

Abstract
Base excision repair (BER) enzymes are attractive targets for antiviral and anticancer agents. A number of nucleotides and nucleotide analogues are potent competitive inhibitors of BER glycosylases when they are incorporated into synthetic oligonucleotides. However, these molecules often are not substrates for DNA polymerases, which limits their utility in cells and as potential therapeutic agents. 1′-Cyano-2′-deoxyuridine (CNdU) is a nanomolar competitive inhibitor of uracil DNA glycosylase. In addition, the respective nucleotide triphosphate is accepted as a substrate by the Klenow fragment (exo−) of DNA polymerase I from E. coli. This is the first competitive inhibitor of UDG that is incorporated into DNA by Klenow exo−, a model replicative polymerase. 1′-Cyano-2′-deoxyuridine (CNdU) and related molecules may prove useful as a new family of therapeutic or experimental agents that target DNA repair by using the cells’ polymerase(s) to incorporate them into DNA. A potential benefit of such a mechanism is that multiple incorporations can occur for longer DNA molecules leading to amplification of the inhibitory effect beyond that seen here with short DNA duplexes.
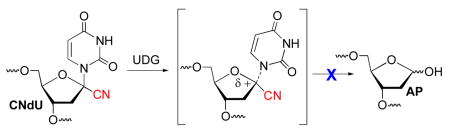
DNA base excision repair (BER) is an essential process in prokaryotes and eukaryotes for protecting the integrity of the genome. The first step in BER of modified nucleotides is carried out by glycosylases, such as uracil DNA glycosylase (UDG), which produces abasic sites (AP) from 2′-deoxyuridines that arise from deamination of 2′-deoxycytidine via an oxocarbenium ion (Scheme 1).1,2 UDG homologues exist in bacteria, viruses (e.g. HIV, HSV-1), as well as humans, and are potential antiviral and anticancer targets.3,4 Small molecule competitive inhibitors that bind UDG in the micromolar range have been discovered.5 In addition, synthetic oligonucleotides containing modified nucleotides and nucleotide analogues that inhibit UDG or other BER glycosylases have also been reported.6–11 However, the most potent of these inhibitors are not incorporated into DNA by polymerases because they lack a nucleobase. In addition, some 2′-deoxynucleotide triphosphates of inhibitors containing 2′-fluoro substituents are substrates for thermostable polymerases used in PCR and Pol α, but not Pol III, which limits their potential as therapeutic candidates.12–14
Scheme 1.

We postulated that 1′-cyano-2′-deoxyuridine (CNdU) would be a UDG inhibitor whose respective nucleotide triphosphate (2) would also be a substrate for DNA polymerase. The strong destabilization of carbocations by α-cyano groups suggested that CNdU would be a potent UDG inhibitor in DNA. Furthermore, molecular modeling of the enzyme-DNA complex in which CNdU was substituted for pseudouridine suggested that the cyano group would only weakly perturb the protein’s structure (Figure 1). The nucleotide triphosphates of various 2′-fluoro substituted nucleotides that inhibit BER glycosylases when incorporated in synthetic oligonucleotides are not accepted as substrates for DNA polymerases because they alter the pucker of the sugar ring. C1′-substituted 2′-deoxyribonucleotide triphosphates (dNTPs) have not been reported as substrates for DNA polymerases. However, the acceptance of C4′-modified dNTPs as substrates by these enzymes encouraged us to investigate 2 in this regard.15
Figure 1.
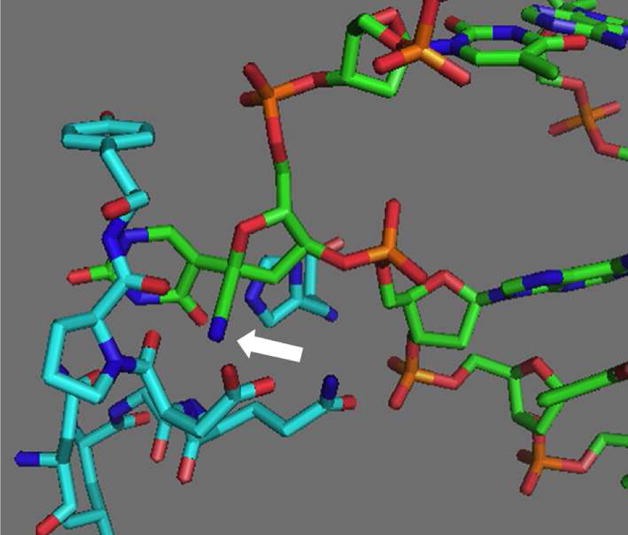
Molecular modeling showing the accommodation of a 1′-cyano substituent on pseudo-2′-deoxyuridine containing DNA co-crystallized with human UDG (PDB ID: 1EMH). The arrow points towards the cyano substituent.
