

Abstract
Background
Ischemia-reperfusion (I/R) injury remains a primary complication of transplant surgery, accounting for ∼80% of liver transplant failures, and a major source of morbidity in other pathologic conditions. Activation of endothelium and inflammatory cell recruitment are central to the initiation and promulgation of I/R injury, which can be limited by the bioactive gas nitric oxide (NO). The discovery that thrombsospondin-1 (TSP1), via CD47, limits NO signaling in vascular cells and ischemic injuries in vivo suggested that I/R injury could be another important target of this signaling pathway.
Methods
Wild type, TSP1 null and CD47 null mice underwent liver I/R injury. Wild type animals were pretreated with CD47 or control antibodies prior to liver I/R injury. Tissue perfusion via laser Doppler imaging, serum enzymes, histology and immunohistology were assessed.
Results
TSP1 null and CD47 null mice subjected to subtotal liver I/R injury showed improved perfusion relative to wild type mice. Null mice subjected to liver I/R had decreased liver enzyme release and less histologic evidence of injury. Elevated TSP1 expression in liver tissue following I/R injury suggested that preventing its interaction with CD47 could be protective. Thus, pretreatment of wild type mice using a blocking CD47 antibody improved recovery of tissue perfusion and preserved liver integrity following I/R injury.
Conclusions
Tissue survival and perfusion after liver I/R injury are limited by TSP1 and CD47. Targeting CD47 prior to I/R injury enhances tissue survival and perfusion in a model of liver I/R injury and suggests therapeutics for enhancing organ survival in transplantation surgery.
Introduction
Ischemia and reperfusion (I/R) injury is a complex process that involves a variety of pathophysiologic mechanisms. Up-regulation of adhesion molecule expression mediates increased adhesion of lymphocytes and neutrophilic granulocytes to organ endothelium and their subsequent extravasation. These in turn release inflammatory cytokines and generate reactive oxygen species that mediate tissue damage. Total body or localized organ damage mediated by I/R injury is relevant in a variety of surgical fields such as transplantation medicine, cardiac surgery, and trauma surgery. Intervals of ischemia are also encountered during solid organ transplantation, myocardial revascularization, shock, and a variety of traumatic situations. Total and subtotal limb and acral part (i.e. scalp, nose, eye lids, lips, ears, digits) amputations create periods of profound ischemia that initiate an I/R response. Microsurgical replantation of devascularized tissues and organs also initiates I/R injury.
The pathophysiology of liver I/R injury includes direct cellular damage as the result of the ischemic insult as well as delayed dysfunction and damage that results from activation of inflammatory pathways 1. Histopathologic changes include cellular swelling, vacuolization, endothelial cell disruption, neutrophil infiltration, and hepatocellular necrosis 2. The distal cascade of inflammatory responses that result in organ damage after I/R injury has been studied extensively 3. Activation of Kupffer cells with production of reactive oxygen species, up-regulation of the inducible nitric oxide synthase and proinflammatory cytokines, and neutrophil accumulation contribute to inflammation-associated damage in the liver 4.
Nitric oxide (NO) is a constitutively produced bioactive gas with wide ranging physiologic properties. At low to moderate levels, NO promotes angiogenesis and tissue survival and directly increases blood flow and tissue perfusion. Inhibition of NO production worsened the outcome in a model of myocardial I/R injury 5. Conversely, administration of NO gas markedly improved the cardiac response to I/R injury 6, 7. L-arginine, the precursor for NO synthesis, added to cardioplegia solution dramatically reduced cardiac injury during cold storage 8. Therapies that increase either endogenous or exogenous NO are also beneficial in protecting other major organs from I/R injury 9. Alterations in serum liver enzymes were decreased in animals treated with L-arginine following liver I/R injury 10. Intestinal I/R injury following pre-treatment with L-arginine was also reduced and was associated with enhanced wound healing 11. Treatment with NO donor compounds in murine models of liver I/R injury dramatically decreased hepatic necrosis 12. The precise role NO plays in the response of tissues to I/R injury depends on the nature of the organ system and injury 13.
Recently, we reported that the secreted matricellular protein thrombospondin-1 (TSP1) potently blocks NO/cGMP signaling in vascular endothelial cells 14, vascular smooth muscle cells 15, and platelets 16 and that this process requires interaction with the cell surface receptor CD47 17. The physiologic implications of this are many. Deletion of TSP1 or CD47 in transgenic mice dramatically increases blood flow following NO challenge and enhanced soft tissue and hindlimb survival of fixed ischemia 18-20. Blocking the TSP1-CD47 signal in wild type mice and pigs similarly confers increased ischemic tissue survival, blood flow, and perfusion. These therapeutic advantages were demonstrated using in vivo models of subtotal and total fixed ischemia. It is not clear, however, whether TSP1 signaling through CD47 has a similar effect on reperfusion injury. In the present report we demonstrate enhanced reperfusion and decreased tissue damage following I/R injury in the absence of TSP1 and CD47. We further show that an antibody targeting CD47 can mitigate the complications of liver I/R injury in wild type mice.
Methods
Animals
Wild type, TSP1 null and CD47 null C57BL/6 mice were housed under pathogen free conditions and had ad libitum access to filtered water and standard rat chow. Handling and care of animals was in compliance with the guidelines established by the Animal Care and Use Committees of the National Cancer Institute.
