

Abstract
Arthrogryposis, Renal dysfunction and Cholestasis (ARC) syndrome is a multi-system autosomal recessive disorder caused by germline mutations in VPS33B. The detection of germline VPS33B mutations removes the need for diagnostic organ biopsies (these carry a >50% risk of life-threatening haemorrhage due to platelet dysfunction); however, VPS33B mutations are not detectable in ∼25% of patients. In order further to define the molecular basis of ARC we performed mutation analysis and mRNA and protein studies in patients with a clinical diagnosis of ARC. Here we report novel mutations in VPS33B in patients from Eastern Europe and South East Asia. One of the mutations was present in 7 unrelated Korean patients. Reduced expression of VPS33B and cellular phenotype was detected in fibroblasts from patients clinically diagnosed with ARC with and without known VPS33B mutations. One mutation-negative patient was found to have normal mRNA and protein levels. This patient's clinical condition improved and he is alive at the age of 2.5 years. Thus we show that all patients with a classical clinical course of ARC had decreased expression of VPS33B whereas normal VPS33B expression was associated with good prognosis despite initial diagnosis of ARC.
Keywords: arthrogryposis, renal tubular dysfunction, neonatal cholestasis, ARC, vesicular trafficking defect
INTRODUCTION
Arthrogryposis, Renal dysfunction and Cholestasis syndrome (ARC; MIM# 208085) is an autosomal recessive disorder associated with germline mutations in VPS33B. The gene encodes VPS33B protein, one of two (−A and −B) homologues of yeast Vps33p which form part of the HOmotypic Protein Sorting (HOPS) complex involved in assisting SNARE proteins in performing membrane fusion. [Gissen et al., 2004]. Characteristic features of ARC include severe failure to thrive, renal tubular acidosis, neonatal cholestasis, ichthyosis and platelet dysfunction. Liver abnormalities (cholestasis, intrahepatocyte deposition of lipofuscin granules and bile duct hypoplasia) are also frequent but the severity of failure to thrive exceeds that expected for the degree of liver and intestinal dysfunction. The identification of mislocalised apical membrane proteins in liver and renal biopsy materials suggests a possibility of a global abnormality in intestinal absorption and renal tubular reabsorption [Gissen et al., 2004; Bull et al., 2006]. Although arthrogryposis in ARC syndrome appears to be at least partially neurogenic in origin, the degree of arthrogryposis may depend on the fetal position and severity of oligohydramnios [Gissen et al., 2006]. Platelet function abnormalities are likely to be secondary to platelet α-granule biosynthesis defect [Lo et al., 2005]. Additional features of ARC include corpus callosum dysgenesis (at least 20%) and congenital cardiac defects (∼10%).
In a previous study of ARC syndrome, mutations in VPS33B were detected in 77% of patients (48/62 individuals, Gissen et al., 2006). Absence of VPS33B mutations in patients with a clinical diagnosis of ARC syndrome might result from locus heterogeneity or the presence of VPS33B mutations that are not easily detectable by direct sequencing analysis. In order to investigate these possibilities further and to develop additional diagnostic methods for ARC syndrome we performed VPS33B mRNA and protein analysis in patients with a clinical diagnosis of ARC syndrome.
MATERIALS AND METHODS
Patients and controls
DNA and fibroblasts from patients referred for molecular investigations of ARC were used. A clinical diagnosis of ARC syndrome was based on a triad of arthrogryposis, renal tubular dysfunction and cholestasis with low serum γ-glutamyl transpeptidase (gGT) activity. Study protocols were approved by the South Birmingham research ethics committee.
Mutation detection
Mutation analysis was performed by direct sequencing of coding exons and flanking sequences as described (Gissen et al. 2004). Segregation of the putative disease-causing mutations was analysed in the affected families. DNA samples from 50 anonymised healthy South Koreans (100 chromosomes) and 100 members from a mixed UK population (200 chromosomes) were used as controls for the South Korean patients. We used 100 anonymised DNA samples from healthy UK Caucasians (200 chromosomes) as controls for the patients with Eastern and Southern European origin. None of the presumed pathological DNA variants was found in the controls.
