shown to increase cAMP levels in αT3-1 cells, resulting in increased α-subunit expression (Attardi and Winters 1998; Schomerus et al. 1994; Tsujii et al. 1995). Interestingly, while GnRH does not alter cAMP levels in αT3-1 cells, GnRH significantly reduces the PACAP-mediated increase in cAMP levels via activation of the protein kinase C (PKC) signaling pathway (McArdle and Counis 1996; McArdle, et al. 1994), emphasizing the importance of crosstalk between distinct intracellular signaling pathways in these gonadotrope-like cells. It is possible that GnRH may also modify the CRH-mediated increases in cAMP in these cells. Similarly, we have initiated preliminary studies of GnRH signaling and phospholipase C (PLC) activation in αT3-1 cells. While GnRH treatment dramatically increases total inositol phosphate levels via PLC activation, CRH treatment alone has no significant effect on inositol phosphate levels (unpublished data, A. Seasholtz). However, if CRH and GnRH are co-administered, CRH significantly decreases the GnRH-mediated increases in inositol phosphates, showing another level of cross-talk between the PKA and PLC/PKC signaling pathways in these gonadotrope-like cells (unpublished data, A. Seasholtz). Functional interactions between the PKA and PLC/PKC systems have also been observed in the LβT2 gonadotrope like cell line, with GnRH stimulating PACAP receptor phosphorylation by PKC (Lariviere, et al. 2008). Hence, while αT3-1 cells represent only a model of gonadotrope function, these studies suggest that CRH receptor signaling on pituitary gonadotropes may contribute, along with other peptide and gonadal hormones, to modulation of GnRH signaling at these cells and resultant gonadotropin synthesis and release.
Finally, it should also be noted that the CRH-binding protein (CRH-BP) is highly expressed in rodent anterior pituitary (Speert, et al. 2002). This secreted glycoprotein binds CRH and UCN with high affinity and is thought to act largely as an inhibitory protein, binding the ligands and decreasing CRH receptor activation (reviewed in (Westphal and Seasholtz 2006)). CRH-BP expression is positively regulated by estrogen, resulting in a sexually dimorphic pattern of expression with CRH-BP detected in corticotropes in male mice, while it is detected in corticotropes, lactotropes, and gonadotropes in female mice (Speert et al. 2002). CRH-BP is also expressed in αT3-1 cells where its expression is regulated by GnRH (Westphal and Seasholtz 2005). Hence, the actions of CRH and UCN on αT3-1 cells or gonadotropes in vivo may be highly regulated by the pituitary CRH-BP.
In conclusion, we have demonstrated that αT3-1 cells express CRH-R1 and CRH-R2α mRNAs and that these CRH receptors are functionally coupled to the Gs signal transduction pathways. These receptors respond to CRH, UCN1, UCN2, or UCN3 treatment with rapid, transient increases in intracellular cAMP levels, and CRH receptor antagonist data confirm the contributions of both CRH-R1 and CRH-R2 to the cAMP inductions. The activation of these receptors also increases the transcriptional activity of the glycoprotein hormone α-subunit promoter, a well-characterized cAMP-responsive promoter. Since αT3-1 cells serve as a model for gonadotrope cells, these data suggest a potential direct role for CRH or the other CRH-like ligands, UCN1, UCN2, and UCN3, in the coordinated control of gonadotropin expression and secretion at the gonadotrope. Studies in primary mouse anterior pituitary cultures and in vivo will allow us to further examine the actions of CRH and urocortins (and the CRH-BP) on both basal and stimulated pituitary hormone release, potentially revealing new roles for these ligands in pituitary function.
Acknowledgments
We would like to thank Linda Harper Gates for her assistance with the αT3-1 cells. The studies were supported by NIH grants DK54323 to R.C.T. and DK42730 and DK57660 to A.F.S. We gratefully acknowledge the postdoctoral support of M.Ö. from the Academy of Finland (grant 52840) and the Finnish Medical Foundation. This work was presented in part at the 85th Annual Meeting of the Endocrine Society, Philadelphia, PA. While this manuscript was under revision, a separate manuscript from our laboratory was published that documents CRH receptor expression in multiple anterior pituitary cells lines (including αT3-1 cells) and in murine corticotropes, lactotropes and gonadotropes (Westphal et al. 2008). The authors have no conflict of interest to report.