Incorporation of 2 by the Klenow fragment of E. coli DNA polymerase I that lacks proofreading capability (Klenow exo−) was examined quantitatively under steady-state conditions using primer-template complex 3 and dNTP 2 that was synthesized from CNdU via standard methods.16,17 The dNTP was accepted as a substrate by Klenow exo+ (apparent Km = 14.8 ± 1.1 μM, Vmax = 6.5 ± 1.2 %•min−1, 0.67 nM), which contains proofreading ability (Figure 2A). CNdU was also incorporated slightly less efficiently by Klenow exo− (apparent Km = 15.6 ± 1.1 μM, Vmax = 6.7 ± 1.0 %•min−1, 2 nM), albeit ~350-fold less efficiently than dT is (Figure 2B). CNdU was not incorporated opposite dG or dC, and only weakly opposite dT in the presence of high concentrations (0.3 mM) of 2. In addition to selective incorporation opposite dA, complete extension was achieved when the primer-template complex (50 nM) was in excess of enzyme (5 nM), indicating that multiple molecules of CNdU can be incorporated.17
Figure 2.

Acceptance of 2 as a substrate by (A) Klenow exo+ (B) Klenow exo− using 3 as a template under steady-state conditions.
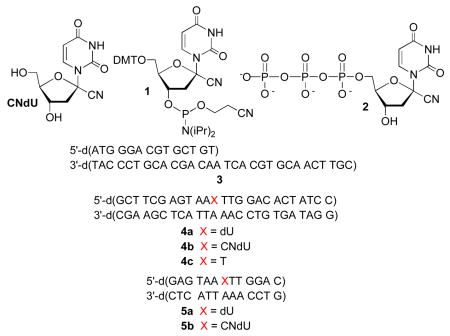
Having established that CNdU can be incorporated into DNA we turned our attention to its ability to inhibit UDG when present in the biopolymer. Oligonucleotides containing CNdU were synthesized by solid-phase synthesis from the respective phosphoramidite (1). DNA containing CNdU (4b) was not a substrate for UDG. Hydrolysis of CNdU by UDG would produce 2-deoxyribonolactone, which like AP is cleaved under mild alkaline conditions.17,18 However, no evidence of reaction of 5′-32P-4b was observed even upon prolonged exposure (24 h, 37 °C) to E. coli UDG. In contrast, 4b was a potent inhibitor. Measurement of the apparent Km (K′m) of E. coli UDG acting on 4a as a function of concentration of an otherwise identical duplex containing CNdU (4b) yielded Ki = 1.4 ± 0.1 nM (Figure 3A). The Ki of 4b was lower than the Km (10.7 ± 0.2 nM) for the substrate. The strength of the inhibition was independently verified using a method in which the ratio of observed rate constants at various concentrations of inhibitor (4b) relative to that in the absence of inhibitor were measured when the substrate is present at a concentration much lower than its Km (Table 1).19 The Ki (4.6 ± 1.2 nM) very close to that determined from the plot of Km,app versus 4b concentration. Control experiments using duplex DNA containing thymidine (4c) in place of CNdU showed that inhibition was not due to nonspecific binding. For instance, addition of 20 nM 4c (> 4 × the Ki of 4b) diminished the hydrolysis of the substrate by <15%. Finally, 4b also effectively inhibits human UDG (Table 1).
Figure 3.