Liver ischemia/reperfusion model
Age and sex matched wild type, TSP1 null and CD47 null mice were maintained with 1.5% isoflurane anesthesia. Using loupe magnification and sterile technique the right and left upper quadrants of the abdomen were entered via a chevron incision, the branches of the portal triad to the left/median lobes of the liver were identified and a 1 gram force microsurgical clamp applied for an ischemic interval of 45 minutes. This approach commonly referred to as a subtotal I/R liver injury model does not induce complete intestinal outflow obstruction as found with occlusion of the vascular flow to all portions of the liver. Prior to triad occlusion, diaphragmatic attachments to the liver were incised allowing for anterior and inferior migration of the liver and minimizing motion artifact on laser Doppler analysis from respiratory activity. Animal core temperature was continuously monitored via rectal probe and maintained at 35.5 °C with a warming surface and heat lamp. Evaporative loss was minimized with sterile plastic wrap applied to the anterior abdominal wall wound. Animals experienced either 60 or 360 minutes of reperfusion. In those animals undergoing 360 minutes of reperfusion, the anterior abdominal wall was closed in a layered manner following clamp removal with interrupted 5-0 nylon suture, animals recovered and when mobile returned to their cages. At the end of the reperfusion interval, animals were again anesthetized with isoflurane, the anterior abdominal wall closure opened, and the liver exposed to allow for laser Doppler analysis. In another group of control wild type, TSP1 null and CD47 null animals sham surgery without I/R injury was performed. Liver blood flow before and after I/R injury was determined via laser Doppler at the indicated time intervals. Serum and tissue samples were processed for enzyme levels, histology and immunohistology as described below. Data represents results of studies performed in a total of 102 animals distributed as follows: untreated liver I/R group – 48 animals including wild type, n = 24; TSP1 null, n = 12; CD47 null, n = 12; treated liver I/R – 30 animals, 15 receiving a CD47 antibody and 15 receiving an isotype matched control antibody; sham groups – 24 animal, 8 each of wild type, TSP1 null and CD47 null.
CD47 antibody treatment
Age and sex matched wild type C57BL/6 mice were randomized in 2 experimental groups and received either a rat anti-mouse CD47 monoclonal antibody (clone 301; 0.4 μg/gram weight i.p. in 100 μl sterile PBS) 90 minutes prior to surgery or a similar dose of an IgG2a isotype matched antibody (Santa Cruz Biotechnology) in 100 μl of sterile PBS. Liver blood flow before and after I/R injury was determined via laser Doppler at the indicated time intervals. Serum and tissue samples were processed for enzyme levels, histology and immunohistology as described below.
Laser Doppler analysis
Liver blood flow was measured using laser Doppler imaging (MoorLD1-2λ, Moor Instruments, Devon, England). Briefly, animals were placed in a supine position on a heating pad, and anesthesia was provided by 1.5% inhalation isoflurane in a 50:50 mixture of oxygen to room air. Core temperature was maintained via heat lamp at 35.5 °C and monitored by rectal probe. The hair of the ventral surface of the anterior abdominal wall or respective hindlimb was clipped and depiliated with Nair™ and a region of interest (ROI) marked. After equilibration to the experimental set-up, analysis of baseline hepatic blood flow was obtained. The following instrument settings were used: override distance 21 cm; scan time 4 msec/pixel. Results are expressed as the percent change from baseline control of the ROI.
Serum analysis of liver function
Blood was obtained from animals following I/R injury with a 1 cc heparin-wetted syringe via direct cardiac puncture, centrifuged at 4 °C, and serum collected and immediately analyzed. To assess hepatic function and cellular injury following liver ischemia serum enzymes including alanine aminotransferase (sALT) and aspartate aminotransferase (sAST) were measured using the Synchron LX 20 System Chemistry Analyzer (Beckman Coulter, Fullerton, CA) by the Department of Laboratory Medicine, Clinical Center, NIH.
Histology
Hepatic tissues (ischemic and normal lobes) were fixed in 10% formalin, embedded in paraffin, 5 μm thick sections cut and stained with hematoxylin-eosin, and examined by a pathologist. Leukocyte infiltration was evaluated to determine the severity of inflammation. Each liver section was divided into 10 subsections, and polymorphonuclear cell infiltration was examined by a trained pathologist blind to strain or treatment administered in each of subsections at a magnification of ×400 with cells found in sinusoids or tissue counted and those in hepatic vessels disregarded.
Immunohistochemistry
Paraffin embedded livers were sectioned at a thickness of 5 μm and applied to charged glass slides and processed for immunohistology. Briefly tissue sections were deparaffinized with xylene and rehydrated in alcohol. Slides were then heat inactivated in 10 mmol/L sodium citrate (pH 6.0) in a microwave for 5 minutes. Cooled slides were rinsed with PBS and then incubated with 3% H2O2 for 30 minutes at room temperature. Sections were then blocked with 5% normal goat serum in PBS for 30 minutes at room temperature followed by 12 hour incubation in a humidified chamber at 37 °C with a rat anti-murine macrophage antibody CD68 (clone FA-11, AbD Serotec, Oxford, England) at a 1:50 dilution, or a TSP1 monoclonal antibody clone A6.1 (LabVision, Freemont, CA) at a 1:100 dilution. Slides were washed and then incubated with goat anti-rabbit IgG-biotin conjugate (BD PharMingen) diluted at 1:100 for 45 minutes, washed with PBS and incubated in pre-diluted Streptavidin-HRP conjugate (BD PharMingen) for 45 minutes at room temperature. Color was developed by DAB substrate kit (BD PharMingen). Slides were counterstained using Mayer's hematoxylin for 2 minutes, dehydrated, and mounted.
Programmed cell death
The ApopTag in situ detection kit (Chemicon, Millipore, MA) was employed following the manufacturer's recommendations In brief, sections underwent deparaffinization, rehydration and washing, followed by treatment with 20 μg/ml of proteinase K for 15 min at room temperature and repeat washing. Endogenous peroxidase activity was quenched with 3% H2O2 in PBS for 5 min. The 3′-hydroxy DNA strand breaks were enzymatically labeled with digoxygenin nucleotide via TdT and incubated for 1 h at 37°C, and the reaction terminated with a stop buffer. Sections were then incubated with anti-digoxygenin peroxidase for 30 min at room temperature, washed, stained with 3-3′ diaminobenzidine substrate, counterstained with methyl green and mounted. Positive and negative control slides provided with the kit were used in each assay to insure consistency.
Statistics
Results are presented as the mean ± SD or where indicated as the mean ± SE with significance calculated by the Student's t test or ANOVA. Significance was assigned a p value ≤ 0.05.
Results
TSP1 and CD47 limit acute reperfusion following liver I/R injury
Wild type and null animals underwent a comparable degree of liver I/R injury which was documented by laser Doppler analysis of blood flow in all groups. However, flow following 60 minutes of reperfusion was significantly greater in the TSP1 null and CD47 null animals (Fig. 1A).
Figure 1. Liver and hind limb reperfusion is limited by TSP1 and CD47.