Footnotes
Publisher's Disclaimer: This is not the definitive version of record of this article. This manuscript has been accepted for publication in Journal of Endocrinology, but the version presented here has not yet been copy edited, formatted or proofed. Consequently, the Society for Endocrinology accepts no responsibility for any errors or omissions it may contain. The definitive version is now freely available at http://dx.doi.org/10.1677/JOE-08-0139
Literature Cited
- Attardi B, Winters SJ. Transcriptional regulation of the glycoprotein hormone alpha-subunit gene by pituitary adenylate cyclase-activating polypeptide (PACAP) in alphaT3-1 cells. Mol Cell Endocrinol. 1998;137:97–107. doi: 10.1016/s0303-7207(98)00006-9. [DOI] [PubMed] [Google Scholar]
- Baldwin HA, Rassnick S, Rivier J, Koob GF, Britton KT. CRF antagonist reverses the “anxiogenic” response to ethanol withdrawal in the rat. Psychopharmacology (Berl) 1991;103:227–232. doi: 10.1007/BF02244208. [DOI] [PubMed] [Google Scholar]
- Bale TL, Vale WW. CRF and CRF receptors: role in stress responsivity and other behaviors. Annu Rev Pharmacol Toxicol. 2004;44:525–557. doi: 10.1146/annurev.pharmtox.44.101802.121410. [DOI] [PubMed] [Google Scholar]
- Blank MS, Fabbri A, Catt KJ, Dufau ML. Inhibition of luteinizing hormone release by morphine and endogenous opiates in cultured pituitary cells. Endocrinology. 1986;118:2097–2101. doi: 10.1210/endo-118-5-2097. [DOI] [PubMed] [Google Scholar]
- Brauns O, Brauns S, Jenke M, Zimmermann B, Dautzenberg F. Secondary structure of antisauvagine analogues is important for CRF receptor antagonism: development of antagonists with increased potency and receptor selectivity. Peptides. 2002;23:1817–1827. doi: 10.1016/s0196-9781(02)00139-0. [DOI] [PubMed] [Google Scholar]
- Brinkmeier ML, Gordon DF, Dowding JM, Saunders TL, Kendall SK, Sarapura VD, Wood WM, Ridgway EC, Camper SA. Cell-specific expression of the mouse glycoprotein hormone alpha-subunit gene requires multiple interacting DNA elements in transgenic mice and cultured cells. Mol Endocrinol. 1998;12:622–633. doi: 10.1210/mend.12.5.0103. [DOI] [PubMed] [Google Scholar]
- Burrin JM, Aylwin SJ, Holdstock JG, Sahye U. Mechanism of action of pituitary adenylate cyclase-activating polypeptide on human glycoprotein hormone alpha-subunit transcription in alphaT3-1 gonadotropes. Endocrinology. 1998;139:1731–1737. doi: 10.1210/endo.139.4.5937. [DOI] [PubMed] [Google Scholar]
- Chalmers DT, Lovenberg TW, De Souza EB. Localization of novel corticotropin-releasing factor receptor (CRF2) mRNA expression to specific subcortical nuclei in rat brain: comparison with CRF1 receptor mRNA expression. J Neurosci. 1995;15:6340–6350. doi: 10.1523/JNEUROSCI.15-10-06340.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalmers DT, Lovenberg TW, Grigoriadis DE, Behan DP, De Souza EB. Corticotrophin-releasing factor receptors: from molecular biology to drug design. Trends Pharmacol Sci. 1996;17:166–172. doi: 10.1016/0165-6147(96)81594-x. [DOI] [PubMed] [Google Scholar]
- Chen A, Perrin M, Brar B, Li C, Jamieson P, Digruccio M, Lewis K, Vale W. Mouse corticotropin-releasing factor receptor type 2alpha gene: isolation, distribution, pharmacological characterization and regulation by stress and glucocorticoids. Mol Endocrinol. 2005;19:441–458. doi: 10.1210/me.2004-0300. [DOI] [PubMed] [Google Scholar]
- Cortright DN, Goosens KA, Lesh JS, Seasholtz AF. Isolation and characterization of the rat corticotropin-releasing hormone (CRH)-binding protein gene: transcriptional regulation by cyclic adenosine monophosphate and CRH. Endocrinology. 1997;138:2098–2108. doi: 10.1210/endo.138.5.5128. [DOI] [PubMed] [Google Scholar]
- Dautzenberg FM, Hauger RL. The CRF peptide family and their receptors: yet more partners discovered. Trends Pharmacol Sci. 2002;23:71–77. doi: 10.1016/s0165-6147(02)01946-6. [DOI] [PubMed] [Google Scholar]