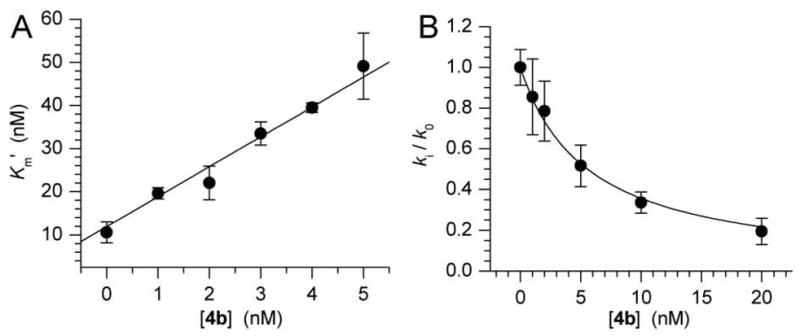
Determination of Ki of E. coli UDG by 4b by (A) determining the apparent Km (K′m) of 4a as a function of inhibitor concentration and (B) measuring the rate constant ratio in the presence of varying [4b] (ki) versus no inhibitor (k0) at [4a] ≪ Km.
Table 1.
UDG inhibition by 1′-cyano-2′-deoxyuridine (CNdU).
| Inhibitor | UDG | Ki (μM)a |
|---|---|---|
| 4b | E. coli | 4.6 ± 1.2 × 10−3 |
| 4b | Human | 13.8 ± 1.7 × 10−3 |
| CNdUb | E. coli | 245.7 ± 19.9 |
| CNdUb | Human | 131.5c |
| dUb | Human | 86.4c |
Data are the average of at least three experiments. Each experiment consists of 3 replicates.
Free nucleoside.
Result of a single experiment.
The importance of the ability of DNA polymerase to incorporate CNdU into DNA is illustrated by inhibition studies using the free nucleoside. Although the free nucleoside of CNdU inhibited UDG, its Ki was more than 10,000 times higher than when it was present in DNA (Table 1). In addition, UDG inhibition by dU and CNdU monomers are comparable, indicating that the uracil ring of the inhibitor is bound within the same enzyme active site as the substrate. We propose that a portion of the improved inhibition is attributable to the inherent electrostatic attraction between the protein and DNA. The 1′-cyano substituent may also indirectly contribute to the ability of DNA containing CNdU to bind to UDG by destabilizing the duplex. Van’t Hoff plots of otherwise identical duplexes containing dU (5a) or CNdU (5b) show that the modified nucleotide decreases the enthalpy of melting and decreases the increase in entropy (Table 2). The thermodynamic differences are consistent with a destabilized duplex, which would be expected to make binding to UDG more favorable by decreasing the energy required to flip the base out of the helix.
Table 2.
Melting thermodynamics of DNA containing CNdU and dU.
| Duplex | TM (°C)a | ΔH (kcal/mol) | ΔS (cal/mol•deg) | ΔG298 (kcal/mol) |
|---|---|---|---|---|
| 5a | 48.9 ± 0.1 | 92.9 ± 0.7 | 260.8 ± 0.1 | 15.5 |
| 5b | 44.7 ± 0.3 | 85.2 ± 4.8 | 240.2 ± 0.1 | 13.6 |
[Duplex] = 2.5 μM
In summary, we have described the first competitive inhibitor of UDG that is incorporated into DNA by the Klenow fragment of DNA polymerase I, a replicative polymerase. The presence of the molecule within the DNA scaffold contributes significantly to its potency. Nucleosides are often useful as therapeutic agents. 1′-Cyano-2′-deoxyuridine (CNdU) and related molecules may prove useful as a new family of therapeutic or experimental agents that target DNA repair by using the cells’ polymerase(s) to incorporate them into DNA. In order to be useful in this way, CNdU or a pro-drug of it will need to be a substrate for cellular kinases, which at this time is unknown. A potential benefit of such a mechanism is that multiple incorporations can occur for longer DNA molecules leading to amplification of the inhibitory effect beyond that seen here with short DNA duplexes. The in vivo effectiveness of such a strategy has been validated for the inhibition of cytosine 5-methyl DNA methyltransferases by the nucleoside prodrugs 5-azadeoxycytidine and deoxyzebularine.20
Supplementary Material
Experimental procedures, autoradiogram of full-length extension using 2, Van’t Hoff plots, and oligonucleotide characterization. This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
We are grateful for generous support from the National Institute of General Medical Sciences (GM-063028 to MMG and GM-056834 to JTS).