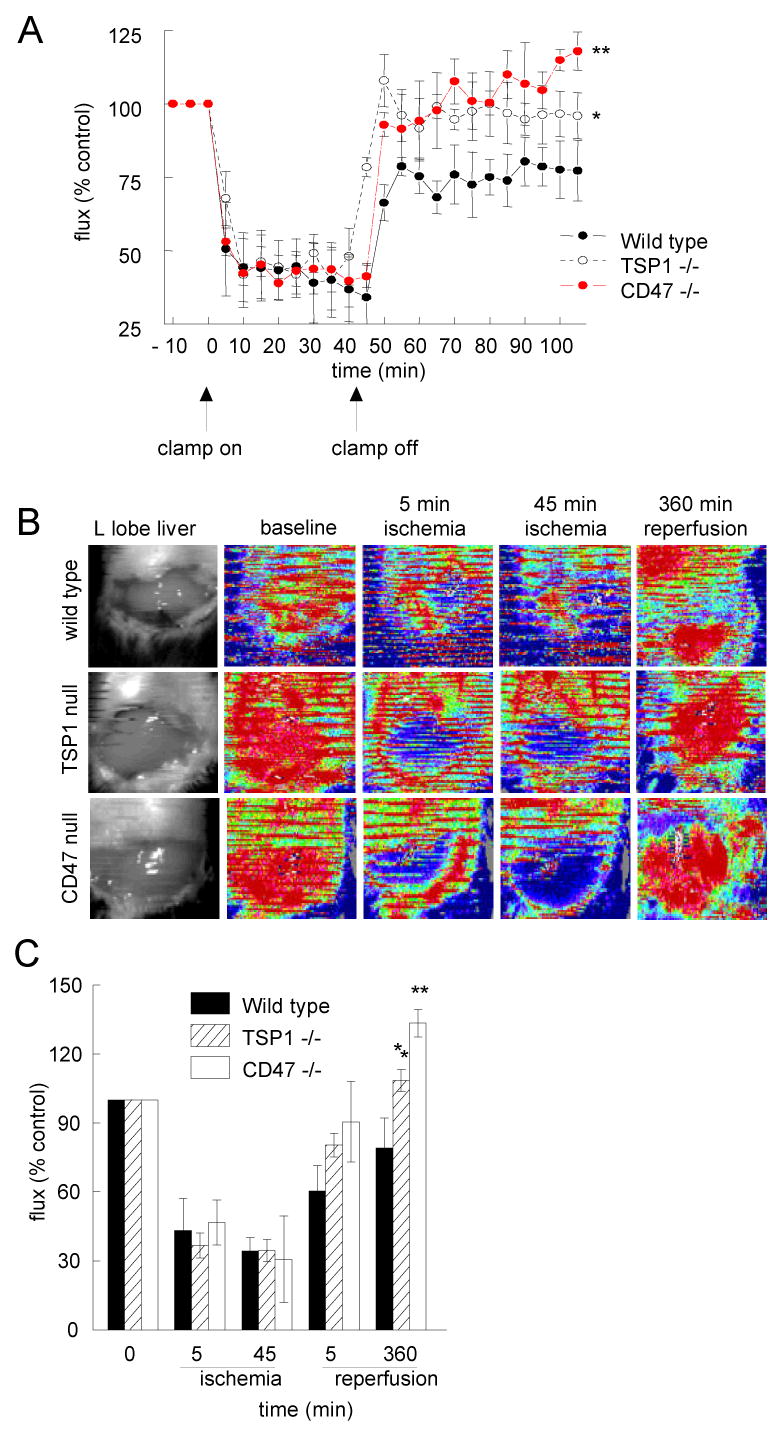
Sex and age matched C57BL/6 wild type, TSP1 null and CD47 null mice underwent 45 min occlusion of the vascular inflow of the left and median hepatic lobes followed by 60 minutes of reperfusion. Laser Doppler analysis of tissue blood flow was performed (A). Results represent the mean ± SD of 12 wild type animals and 6 each of TSP1 null and CD47 null. Data from sham animals is not shown. Sex and age matched C57BL/6 wild type, TSP1 null and CD47 null mice underwent 45 min occlusion of the vascular inflow of the left and median hepatic lobes followed by 360 minutes of reperfusion. Laser Doppler analysis of tissue blood flow was performed. Representative color Doppler images of the liver at baseline, following 5 and 45 minutes of ischemia and 360 minutes of reperfusion are presented (B). Red coloration of laser Doppler images indicates maximum and blue coloration minimum blood flow. Changes in liver perfusion at indicated time points are presented as flux normalized to baseline values (% control, A & C). Results represent the mean ± SD of 12 wild type animals and 6 each of TSP1 null and CD47 null. p < 0.05 TSP1 versus wild type indicated by * and CD47 null versus wild type indicated by **.
TSP1 and CD47 limit subacute reperfusion flux following liver I/R injury
We next extended the reperfusion time to 360 minutes. Despite comparable degrees of temporary ischemia in all groups, laser Doppler analysis of blood flow demonstrated that liver reperfusion was significantly greater following 360 minutes of reperfusion in CD47 and TSP1 null animals than in the wild type controls (Fig. 1B, C).
Liver I/R injury is decreased in the absence of TSP1 and CD47
Following liver I/R injury and both 60 and 360 minutes of reperfusion we assessed liver enzyme levels. Serum ALT was significantly lower in CD47 null animals and serum AST significantly lower in both TSP1 null and CD47 null animals compared to wild type as early as 60 minutes following clamp removal and flow restoration (Fig. 2A). With extended reperfusion of 360 minutes, decreased serum levels of AST and ALT became significant for both CD47 null and TSP1 null animals (Fig. 2B).
Figure 2. Serum enzyme markers are reduced in null animals flowing acute and sub-acute reperfusion.

Following liver ischemia and 60 (A) or 360 (B) minutes of reperfusion as described and while maintained at a constant core temperature and under isoflurane anesthesia sex and age matched wild type, TSP1 null and CD47 null mice underwent direct cardiac puncture. Blood was collected in heparin coated syringes, centrifuged, serum separated and immediately processed for serum and liver enzymes. Results represent the mean ± SD of 18 animals, six of each genotype. p < 0.05 CD47 null sALT versus wild type indicated by * and TSP1 null and CD47 null sAST null versus wild type indicated by ** (A) or TSP1 null and CD47 null sALT and sALT versus wild type indicated by * (B).