Mutation nomenclature
Mutation nomenclature is based on GenBank entry NM_018668.3, with +1 corresponding to the A of the translation initiation codon ATG in the cDNA nomenclature, according to HGVS nomenclature guidelines (http://www.hgvs.org/mutnomen) (den Dunnen and Antonarakis, 2000).
Cell culture
Control and patients' skin fibroblasts were grown in Dulbecco's Modified Eagle's Medium supplemented with 10% fetal calf serum, 2 mM L-glutamine, MEM Non-Essential Amino Acid solution and penicillin-streptomycin (all purchased from Sigma-Aldrich, Poole, UK). Skin fibroblasts from an anonymous patient not affected with ARC were used as controls.
RNA Extraction and cDNA Synthesis
Fibroblast cells were grown to confluence in 75 cm2 flasks before RNA was extracted using RNAzol B reagent (Campro Scientific, UK) according to manufacturer's instructions. For cDNA synthesis, 1μg of total RNA was reverse transcribed using ImProm-II Reverse Transcription Systems and oligo dT primers (Promega, Southampton, UK) according to manufacturer's protocols.
Quantitative Real Time PCR Analysis
To assess the level of VPS33A and VPS33B RNA expression in patients' fibroblasts versus control fibroblasts, qRT-PCR was carried out essentially as described by Maina et al., 2005. One μl of cDNA obtained previously was used as a template for PCR amplification and primers used were denaturing HPLC-purified and specific to VPS33A and VPS33B (Thermo Fisher Scientific, Ulm, Germany). β-Actin (ACTB) was also amplified as a normalising control. Primer sequences are available upon request. After optimisation to eliminate primer-dimers, three sets of independent assays were run in triplicate for each patient and control. A negative control without cDNA template was run with every assay to assess overall specificity. An annealing temperature of 60°C and a final primer concentration of 0.1 μM were used for all reactions. Amplimer levels were continuously quantified using the ABI Power SYBR Green system and an ABI 7500 instrument (Applied Biosystems, Warrington, UK). After the assay was complete a dissociation curve analysis was carried out, and all samples were run out on a 4% agarose gel to assess the specificity of the reaction and to ensure that no primer-dimers were present.
Protein Extraction
Fibroblast cells were grown to confluence in 75 cm2 flasks before proteins were extracted. The cells were washed twice with ice cold PBS before being scraped into 1 ml of RIPA lysis buffer (50 mM Tris-HCl, 150 mM NaCl, 1 mM EDTA, 1% Igepal CA 630, 0.5% Na-deoxycholate, 0.1% SDS and complete mini protease inhibitor cocktail (Roche, Welwyn Garden City, UK). The cell lysates were then centrifuged at 17,000 RPM for 15 minutes at 4°C and the supernatants removed for immunoblotting.
Immunoblotting
For immunoblotting, 20 μg of the extracted proteins were separated using 10% SDS - PAGE and transferred to transblot polyvinylidene difluoride membranes (Hybond-P; Amersham Biosciences, Little Chalfont, UK). Proteins were immunodetected by standard protocols [Harlow and Lane, 1998]. The antibodies used were: monoclonal mouse anti-β-actin (Clone AC-15; Sigma-Aldrich, Poole, UK), polyclonal rabbit anti-VPS33B (a kind gift from Dr B. Lo, Yale University), rabbit anti-mouse IgG and goat anti-rabbit IgG HRP conjugates (Dako, Ely, UK). For detection of VPS33B, the primary and secondary antibodies were both used at a dilution of 1/2500. The bands were detected using enhanced chemoluminescence plus western blotting detection system (GE Healthcare UK, Amersham, UK). After detection of VPS33B the membranes were stripped of the first antibodies, using 0.2M NaOH for 20 minutes, and were re-blocked overnight. β-actin was then immunodetected on the same membrane, to check equal loading of proteins, using the primary antibody at a dilution of 1/15,000 and the appropriate secondary antibody at a dilution of 1/10,000.