References
- 1.David SS, Williams SD. Chem Rev. 1998;98:1221–1261. doi: 10.1021/cr980321h. [DOI] [PubMed] [Google Scholar]
- 2.Stivers JT, Jiang YL. Chem Rev. 2003;103:2729–2759. doi: 10.1021/cr010219b. [DOI] [PubMed] [Google Scholar]
- 3.Krusong K, Carpenter EP, Bellamy SRW, Savva R, Baldwin GS. J Biol Chem. 2006;281:4983–4992. doi: 10.1074/jbc.M509137200. [DOI] [PubMed] [Google Scholar]
- 4.Liu L, Nakatsuru Y, Gerson SL. Clin Canc Res. 2002;8:2985–2991. [PubMed] [Google Scholar]
- 5.Krosky DJ, Bianchet MA, Seiple L, Chung S, Amzel LM, Stivers JT. Nucleic Acids Res. 2006;34:5872–5879. doi: 10.1093/nar/gkl747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chepanoske CL, Porello SL, Fujiwara T, Sugiyama H, David SS. Nucleic Acids Res. 1999;27:3197–3204. doi: 10.1093/nar/27.15.3197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Doi Y, Katafuchi A, Fujiwara Y, Hitomi K, Tainer JA, Ide H, Iwai S. Nucleic Acids Res. 2006;34:1540–1551. doi: 10.1093/nar/gkl061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jiang YL, Ichikawa Y, Stivers JT. Biochemistry. 2002;41:7116–7124. doi: 10.1021/bi025694y. [DOI] [PubMed] [Google Scholar]
- 9.Deng L, Schärer OD, Verdine GL. J Am Chem Soc. 1997;119:7865–7866. [Google Scholar]
- 10.Sekino Y, Bruner SD, Verdine GL. J Biol Chem. 2000;275:36506–36508. doi: 10.1074/jbc.C000585200. [DOI] [PubMed] [Google Scholar]
- 11.Schärer OD, Nash HM, Jiricny J, Laval J, Verdine GL. J Biol Chem. 1998;273:8592–8597. doi: 10.1074/jbc.273.15.8592. [DOI] [PubMed] [Google Scholar]
- 12.Ono T, Scalf M, Smith LM. Nucleic Acids Res. 1997;25:4581–4588. doi: 10.1093/nar/25.22.4581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Richardson FC, Kuchta RD, Mazurkiewicz A, Richardson KA. Biochem Pharm. 2000;59:1045–1052. doi: 10.1016/s0006-2952(99)00414-1. [DOI] [PubMed] [Google Scholar]
- 14.Wright GE, Brown NC. Pharmac Ther. 1990;47:447–497. doi: 10.1016/0163-7258(90)90066-b. [DOI] [PubMed] [Google Scholar]
- 15.Strerath M, Cramer J, Restle T, Marx A. J Am Chem Soc. 2002;124:11230–11231. doi: 10.1021/ja027060k. [DOI] [PubMed] [Google Scholar]
- 16.Creighton S, Bloom LB, Goodman MF. Methods Enzymol. 1995;262:232–256. doi: 10.1016/0076-6879(95)62021-4. [DOI] [PubMed] [Google Scholar]
- 17.Supporting Information.
- 18.Roupioz Y, Lhomme J, Kotera M. J Am Chem Soc. 2002;124:9129–9135. doi: 10.1021/ja025688p. [DOI] [PubMed] [Google Scholar]
- 19.Krosky DJ, Schwarz FP, Stivers JT. Biochemistry. 2004;43:4188–4195. doi: 10.1021/bi036303y. [DOI] [PubMed] [Google Scholar]
- 20.Lyko F, Brown R. J Natl Cancer Institute. 2005;97:1498–1506. doi: 10.1093/jnci/dji311. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Experimental procedures, autoradiogram of full-length extension using 2, Van’t Hoff plots, and oligonucleotide characterization. This material is available free of charge via the Internet at http://pubs.acs.org.