The absence of TSP1 or CD47 preserves normal liver cyto-architecture following I/R injury
H&E staining of normal and ischemic liver was performed in wild type and null animals. Wild type ischemic liver sections demonstrated significant changes in histology at both 60 and 360 minutes of reperfusion with peri-lobular swelling, cell vacuolation, necrosis of hepatocytes and destruction of parenchymal chords (Fig 3A). In contrast, TSP1 null and CD47 null ischemic livers demonstrated preservation of normal hepatic cytoarchitecture and showed markedly less necrotic areas, cell swelling and vacuolation.
Figure 3. TSP1 and CD47 null liver sections demonstrate minimal damage following I/R injury.
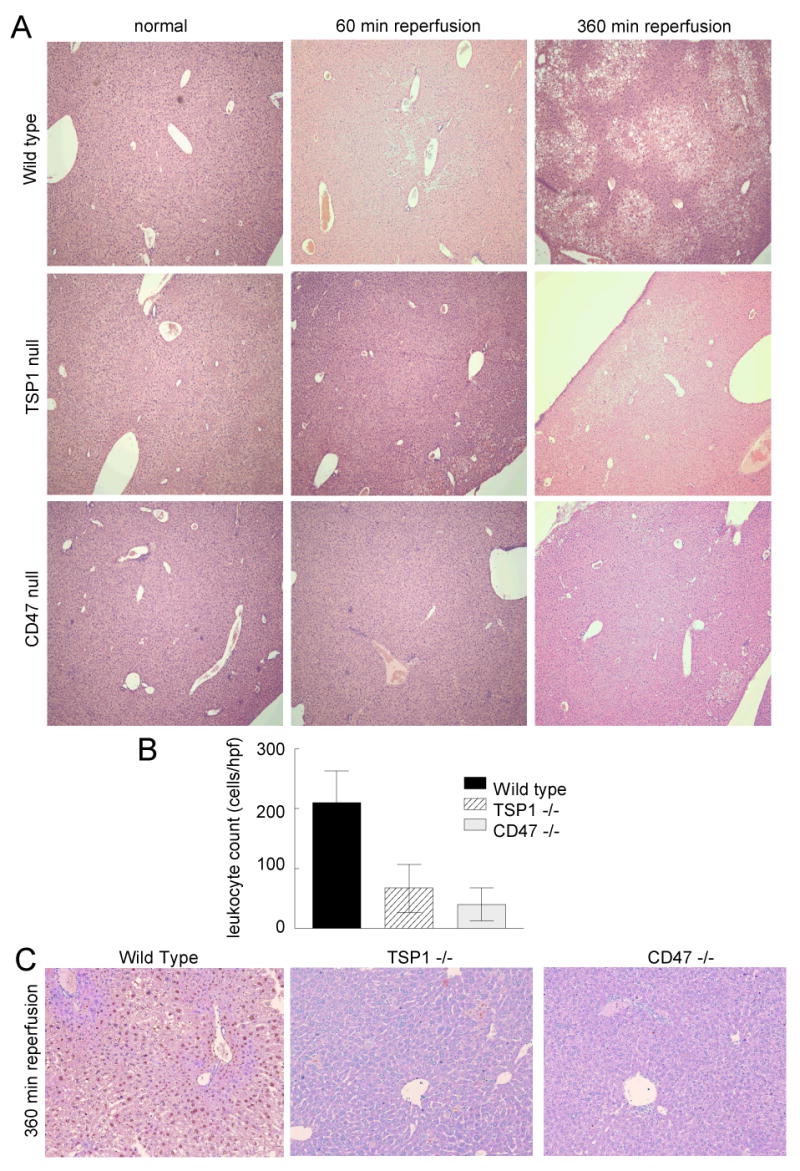
Following 45 min of liver ischemia and either 60 or 360 minutes of reperfusion age and sex matched wild type, TSP1 null and CD47 null mice were euthanized and the left and middle hepatic lobes excised, processed and stained with hematoxylin-eosin (A) (original magnification ×10). Liver sections from sham operated animals were treated similarly. Images are representative liver sections from 18 mice, six of each strain. Inflammatory cell infiltrate was determined in 10 high power fields from wild type, TSP1 null and CD47 null sections (B). Results are from analysis of 72 total sections, 4 sections each from 18 liver lobes, 6 of each genotype. Following 45 min of ischemia and 360 minutes of reperfusion wild type, TSP1 null and CD47 null liver sections were stained for evidence of tissue apoptosis/necrosis (C) (original magnification ×20). Images are representative of sections from 6 mice of each genotype.
Inflammatory infiltration of hepatic tissue is markedly reduced in TSP1 and CD47 null animals following I/R injury
Evidence of inflammatory cell infiltration was found in all ischemic liver sections after 360 minutes of reperfusion regardless of strain. However, leukocyte counts were significantly reduced in TSP1 null and CD47 null livers (Fig. 3B). Immunohistochemical staining of normal and ischemic liver for intra-hepatic macrophages (CD68) was performed. Occasional CD68 positive cells were located in sections of non-ischemic liver from all animals, though after 360 minutes of reperfusion minimal increase in macrophage infiltration was found in ischemic liver sections regardless of strain (data not shown).
Hepatocyte programmed cell death following I/R injury is decreased in the absence of TSP1 and CD47
Apoptosis and necrosis, as manifestations of programmed cell death, have been reported as a major feature of liver I/R injury 21. Analysis of tissue sections from wild type, TSP1 null and CD47 null livers following 6 hours of reperfusion demonstrated minimal to no programmed cell death in null sections (brown nuclear staining, Fig. 3C). In contrast wild type sections had substantial numbers of apoptotic and necrotic cells.
Pretreatment with a CD47 antibody protects against liver I/R injury
To investigate the potential for targeting CD47 to protect liver from damage secondary to I/R injury, a rat anti-mouse CD47 antibody (clone 301) with demonstrated protective effects in a soft tissue model of fixed ischemia 19 was administered to age and sex matched wild type mice 90 minutes prior to ischemia. Other age and sex matched wild type animals received a comparable dose of an istotype matched IgG2a control antibody 90 minutes pre-ischemia. Immunoglobulin antibodies persist for extended intervals in vivo with the major IgG subclasses lasting up to 3 weeks in people 22. Laser Doppler analysis of tissue perfusion following 360 minutes of reperfusion demonstrated significantly more robust blood flow in animals pre-treated with the CD47 targeting antibody versus the isotype matched control (Fig. 4A, p < 0.05). Serum ALT and AST levels in animals receiving CD47 antibody were also significantly decreased relative to levels obtained from animals treated with the isotype-matched control IgG (Fig. 4B).