Electron microscopy
Cells were fixed overnight at 4°C in 2.5% glutaraldehyde in 0.1 M sodium cacodylate buffer. Cells were then rinsed and postfixed for 1 hour at room temperature in reduced osmium tetraoxide (1:1 mixture of 2% aqueous potassium ferrocyanide and 3% aqueous potassium ferrocyanide) as described by Karnovsky,1971. After postfixation, cells were pre-embedded in agar (1%), dehydrated in ethanol, and processed for Epon embedding. Thin (80nm) sections were cut and collected on a copper grid and contrasted with lead citrate (2 min). Sections were then examined with a Technai 20 electron microscope operating at 120 kV.
RESULTS
Mutation analysis was performed in 16 previously unreported patients with clinical features of ARC syndrome. Eight novel mutations were identified in 12 probands (see Table 1). Strikingly, a splice site mutation (IVS6 ds+2 T>A) was detected in 7 of 9 apparently unrelated patients from South Korea. Although haplotype analysis was not possible (due to insufficient DNA) all the Korean patients were homozygous for the T allele of the rs11073964 (http://www.ensembl.org/Homo_sapiens/snpview?source=dbSNP;snp=rs11073964), which has a prevalence of 100% in the Asian population in contrast to its prevalence of 10-30% in Europeans. Among the novel mutations there were 4 insertions, one deletion, one nonsense substitution, one splice site mutation and one missense substitution. The c.728C>T (p.Ser243Phe) mutation was not present in 300 control chromosomes. The hydrophilic serine at position 243 is highly conserved in evolution [Gissen et al., 2005] and the change for a strongly hydrophobic phenylalanine may affect protein conformation and subsequently the effect of VPS33B on SNARE complex formation. Four Polish families were analysed and 2 apparently unrelated probands were homozygous for a novel c.1235_1236delCCinsG frameshift mutation. In a further Polish family a c.1594C>T mutation that was previously identified in a large consanguineous UK Pakistani family was detected.
Table 1.
Ethnicity and VPS33B mutation data in ARC patients
| Patient | Ethnicity | Nucleotide alteration | Coding-sequence alteration | Exon | Status |
|---|---|---|---|---|---|
| Pt 1 | Korean | NI | |||
| Pt 2 | Korean |
c.403+2 T>A c.1509_1510insG |
p.Lys504GlufsX23 | 6 20 |
Het Het |
| Pt 3 | Korean | c.740_741delAT | p.Tyr247X | 10 | Het |
| Pt 4 | Korean | c.403+2 T>A | 6 | Hom | |
| Pt 5 | Korean | c.403+2 T>A | 6 | Het | |
| Pt 6 | Korean |
c.403+2 T>A c.728C>T |
p.Ser243Phe | 6 10 |
Het Het |
| Pt 7 | Korean | c.403+2 T>A | 6 | Hom | |
| Pt 8 | Korean |
c.403+2 T>A c.1803_1804insA |
p.Val602SerfsX13 | 6 23 |
Het Het |
| Pt 9 | Korean | c.403+2 T>A | 6 | Hom | |
| Pt 10 | Polish | c.1235_1236delCCinsG | p.Pro12ArgfsX7 | 17 | Hom |
| Pt 11 | Polish | c.1235_1236delCCinsG | p.Pro12ArgfsX7 | 17 | Hom |
| Pt 12 | Polish | c.1576_1577insT | p.Glu525ValfsX13 | 21 | Hom |
| Pt 13* | Polish | c.1594C>T | p.Arg532X | 21 | Het |
| Pt 14 | Turkish | c.352C>T | pGln118X | 5 | Hom |
| Pt 15* | Thai | NI | |||
| Pt 16* | Portuguese | c.853-2 A>G/ c.1519 C>T |
p.Arg507X | 12-2 21 |
Het Het |
| Pt 17* | Israeli Arab | NI | |||
| Pt 18* | Pakistani | c.1312C>T | p.Arg438X | 18 | Hom |
| Pt 19* | Pakistani | c.1594 C>T | p.Arg532X | 21 | Hom |
| Pt 20* | Swedish | c.498+1 G>A | 7+1 | Het | |
| Pt 21 | Afro- Caribbean |
NI | |||
| Pt 22 | Turkish | NI |
Het - heterozygous, Hom - homozygous. NI- DNA alterations not identified. Novel mutations are indicated in boldface.