- Donaldson C, Sutton S, Perrin M, Corrigan A, Lewis K, Rivier J, Vaughan J, Vale W. Cloning and characterization of human urocortin. Endocrinology. 1996;137:2167–2170. doi: 10.1210/endo.137.5.8612563. [published erratum appears in Endocrinology 1996 Sep;137(9)3896] [DOI] [PubMed] [Google Scholar]
- Dunn AJ, Berridge CW. Physiological and behavioral responses to corticotropin-releasing factor administration: is CRF a mediator of anxiety or stress responses? Brain Res Brain Res Rev. 1990;15:71–100. doi: 10.1016/0165-0173(90)90012-d. [DOI] [PubMed] [Google Scholar]
- Evans JJ. Modulation of gonadotropin levels by peptides acting at the anterior pituitary gland. Endocr Rev. 1999;20:46–67. doi: 10.1210/edrv.20.1.0355. [DOI] [PubMed] [Google Scholar]
- Grammatopoulos DK, Chrousos GP. Functional characteristics of CRH receptors and potential clinical applications of CRH-receptor antagonists. Trends in Endocrinology and Metabolism. 2002;13:436–444. doi: 10.1016/s1043-2760(02)00670-7. [DOI] [PubMed] [Google Scholar]
- Hauger RL, Risbrough V, Brauns O, Dautzenberg FM. Corticotropin releasing factor (CRF) receptor signaling in the central nervous system: new molecular targets. CNS Neurol Disord Drug Targets. 2006;5:453–479. doi: 10.2174/187152706777950684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heldwein KA, Redick DL, Rittenberg MB, Claycomb WC, Stenzel-Poore MP. CRH receptor expression and functional coupling in neonatal cardiac myocytes and AT-1 cells. Endocrinology. 1996;137:3631–3639. doi: 10.1210/endo.137.9.8756527. [DOI] [PubMed] [Google Scholar]
- Hillhouse E, Grammatopoulos D. The molecular mechanisms underlying the regulation of the biological activity of CRH receptors: Implications for physiology and pahthophysiology. Endocr Rev. 2006;27:260–286. doi: 10.1210/er.2005-0034. [DOI] [PubMed] [Google Scholar]
- Horn F, Bilezikjin LM, Perrin MH, Bosma MM, Windle JJ, Huber KS, Blount A, Hille B, Vale W, Mellon P. Intracellular responses to gonadotropin-releasing hormone in a clonal cell line of the gonadotrope lineage. Mol Endocrinology. 1991;5:347–355. doi: 10.1210/mend-5-3-347. [DOI] [PubMed] [Google Scholar]
- Hsu S, Hsueh AJW. Human stresscopin and stresscopin-related peptide are selective ligands for the type 2 corticotropin-releasing hormone receptor. Nature Medicine. 2001;7:605–611. doi: 10.1038/87936. [DOI] [PubMed] [Google Scholar]
- Jahn O, Tezval H, van Werven L, Eckart K, Spiess J. Three-amino acid motifs of urocortin II and III determine their CRF receptor subtype selectivity. Neuropharmacology. 2004;47:233–242. doi: 10.1016/j.neuropharm.2004.03.018. [DOI] [PubMed] [Google Scholar]
- Kageyama K, Li C, Vale W. CRF receptor type 2 mRNA in rat pituitary: localization and regulation by immune challenge, restraint stress, and glucocorticoids. Endocrinology. 2003;144:1524–1532. doi: 10.1210/en.2002-221046. [DOI] [PubMed] [Google Scholar]
- Lariviere S, Garrel-Lazayres G, Simon V, Shintani N, Baba A, Counis R, Cohen-Tannoudji J. Gonadotropin-Releasing Hormone inhibits Pituitary Adenylyl Cyclase-Activating Polypeptide coupling to 3′, 5′-Cyclic Adenosine-5′-Monophosphate pathway in L{beta}T2 gonadotrope cells through novel Protein Kinase C isoforms and phosphorylation of Pituitary Adenylyl Cyclase-Activating Polypeptide type I receptor. Endocrinology. 2008 doi: 10.1210/en.2008-0504. [DOI] [PubMed] [Google Scholar]
- Lewis K, Li C, Perrin MH, Blount A, Kunitake J, Donaldson C, Vaughan J, Reyes TM, Gulyas J, Fischer W, et al. Identification of urocortin III, an additional member of the corticotropin-releasing factor (CRF) family with high affinity for the CRF2 receptor. Proc Natl Acad Sci USA. 2001;98:7570–7575. doi: 10.1073/pnas.121165198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li XF, Bowe JE, Kinsey-Jones JS, Brain SD, Lightman SL, O’Byrne KT. Differential role of corticotrophin-releasing factor receptor types 1 and 2 in stress-induced suppression of pulsatile luteinising hormone secretion in the female rat. J Neuroendocrinol. 2006;18:602–610. doi: 10.1111/j.1365-2826.2006.01450.x. [DOI] [PubMed] [Google Scholar]