Figure 4. Monoclonal antibody targeting of CD47 decreases I/R liver damage.
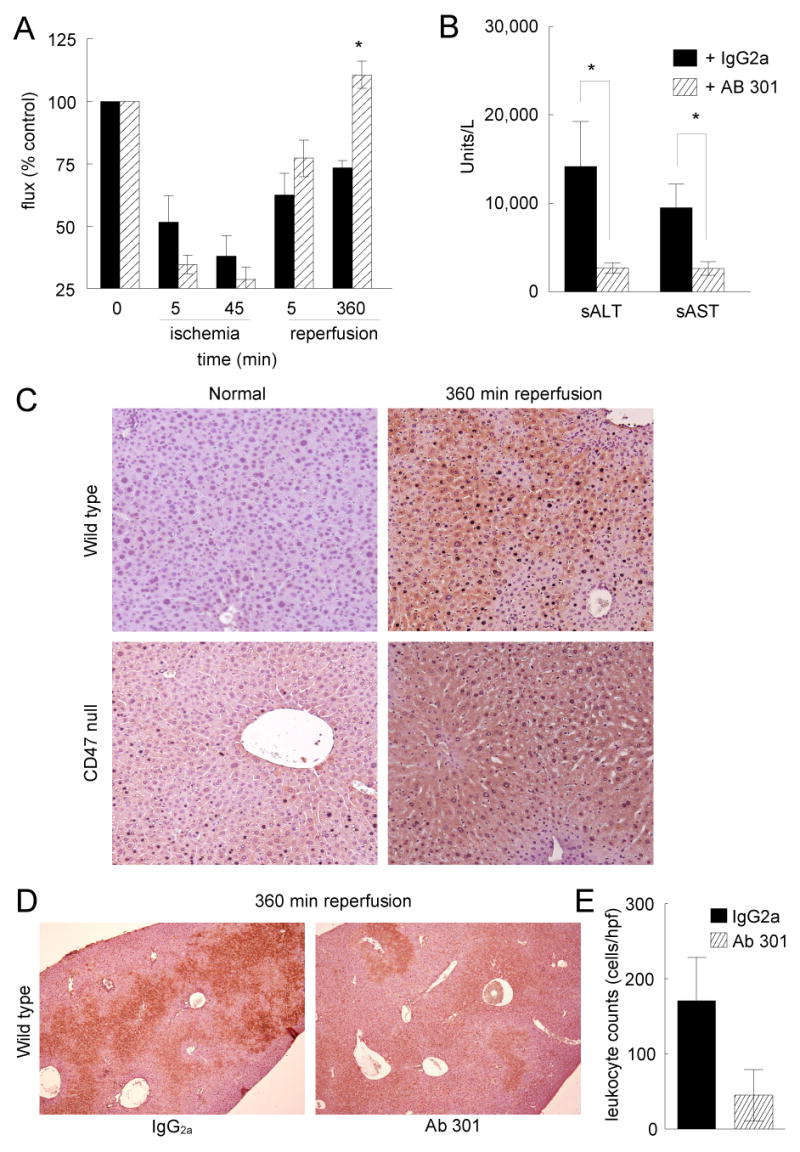
Age and sex matched wild type C57BL/6 mice received either a monoclonal CD47 antibody (clone 301) or an isotype matched control IgG2a antibody 90 minutes pre-operatively. Laser Doppler analysis of liver tissue perfusion was performed pre-operatively, after 45 minutes of ischemia, and following 360 minutes of reperfusion (A). Results represent the mean ± SD of 30 animals, 15 in each treatment group. Blood was collected for analysis of serum liver enzymes (B). Results represent the mean ± SE of 12 animals, six in each treatment group. p < 0.05 IgG2a versus 301 indicated by *. Liver tissue sections were processed for immunohistology. Normal and post I/R injury liver sections (360 minutes reperfusion) from wild type and CD47-null animals (C) (original magnification ×20) or liver sections form wild type animals pretreated with a CD47 antibody (301) or an isotype control IgG2a control antibody prior to I/R injury (360 minutes reperfusion) (D) were stained with a monoclonal TSP1 antibody (clone A6.1) (original magnification ×10). Inflammatory cell infiltrate was determined in 10 high powered fields from wild type, TSP1 null and CD47 null sections (E). Results are from analysis of 48 total sections, 4 sections each from 12 liver lobes, 6 animals in each group.
Antibody 301 is known to block CD47-dependent responses to TSP1 in fixed ischemic injuries and full thickness skin grafts 19, 23, so we wanted to confirm that TSP1 is present in the liver to engage CD47 following I/R injury. TSP1 expression was minimal in livers of control (sham surgery) wild type mice but was markedly increased following I/R injury (Fig. 4C). Post I/R injury, TSP1 expression was qualitatively less in liver sections from CD47 null animals than in wild type, although basal TSP1 expression was somewhat higher in the CD47 null livers from sham operated animals (Fig. 4C). After I/R injury, TSP1 staining in the liver was markedly less in wild type animals treated with the CD47 targeting antibody (clone 301) vs the control antibody (Fig. 4D).
Liver necrosis, peri-lobular swelling and inflammatory cell infiltration were also decreased following pretreatment with a CD47 antibody compared to controls treated with the isotype matched IgG2a antibody (data not shown). These findings correlated with both decreased TSP1 expression and inflammatory cell infiltration in post I/R injury sections from mice pre-treated with a CD47 monoclonal antibody compared to sections from animals treated with the control antibody (Fig. 4D, E).
Discussion
We demonstrate here a limiting role for TSP1 and CD47 in liver I/R injury. TSP1 null and CD47 null animals show markedly enhanced flow responses following I/R injury in a standard liver model. The absence of either protein is associated with significant increases in flow immediately and after 1 and 6 hours of reperfusion. TSP1 expression increases following I/R injury. Blocking TSP1 signaling via CD47 using a monoclonal antibody recognizing CD47 significantly enhances tissue reperfusion following I/R injury, essentially converting the wild type phenotype to that of the null.