patients whose fibroblasts were used for expression analysis and who were reported previously by Gissen et al, 2006. Mutation nomenclature is based on GenBank entry NM_018668.3, with +1 corresponding to the A of the translation initiation codon ATG in the cDNA nomenclature, according to HGVS nomenclature guidelines (http://www.hgvs.org/mutnomen) (den Dunnen and Antonarakis, 2000).
To determine the potential diagnostic value of VPS33B mRNA and protein studies in cultured skin fibroblasts, expression was analysed in 7 patients with classical features of ARC (5 with a detectable VPS33B mutation) and one with a milder phenotype (Patient 21). QRT-PCR and western blotting experiments demonstrated that patients with at least one VPS33B mutation had minimal VPS33B protein expression. Of the 3 patients without identified mutations, patient 15 also had minimal VPS33B levels, patient 17 had mRNA levels of <50% compared with the control sample and patient 21 had normal levels of mRNA and protein. Patient 21 initially presented with a triad of typical ARC features as well as some additional clinical characteristics (see below); however, his disease appeared transient.
VPS33A is another homologue of yeast Vps33p in mammalian cells which may bind to HOPS complex proteins in a similar way to VPS33B. As it is not known whether VPS33A can partially substitute for the VPS33B function in ARC patients we, therefore, tested the expression of the −A homologue in VPS33B-deficient cell lines. No change in expression of VPS33A was detected in ARC fibroblasts compared with the control samples.
Cellular phenotype of skin fibroblasts from patient 13 was assessed by transmission electron microscopy (Figure 1C and 1D). Multiple heterogeneous granules were identifiable in almost all of the patient's cells. These granules were found to be osmiophilic in nature and contained multilamellar membranes and multiple vesicular aggregates.
Figure 1. Expression analysis of VPS33A and VPS33B in skin fibroblasts.
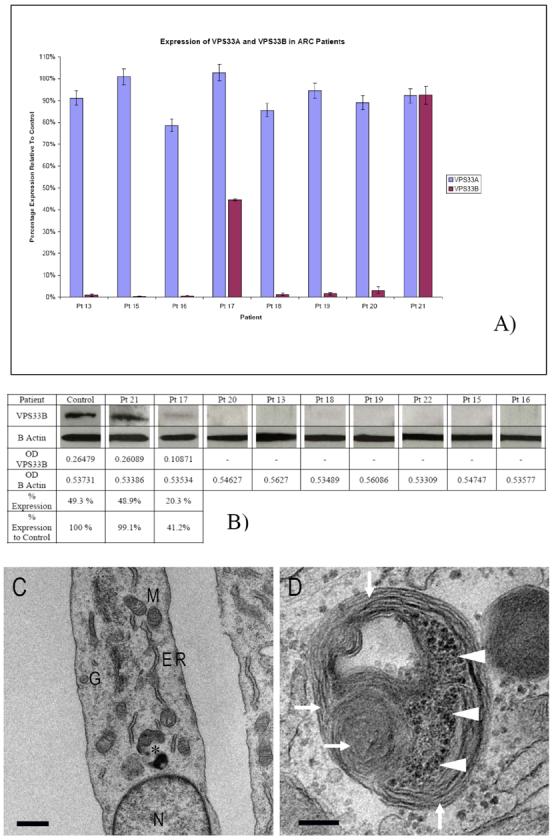
A) Bar chart showing the results from the quantitative real-time PCR experiments. Standard error of the mean shown using error bars. Pt- patient. B) Immunoblotting using anti-VPS33B primary antibody. Loading controls with anti-β-actin antibody. C) Transmission electron micrograph of patient 13 cultured skin fibroblasts. Nucleus (N), Golgi apparatus (G), endoplasmic reticulum (ER) and mitochondria (M) appear normal, bar 1μM. Atypical granule (asterisk) enlarged in D) filled with lamellar membranes (arrowheads) and vesicular aggregates (arrows), bar 200 nm.