- Li XF, Bowe JE, Lightman SL, O’Byrne KT. Role of corticotropin-releasing factor receptor-2 in stress-induced suppression of pulsatile luteinizing hormone secretion in the rat. Endocrinology. 2005;146:318–322. doi: 10.1210/en.2004-0950. [DOI] [PubMed] [Google Scholar]
- Lovenberg TW, Chalmers DT, Liu C, DeSouza EB. CRF2a and CRF2b receptor mRNAs are differentially distributed between the rat central nervous system and peripheral tissues. Endocrinology. 1995a;136:4139–4142. doi: 10.1210/endo.136.9.7544278. [DOI] [PubMed] [Google Scholar]
- Lovenberg TW, Liaw CW, Grigoriadis DE, Clevanger W, Chalmers DT, De Souza EB, Oltersdorf T. Cloning and characterization of a functionally distinct CRF receptor subtype from rat brain. PNAS. 1995b;92:836–840. doi: 10.1073/pnas.92.3.836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda K, Cagampang FR, Coen CW, Tsukamura H. Involvement of the catecholaminergic input to the PVN and of CRH in the fasting induced suppression of LH release in female rats. Endocrinology. 1994;134:1718–1722. doi: 10.1210/endo.134.4.8137735. [DOI] [PubMed] [Google Scholar]
- McArdle CA, Counis R. GnRH and PACAP action in gonadotropes Cross-talk between phosphoinositidase C and adenylyl cyclase mediated signaling pathways. Trends Endocrinol Metab. 1996;7:168–175. doi: 10.1016/1043-2760(96)00051-3. [DOI] [PubMed] [Google Scholar]
- McArdle CA, Poch A, Schomerus E, Kratzmeier M. Pituitary adenylate cyclase-activating polypeptide effects in pituitary cells: modulation by gonadotropin-releasing hormone in alpha T3-1 cells. Endocrinology. 1994;134:2599–2605. doi: 10.1210/endo.134.6.7515005. [DOI] [PubMed] [Google Scholar]
- Nunez L, Villalobos C, Senovilla L, Garcia-Sancho J. Multifunctional cells of mouse anterior pituitary reveal a striking sexual dimorphism. J Physiol. 2003;549:835–843. doi: 10.1113/jphysiol.2003.040758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada R, Miller MF, Yamamoto K, De Groef B, Denver RJ, Kikuyama S. Involvement of the corticotropin-releasing factor (CRF) type 2 receptor in CRF-induced thyrotropin release by the amphibian pituitary gland. Gen Comp Endocrinol. 2007;150:437–444. doi: 10.1016/j.ygcen.2006.11.002. [DOI] [PubMed] [Google Scholar]
- Owens MJ, Nemeroff CB. Physiology and pharmacology of corticotropin-releasing factor. Pharmacol Rev. 1991;43:425–473. [PubMed] [Google Scholar]
- Potter E, Sutton S, Donaldson C, Chen R, Perrin M, Lewis K, Sawchenko PE, Vale W. Distribution of corticotropin-releasing factor receptor mRNA expression in the rat brain and pituitary. Proc Natl Acad Sci USA. 1994;91:8777–8781. doi: 10.1073/pnas.91.19.8777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes TM, Lewis K, Perrin M, Kunitake KS, Vaughan J, Arias CA, Hogenesch JB, Gulyas J, Rivier J, Vale WW, et al. Urocortin II: A member of the corticotropin-releasing factor (CRF) neuropeptide family that is selectively bound by type 2 CRF receptors. Proc Natl Acad Sci USA. 2001;98:2843–2848. doi: 10.1073/pnas.051626398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivest S, Rivier C. The role of CRF and interleukin-1 in the regulation of neurons controlling reproductive functions. Endocr Rev. 1995;16:177–200. doi: 10.1210/edrv-16-2-177. [DOI] [PubMed] [Google Scholar]
- Rivier C, Vale W. Influence of corticotropin-releasing factor on reproductive functions in the rat. Endocrinology. 1984;114:914–919. doi: 10.1210/endo-114-3-914. [DOI] [PubMed] [Google Scholar]