The protective role of blocking TSP1/CD47 signaling in liver I/R injury response may involve several mechanisms. TSP1/CD47 interactions are known to increase platelet activation, aggregation and thrombosis in inflamed vasculature 24. Since TSP1/CD47 signaling blocks the anti-thrombotic activity of NO 16, enhanced flow dynamics in null animals following I/R injury may arise in part by augmenting the anti-thrombotic activity of NO.
Programmed cell death, manifesting as either cell necrosis or apoptosis, is a second important component of I/R injury 25-27. Cultured kidney cells demonstrated apoptotic changes following treatment with exogenous TSP1 28. Thyroid cells are protected from apoptotic cell death by a TSP1 peptide that binds to CD47 29 and both a gene silencing CD47 morpholino and a CD47 monoclonal antibody increased ischemic tissue survival 19, 20. Conversely, TSP1-CD47 interactions have been found to suppress non-I/R driven inflammation induced by topical application of oxazolone due to increased apoptosis of inflammatory cells 30. Compared to wild type, TSP1 null and CD47 null liver sections demonstrated dramatically less necrosis/apoptosis following I/R injury and 6 hours of reperfusion.
Recruitment of inflammatory leukocytes is another hallmark of I/R injury 31. Both TSP1 and CD47 are known to support the recruitment of inflammatory neutrophils, monocytes, and T cells 32-35. However, this is the first report of a specific role of CD47 in I/R injury. A CD47 agonist peptide sequence from the C-terminal domain of TSP1 increases monocyte adhesion to endothelial cells 36, while a CD47 monoclonal antibody (B6H12) blocks neutrophil transendothelial migration 37. Additionally, CD47 expression levels on endothelium correlated with increased neutrophil transmigration 38, and a monoclonal antibody to CD47 decreased PMN transmigration across intestinal epithelial layers in vitro 39. Therefore, the decreased inflammatory cell infiltration we documented in TSP1 and CD47 null liver sections following I/R injury compared to wild type could reflect their specific requirement for leukocyte recruitment in response to I/R stimuli. Importantly, inflammatory cell infiltration after I/R injury in wild type livers from animals pre-treated with a CD47 antibody (clone 301) was substantially less compared to sections from animals pre-treated with a matched IgG2a control antibody. Therefore, blocking CD47 may limit inflammatory cell recruitment via activated endothelium, and the absence of TSP1 may eliminate an important chemoattractant that mediates leukocyte recruitment to sites of I/R injury. Also, blocking TSP1-CD47 signaling has been found to decrease the generation of T-regulatory cells. 40 Then long term suppression of TSP1-CD47 signaling could potentially decrease tolerance development to transplanted organs and would need to be considered in designing therapeutics targeting this pathway.
Our data showing increased TSP1 expression following I/R injury in the liver is consistent with several previous studies in other I/R injury models. In a rat model of middle cerebral artery I/R injury, TSP1 expression peaked at one hour and again at 72 hours 41. I/R injury in myocardial tissue was associated with increased TSP1 mRNA expression 42. TSP1 expression was increased in areas of cell apoptosis in a model of kidney I/R injury 43. Thus, elevated TSP1 expression is a common finding in tissue subjected to I/R injury and may be associated with increased programmed cell death of damaged cells. The mechanism for induction of TSP1 in I/R injury remains to be determined, but the known induction of TSP1 by hypoxic signaling 44, 45 and by changes in NO levels should be considered 46, 47. Regarding the latter pathway, it is interesting that our immunohistochemical studies demonstrated lower TSP1 expression in liver sections following I/R injury in CD47 null mice. This implies that CD47 is both a target of TSP1 and an important mediator of the signals that control TSP1 expression.
The above results suggest that therapeutic targeting of TSP or CD47 may enhance tissue survival after I/R injury. The applications of such therapeutics would include any ischemic injury that could be treated with agents to block TSP1 interactions with CD47 or lower their expression prior to reperfusion. Specific applications would include preservation of whole organs for transplantation, microsurgery and replantation, prevention of I/R injury after open heart surgeries, and stroke. It remains to be determined whether anti-CD47 therapeutics will also be beneficial when used post I/R injury.
Acknowledgments
This research was supported by the Intramural Research Program of the NIH, NCI (M.T., D.D.R.) and NIH grants HL54390 and GM57573 (W.A.F.).