Patient 21
Patient 21 is male, born at term to non-consanguineous Afro-Caribbean parents. He presented with arthrogryposis (both knees), spina bifida (with a lipomeningocele and tethered cord), cholestatic jaundice and renal tubular acidosis. Low gGT typical of ARC was found and liver biopsy findings were consistent with ARC, including abnormal distribution of apical canalicular markers such as carcinoembryonic antigen and alanyl aminopeptidase. Additional features included bilateral sensorineural deafness, severe diarrhoea, pancreatic insufficiency and malabsorption, which was initially treated with parenteral nutrition. Jaundice resolved by the age of 12 months and the child is now fed enterally with pancreatic enzyme supplementation. He is not facially dysmorphic but has bilateral accessory auricles. He continues to have mild metabolic acidosis, which is treated by sodium bicarbonate supplementation. At the age of 2.5 years the patient growth is parallel to 0.4th centile for his weight and height. His arthrogryposis has resolved. He has global developmental delay.
The results of investigations for ARC included negative mutation screening of VPS33B. No abnormalities were found on quantitative real-time PCR and western blotting (see Figure 1A and 1B).
DISCUSSION
ARC syndrome is a severe multisystem disorder leading to death in infancy. Early diagnosis is important for correct clinical management. However, the traditional method of diagnosis (liver biopsy) is associated in ARC with a substantial risk of morbidity and mortality. Direct sequencing of VPS33B is a good method to provide molecular diagnosis in ARC patients. However, in routine clinical practice, mutation analysis results may not be available for months and substantial numbers of patients do not have a detectable mutation. We found markedly reduced levels of VPS33B in skin fibroblasts from all patients with classical ARC phenotype and disease course. The study of patient skin fibroblasts using transmission electron microscopy identified multiple granules containing lamellar and vesicular structures. Careful examination of their identity using specific markers will provide us with better insight into the origin of these granules. This finding is in line with the accumulation of hepatocyte lipofuscin granules and skin keratinocyte lamellar bodies in ARC (Horslen et al, 1994, Hershkovitz et al, 2008) and may reflect abnormal processes of vesicular trafficking and membrane biogenesis.
The only patient with normal VPS33B expression had a milder clinical course and transient liver abnormalities. This child is alive and well on nasogastric tube feeding at 2.5 years despite having an initial clinical diagnosis of ARC syndrome. This suggests that analysis of VPS33B protein expression in cultured fibroblasts may provide a useful initial diagnostic screening investigation to facilitate acute medical management.
We identified novel mutations in our Turkish, South Korean and Polish patients with ARC. One of the mutations was present in 7 South Korean patients. This is the first report of VPS33B mutations in the East European and South East Asian populations. Identification of population-specific mutations improves the ability to provide rapid molecular diagnosis and makes molecular studies more affordable.
We hypothesized that low expression of VPS33B may trigger a cellular feedback mechanism and stimulate VPS33A expression as some functional overlap between the 2 homologues was previously identified. Thus we quantified VPS33A mRNA levels in ARC patients' fibroblasts. No change in VPS33A was found indicating that this mechanism does not spontaneously come into play. However, future attempts at cellular therapies in this disorder may include compensatory overexpression of VPS33A.
ACKNOWLEDGMENTS
PG is a GSK Clinician Scientist. Authors thank all the families and clinicians who contributed to this research. Authors also wish to thank Children's Liver Disease Foundation (CLDF), ARC syndrome association, Children Living with Inherited Metabolic Diseases (CLIMB), the Intramural Program of Eunice Kennedy Shriver National Institute of Child Health and Human Development, National Institutes of Health and Birmingham Children's Hospital Research Foundation (BCHRF) for their generous support.