- Roseboom PH, Urben CM, Kalin NH. Persistent corticotropin-releasing factor(1) receptor desensitization and downregulation in the human neuroblastoma cell line IMR-32. Brain Res Mol Brain Res. 2001;92:115–127. doi: 10.1016/s0169-328x(01)00162-0. [DOI] [PubMed] [Google Scholar]
- Ruhmann A, Bonk I, Lin CR, Rosenfeld MG, Spiess J. Structural requirements for peptidic antagonists of the corticotropin-releasing factor receptor (CRFR): Develoment of CRFR2b-selective antisauvagine-30. Proc Natl Acad Sci USA. 1998;95:15264–15269. doi: 10.1073/pnas.95.26.15264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoderbek WE, Kim KE, Ridgway EC, Mellon PL, Maurer RA. Analysis of DNA sequences required for pituitary-specific expression of the glycoprotein hormone alpha-subunit gene. Mol Endocrinol. 1992;6:893–903. doi: 10.1210/mend.6.6.1379672. [DOI] [PubMed] [Google Scholar]
- Schomerus E, Poch A, Bunting R, Mason WT, McArdle CA. Effects of pituitary adenylate cyclase-activating polypeptide in the pituitary: activation of two signal transduction pathways in the gonadotrope-derived alpha T3-1 cell line. Endocrinology. 1994;134:315–323. doi: 10.1210/endo.134.1.7903932. [DOI] [PubMed] [Google Scholar]
- Speert DBSJMC, Seasholtz AF. Sexually dimorphic expression of corticotropin-releasing hormone-binding protein in the mouse pituitary. Endocrinology. 2002;143:4730–4741. doi: 10.1210/en.2002-220556. [DOI] [PubMed] [Google Scholar]
- Teli T, Markovic D, Levine MA, Hillhouse EW, Grammatopoulos DK. Regulation of corticotropin-releasing hormone receptor type 1alpha signaling: structural determinants for G protein-coupled receptor kinase-mediated phosphorylation and agonist-mediated desensitization. Mol Endocrinol. 2005;19:474–490. doi: 10.1210/me.2004-0275. [DOI] [PubMed] [Google Scholar]
- Tsujii T, Attardi B, Winters SJ. Regulation of alpha-subunit mRNA transcripts by pituitary adenylate cyclase-activating polypeptide (PACAP) in pituitary cell cultures and alpha T3-1 cells. Mol Cell Endocrinol. 1995;113:123–130. doi: 10.1016/0303-7207(95)03613-c. [DOI] [PubMed] [Google Scholar]
- Van Pett K, Viau V, Bittencourt JC, Chan RK, Li HY, Arias C, Prins GS, Perrin M, Vale WW, Sawchenko PE. Distribution of mRNAs encoding CRF receptors in brain and pituitary of rat and mouse. J Comparative Neurology. 2000;428:191–212. doi: 10.1002/1096-9861(20001211)428:2<191::aid-cne1>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- Vaughan J, Donaldson C, Bittencourt J, Perrin MH, Lewis K, Sutton S, Chan R, Turnbull AV, Lovejoy D, Rivier C, et al. Urocortin, a mammalian neuropeptide related to fish urotensin I and to corticotropin-releasing factor. Nature. 1995;378:287–292. doi: 10.1038/378287a0. [DOI] [PubMed] [Google Scholar]
- Villalobos C, Nunez L, Garcia-Sancho J. Anterior pituitary thyrotropes are multifunctional cells. Am J Physiol Endocrinol Metab. 2004;287:E1166–1170. doi: 10.1152/ajpendo.00194.2004. [DOI] [PubMed] [Google Scholar]
- Westphal NJ, Evans RT, Seasholtz AF. Novel expression of type 1 CRH receptor in multiple endocrine cell types in the murine anterior pituitary. Endocrinology. 2008 doi: 10.1210/en.2008-0630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westphal NJ, Seasholtz AF. Gonadotropin-releasing hormone (GnRH) positively regulates corticotropin-releasing hormone-binding protein expression via multiple intracellular signaling pathways and a multipartite GnRH response element in alphaT3-1 cells. Mol Endocrinol. 2005;19:2780–2797. doi: 10.1210/me.2004-0519. [DOI] [PubMed] [Google Scholar]
- Westphal NJ, Seasholtz AF. CRH-BP: the regulation and function of a phylogenetically conserved binding protein. Front Biosci. 2006;11:1878–1891. doi: 10.2741/1931. [DOI] [PubMed] [Google Scholar]
- Windle JJ, Weiner RI, Mellon PL. Cell lines of the pituitary gonadotrope lineage derived by targeted oncogenesis in transgenic mice. Mol Endocrinol. 1990;4:597–603. doi: 10.1210/mend-4-4-597. [DOI] [PubMed] [Google Scholar]