References
- 1.Kupiec-Weglinski JW, Busuttil RW. Ischemia and reperfusion injury in liver transplantation. Transplant Proc. 2005;37(4):1653–6. doi: 10.1016/j.transproceed.2005.03.134. [DOI] [PubMed] [Google Scholar]
- 2.Kim YI. Ischemia-reperfusion injury of the human liver during hepatic resection. J Hepatobiliary Pancreat Surg. 2003;10(3):195–9. doi: 10.1007/s00534-002-0730-x. [DOI] [PubMed] [Google Scholar]
- 3.Arii S, Teramoto K, Kawamura T. Current progress in the understanding of and therapeutic strategies for ischemia and reperfusion injury of the liver. J Hepatobiliary Pancreat Surg. 2003;10(3):189–94. doi: 10.1007/s00534-002-0720-z. [DOI] [PubMed] [Google Scholar]
- 4.Fondevila C, Busuttil RW, Kupiec-Weglinski JW. Hepatic ischemia/reperfusion injury--a fresh look. Exp Mol Pathol. 2003;74(2):86–93. doi: 10.1016/s0014-4800(03)00008-x. [DOI] [PubMed] [Google Scholar]
- 5.Yin H, Chao L, Chao J. Nitric oxide mediates cardiac protection of tissue kallikrein by reducing inflammation and ventricular remodeling after myocardial ischemia/reperfusion. Life Sci. 2007 doi: 10.1016/j.lfs.2007.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu X, Huang Y, Pokreisz P, et al. Nitric oxide inhalation improves microvascular flow and decreases infarction size after myocardial ischemia and reperfusion. J Am Coll Cardiol. 2007;50(8):808–17. doi: 10.1016/j.jacc.2007.04.069. [DOI] [PubMed] [Google Scholar]
- 7.Iwase H, Robin E, Guzy RD, et al. Nitric oxide during ischemia attenuates oxidant stress and cell death during ischemia and reperfusion in cardiomyocytes. Free Radic Biol Med. 2007;43(4):590–9. doi: 10.1016/j.freeradbiomed.2007.05.017. [DOI] [PubMed] [Google Scholar]
- 8.Hiramatsu T, Forbess JM, Miura T, Mayer JE., Jr Effect of L-arginine cardioplegia on recovery of neonatal lamb hearts after 2 hours of cold ischemia. Ann Thorac Surg. 1995;60(5):1187–92. doi: 10.1016/0003-4975(95)00698-K. [DOI] [PubMed] [Google Scholar]
- 9.Katsumi H, Nishikawa M, Yamashita F, Hashida M. Prevention of hepatic ischemia/reperfusion injury by prolonged delivery of nitric oxide to the circulating blood in mice. Transplantation. 2008;85(2):264–9. doi: 10.1097/TP.0b013e31815e902b. [DOI] [PubMed] [Google Scholar]
- 10.Tuncer MC, Ozturk H, Buyukbayram H, Ozturk H. Interaction of L-arginine-methyl ester and Sonic hedgehog in liver ischemia-reperfusion injury in the rats. World J Gastroenterol. 2007;13(28):3841–6. doi: 10.3748/wjg.v13.i28.3841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fotiadis C, Adamis S, Misiakos EP, et al. The prophylactic effect of L-arginine in acute ischaemic colitis in a rat model of ischaemia/reperfusion injury. Acta Chir Belg. 2007;107(2):192–200. [PubMed] [Google Scholar]
- 12.Rogers H, 3rd, Zibari GB, Roberts J, et al. Nitric oxide attenuates ischaemia-reperfusion (I/R) injury in the diabetic liver. Clin Transplant. 2004;18 12:7–11. doi: 10.1111/j.1399-0012.2004.00210. [DOI] [PubMed] [Google Scholar]
- 13.Chen T, Zamora R, Zuckerbraun B, Billiar TR. Role of nitric oxide in liver injury. Curr Mol Med. 2003;3(6):519–26. doi: 10.2174/1566524033479582. [DOI] [PubMed] [Google Scholar]
- 14.Isenberg JS, Ridnour LA, Perruccio EM, et al. Thrombospondin-1 inhibits endothelial cell responses to nitric oxide in a cGMP-dependent manner. Proc Natl Acad Sci U S A. 2005;102(37):13141–6. doi: 10.1073/pnas.0502977102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Isenberg JS, Wink DA, Roberts DD. Thrombospondin-1 antagonizes nitric oxide-stimulated vascular smooth muscle cell responses. Cardiovasc Res. 2006;71(4):785–93. doi: 10.1016/j.cardiores.2006.05.024. [DOI] [PubMed] [Google Scholar]
- 16.Isenberg JS, Romeo MJ, Yu C, et al. Thrombospondin-1 stimulates platelet aggregation by blocking the antithrombotic activity of nitric oxide/cGMP signaling. Blood. 2008;111(2):613–23. doi: 10.1182/blood-2007-06-098392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Isenberg JS, Ridnour LA, Dimitry J, et al. CD47 is necessary for inhibition of nitric oxide-stimulated vascular cell responses by thrombospondin-1. J Biol Chem. 2006;281(36):26069–80. doi: 10.1074/jbc.M605040200. [DOI] [PubMed] [Google Scholar]
- 18.Isenberg JS, Hyodo F, Matsumoto K, et al. Thrombospondin-1 limits ischemic tissue survival by inhibiting nitric oxide-mediated vascular smooth muscle relaxation. Blood. 2007;109(5):1945–52. doi: 10.1182/blood-2006-08-041368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Isenberg JS, Romeo MJ, Abu-Asab M, et al. Increasing survival of ischemic tissue by targeting CD47. Circ Res. 2007;100(5):712–20. doi: 10.1161/01.RES.0000259579.35787.4e. [DOI] [PubMed] [Google Scholar]
- 20.Isenberg JS, Hyodo F, Pappan LK, et al. Blocking thrombospondin-1/CD47 signaling alleviates deleterious effects of aging on tissue responses to ischemia. Arterioscler Thromb Vasc Biol. 2007;27(12):2582–8. doi: 10.1161/ATVBAHA.107.155390. [DOI] [PubMed] [Google Scholar]
- 21.Eum HA, Cha YN, Lee SM. Necrosis and apoptosis: sequence of liver damage following reperfusion after 60 min ischemia in rats. Biochem Biophys Res Commun. 2007;358(2):500–5. doi: 10.1016/j.bbrc.2007.04.153. [DOI] [PubMed] [Google Scholar]
- 22.Ghetie V, Popov S, Borvak J, et al. Increasing the serum persistence of an IgG fragment by random mutagenesis. Nat Biotechnol. 1997;15(7):637–40. doi: 10.1038/nbt0797-637. [DOI] [PubMed] [Google Scholar]
- 23.Isenberg JS, Pappan LK, Romeo MJ, et al. Blockade of thrombospondin-1-CD47 interactions prevents necrosis of full thickness skin grafts. Ann Surg. 2008;247(1):180–90. doi: 10.1097/SLA.0b013e31815685dc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lagadec P, Dejoux O, Ticchioni M, et al. Involvement of a CD47-dependent pathway in platelet adhesion on inflamed vascular endothelium under flow. Blood. 2003;101(12):4836–43. doi: 10.1182/blood-2002-11-3483. [DOI] [PubMed] [Google Scholar]