REFERENCES
- Bull LN, Mahmoodi V, Baker AJ, Jones R, Strautnieks SS, Thompson RJ, Knisely AS. VPS33B mutation with ichthyosis, cholestasis, and renal dysfunction but without arthrogryposis: Incomplete ARC syndrome phenotype. J Pediatr. 2006;148:269–271. doi: 10.1016/j.jpeds.2005.10.005. [DOI] [PubMed] [Google Scholar]
- Gissen P, Johnson CA, Morgan NV, Stapelbroek J, Forshew T, Cooper WN, McKiernan PJ, Klomp LW, Morris AA, Wraith JE, McClean P, Lynch SA, Thompson RJ, Lo B, Quarrell OW, Di Rocco M, Trembath RC, Mandel H, Wali S, Karet FE, Knisely AS, Houwen RH, Kelly DA, Maher ER. Mutations in VPS33B, encoding a regulator of SNARE-dependent membrane fusion, cause arthrogryposis - renal dysfunction - cholestasis (ARC) syndrome. Nature Genetics. 2004;36:400–404. doi: 10.1038/ng1325. [DOI] [PubMed] [Google Scholar]
- Gissen P, Johnson CA, Gentle D, Hurst LD, Doherty AJ, O'Kane CJ, Kelly DA, Maher ER. Comparative Evolutionary Analysis of VPS33 Homologues: Genetic and Functional Insights. Hum Mol Genet. 2005;14:1261–1270. doi: 10.1093/hmg/ddi137. [DOI] [PubMed] [Google Scholar]
- Gissen P, Tee L, Johnson CA, Genin E, Caliebe A, Chitayat D, Clericuzio C, Denecke J, Di Rocco M, Fischler B, Fitzpatrick D, Garcia-Cazorla A, Guyot D, Jacquemont S, Koletzko S, Leheup B, Mandel H, Sanseverino MT, Houwen R, McKiernan PJ, Kelly DA, Maher ER. Clinical and molecular genetic features of ARC syndrome. Hum Genet. 2006;120:396–409. doi: 10.1007/s00439-006-0232-z. [DOI] [PubMed] [Google Scholar]
- Harlow E, Lane D. Using Antibodies: A Laboratory Manual. Cold Spring Harbor Laboratory Press; NY, USA: 1998. [Google Scholar]
- Hershkovitz D, Mandel H, Ishida-Yamamoto A, Chefetz I, Hino B, Luder A, Indelman M, Bergman R, Sprecher E. Defective lamellar granule secretion in arthrogryposis, renal dysfunction, and cholestasis syndrome caused by a mutation in VPS33B. Arch Dermatol. 2008;144:334–40. doi: 10.1001/archderm.144.3.334. [DOI] [PubMed] [Google Scholar]
- Horslen SP, Quarrell OW, Tanner MS. Liver histology in the arthrogryposis multiplex congenita, renal dysfunction, and cholestasis (ARC) syndrome: report of three new cases and review. J Med Genet. 1994;31:62–64. doi: 10.1136/jmg.31.1.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karnovsky MJ. Use of ferrocyanide-reduced osmium tetroxide in electron microscopy. Proc. 11th Meet. Am. Soc. Cell Biol. Abstr. 1971;284:146. [Google Scholar]
- Lo B, Li L, Gissen P, Christensen H, McKiernan PJ, Ye C, Abdelhaleem M, Hayes JA, Williams MD, Chitayat D, Kahr WHA. Requirement of VPS33B, a member of the Sec1/Munc18 protein family, in megakaryocyte and platelet alpha-granule biogenesis. Blood. 2005;106:4159–4166. doi: 10.1182/blood-2005-04-1356. [DOI] [PubMed] [Google Scholar]
- Maina EN, Morris MR, Zatyka M, Raval RR, Banks RE, Richards FM, Johnson CM, Maher ER. Identification of new VHL target genes and relationship to hypoxic response pathways. Oncogene. 2005;24:4549–4558. doi: 10.1038/sj.onc.1208649. [DOI] [PubMed] [Google Scholar]