- 25.Kunduzova OR, Escourrou G, Seguelas MH, et al. Prevention of apoptotic and necrotic cell death, caspase-3 activation, and renal dysfunction by melatonin after ischemia/reperfusion. Faseb J. 2003;17(8):872–4. doi: 10.1096/fj.02-0504fje. [DOI] [PubMed] [Google Scholar]
- 26.Jaeschke H, Lemasters JJ. Apoptosis versus oncotic necrosis in hepatic ischemia/reperfusion injury. Gastroenterology. 2003;125(4):1246–57. doi: 10.1016/s0016-5085(03)01209-5. [DOI] [PubMed] [Google Scholar]
- 27.Hamacher-Brady A, Brady NR, Gottlieb RA. The interplay between pro-death and pro-survival signaling pathways in myocardial ischemia/reperfusion injury: apoptosis meets autophagy. Cardiovasc Drugs Ther. 2006;20(6):445–62. doi: 10.1007/s10557-006-0583-7. [DOI] [PubMed] [Google Scholar]
- 28.Khera TK, Martin J, Riley SG, et al. Glucose modulates handling of apoptotic cells by mesangial cells: involvement of TGF-beta1. Lab Invest. 2007;87(7):690–701. doi: 10.1038/labinvest.3700555. [DOI] [PubMed] [Google Scholar]
- 29.Rath GM, Schneider C, Dedieu S, et al. Thrombospondin-1 C-terminal-derived peptide protects thyroid cells from ceramide-induced apoptosis through the adenylyl cyclase pathway. Int J Biochem Cell Biol. 2006;38(12):2219–28. doi: 10.1016/j.biocel.2006.07.004. [DOI] [PubMed] [Google Scholar]
- 30.Lamy L, Foussat A, Brown EJ, et al. Interactions between CD47 and thrombospondin reduce inflammation. J Immunol. 2007;178(9):5930–9. doi: 10.4049/jimmunol.178.9.5930. [DOI] [PubMed] [Google Scholar]
- 31.Cassie S, Masterson MF, Polukoshko A, et al. Ischemia/reperfusion induces the recruitment of leukocytes from whole blood under flow conditions. Free Radic Biol Med. 2004;36(9):1102–11. doi: 10.1016/j.freeradbiomed.2004.02.007. [DOI] [PubMed] [Google Scholar]
- 32.Lindberg FP, Bullard DC, Caver TE, et al. Decreased resistance to bacterial infection and granulocyte defects in IAP-deficient mice. Science. 1996;274(5288):795–8. doi: 10.1126/science.274.5288.795. [DOI] [PubMed] [Google Scholar]
- 33.Ticchioni M, Raimondi V, Lamy L, et al. Integrin-associated protein (CD47/IAP) contributes to T cell arrest on inflammatory vascular endothelium under flow. Faseb J. 2001;15(2):341–50. doi: 10.1096/fj.99-0833com. [DOI] [PubMed] [Google Scholar]
- 34.Li Z, Calzada MJ, Sipes JM, et al. Interactions of thrombospondins with alpha4beta1 integrin and CD47 differentially modulate T cell behavior. J Cell Biol. 2002;157(3):509–19. doi: 10.1083/jcb.200109098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mansfield PJ, Suchard SJ. Thrombospondin promotes chemotaxis and haptotaxis of human peripheral blood monocytes. J Immunol. 1994;153(9):4219–29. [PubMed] [Google Scholar]
- 36.Narizhneva NV, Razorenova OV, Podrez EA, et al. Thrombospondin-1 up-regulates expression of cell adhesion molecules and promotes monocyte binding to endothelium. Faseb J. 2005;19(9):1158–60. doi: 10.1096/fj.04-3310fje. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cooper D, Lindberg FP, Gamble JR, et al. Transendothelial migration of neutrophils involves integrin-associated protein (CD47) Proc Natl Acad Sci U S A. 1995;92(9):3978–82. doi: 10.1073/pnas.92.9.3978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu Y, Merlin D, Burst SL, et al. The role of CD47 in neutrophil transmigration. Increased rate of migration correlates with increased cell surface expression of CD47. J Biol Chem. 2001;276(43):40156–66. doi: 10.1074/jbc.M104138200. [DOI] [PubMed] [Google Scholar]
- 39.Friedman GB, Taylor CT, Parkos CA, Colgan SP. Epithelial permeability induced by neutrophil transmigration is potentiated by hypoxia: role of intracellular cAMP. J Cell Physiol. 1998;176(1):76–84. doi: 10.1002/(SICI)1097-4652(199807)176:1<76::AID-JCP9>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 40.Grimbert P, Bouguermouh S, Baba N, et al. Thrombospondin/CD47 interaction: a pathway to generate regulatory T cells from human CD4+ CD25- T cells in response to inflammation. J Immunol. 2006;177(6):3534–41. doi: 10.4049/jimmunol.177.6.3534. [DOI] [PubMed] [Google Scholar]
- 41.Lin TN, Kim GM, Chen JJ, et al. Differential regulation of thrombospondin-1 and thrombospondin-2 after focal cerebral ischemia/reperfusion. Stroke. 2003;34(1):177–86. doi: 10.1161/01.str.0000047100.84604.ba. [DOI] [PubMed] [Google Scholar]
- 42.Sezaki S, Hirohata S, Iwabu A, et al. Thrombospondin-1 is induced in rat myocardial infarction and its induction is accelerated by ischemia/reperfusion. Exp Biol Med (Maywood) 2005;230(9):621–30. doi: 10.1177/153537020523000904. [DOI] [PubMed] [Google Scholar]
- 43.Thakar CV, Zahedi K, Revelo MP, et al. Identification of thrombospondin 1 (TSP-1) as a novel mediator of cell injury in kidney ischemia. J Clin Invest. 2005;115(12):3451–9. doi: 10.1172/JCI25461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chan CK, Pham LN, Zhou J, et al. Differential expression of pro- and antiangiogenic factors in mouse strain-dependent hypoxia-induced retinal neovascularization. Lab Invest. 2005;85(6):721–33. doi: 10.1038/labinvest.3700277. [DOI] [PubMed] [Google Scholar]
- 45.Distler JH, Jungel A, Pileckyte M, et al. Hypoxia-induced increase in the production of extracellular matrix proteins in systemic sclerosis. Arthritis Rheum. 2007;56(12):4203–15. doi: 10.1002/art.23074. [DOI] [PubMed] [Google Scholar]
- 46.Ridnour LA, Isenberg JS, Espey MG, et al. Nitric oxide regulates angiogenesis through a functional switch involving thrombospondin-1. Proc Natl Acad Sci U S A. 2005;102(37):13147–52. doi: 10.1073/pnas.0502979102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Murphy-Ullrich JE, Poczatek M. Activation of latent TGF-beta by thrombospondin-1: mechanisms and physiology. Cytokine Growth Factor Rev. 2000;11(12):59–69. doi: 10.1016/s1359-6101(99)00029-5. [DOI] [PubMed] [Google Scholar]