

Abstract
The β cell-specific glucose-sensitive factor (GSF), which binds the A3 motif of the rat I and human insulin promoters, is modulated by extracellular glucose. A single mutation in the GSF binding site of the human insulin promoter abolishes the stimulation by high glucose only in normal islets, supporting the suggested physiological role of GSF in the glucose-regulated expression of the insulin gene. GSF binding activity was observed in all insulin-producing cells. We have therefore purified this activity from the rat insulinoma RIN and found that a single polypeptide of 45 kDa was responsible for DNA binding. Its amino acid sequence, determined by microsequencing, provided direct evidence that GSF corresponds to insulin promoter factor 1 (IPF-1; also known as PDX-1) and that, in addition to its essential roles in development and differentiation of pancreatic islets and in β cell-specific gene expression, it functions as mediator of the glucose effect on insulin gene transcription in differentiated β cells. The human cDNA coding for GSF/IPF-1 has been cloned, its cell and tissue distribution is described. Its expression in the glucagon-producing cell line αTC1 transactivates the wild-type human insulin promoter more efficiently than the mutated construct. It is demonstrated that high levels of ectopic GSF/IPF-1 inhibit the expression of the human insulin gene in normal islets, but not in transformed βTC1 cells. These results suggest the existence of a control mechanism, such as requirement for a coactivator of GSF/IPF-1, which may be present in limiting amounts in normal as opposed to transformed β cells.
Keywords: pancreatic islets, glucose regulation, insulin gene expression
The insulin gene contains several cis-acting regulatory elements located within its 5′-flanking region that are recognized by trans-acting factors, some found ubiquitously, others more restricted to the β cell. These interactions determine the temporal expression of the gene and its inducibility by physiological stimuli (for review see refs. 1 and 2). Important transcriptional regulatory elements have been described in the promoter regions of various insulin genes, such as the E and A boxes (3). The motifs E1 and E2, with the consensus CANNTG, were found to be implicated in tissue-specific expression of insulin by transfection studies in insulin-producing cells. These bind transcription factors of the helix–loop–helix family (4, 5). However, combinations of E1 and E2 fail to confer tissue specificity in transgenic mice (6). Additional important regulatory elements containing A+T-rich sequences are the A boxes (A1–A5; ref. 3). The proximal A1 box in the rat insulin I promoter (around −80) binds a protein selectively expressed in insulin-producing cell lines, insulin promoter factor 1 (IPF-1), which is a homeodomain-containing transactivator of the insulin gene (7, 8). By gene disruption in mice, it was shown that IPF-1 is of crucial importance for normal development of the pancreas (9). The distal A3 and A4 boxes bind several homeodomain-containing proteins, such as isl-1 (10), cdx-3 and lmx-1 (11), HNF1 α (12), and Oct (unpublished results). The A3–A4 boxes of the rat insulin I gene function synergistically with the E2 box in transfected fetal islets as well as β-cell lines (13, 14), constituting the FF minienhancer element of the rat insulin I gene (around −200 to −250).
The rise in preproinsulin message in islets cultured at high glucose concentrations results from both increased mRNA stability and stimulation of the transcription of the gene (15–21). Glucose-stimulated transcription of the rat insulin I gene was shown to be mediated by the FF minienhancer element in fetal islets (18). Using normal adult rat and human islets, we have further mapped the glucose-sensitive element to the sequences −193 to −227, which contain the highly conserved A3 motif. We also demonstrated the existence of an islet-specific nuclear protein whose DNA binding to the A3 sequence of the rat I and human genes was modulated by extracellular glucose in a dose-dependent manner; we named it GSF for glucose-sensitive factor (22).
In this study, we report the purification of the protein corresponding to GSF from a rat insulinoma cell line. A single polypeptide of 45 kDa is responsible for the DNA binding activity; it was identified by microsequencing as the IPF-1 protein, originally described to bind to the A1 motif (7). We confirm the importance of the A3 site for mediating the effect of glucose on insulin gene expression in normal adult islets. In addition, we present evidence that suggests the existence of a coactivator for GSF/IPF-1 in normal as opposed to transformed insulin-producing cells, necessary for the β cell-restricted expression of the human insulin gene.
MATERIALS AND METHODS
Cell Cultures.
Rat insulinoma cells (RINm5F) were grown in RPMI 1640 medium supplemented with 10% fetal calf serum (FCS); hamster insulinoma cells (HIT) and mouse glucagonoma cells (αTC1) were grown in Dulbecco’s modified Eagle’s medium (DMEM) with 15% horse serum and 2.5% FCS; and mouse insulinoma βTC1 line and Hela cells were grown in DMEM with 10% FCS. Penicillin (100 units/ml) and streptomycin (100 mg/ml) were added to the medium.
Islet Isolation.
Male adult Sprague–Dawley rats were used. Islets were isolated and plated as described (22).
Cell Transfections.
Luciferase plasmids (10 μg) and the internal control β-galactosidase DNA plasmid (3 μg) were used to transfect both rat islets [as described (22)] and βTC1 cells [using the cationic lipid DOTAP (Boehringer Mannheim) as recommended by the manufacturer for 12 h]. The medium was then replaced with fresh medium containing the indicated glucose concentrations for 48 h. The cells were harvested, and ≈100 μg of protein extracts was used for luciferase assay employing Promega’s system, and 10 μg were used for the β-galactosidase assay using the Galacto-Light assay system (Tropix, Bedford, MA); enzyme activities were measured using a luminometer. For transactivation experiments, typically 12 μg of luciferase reporter plasmid and indicated quantities of expression vectors were cotransfected with 2 μg of β-galactosidase reporter gene driven by either cytomegalovirus (CMV) or rat β-actin promoters as internal control. Total amount of transfected DNA was adjusted to 19 μg with pcDNA3 vector (Invitrogen). Cells were grown for 48 h before assayed.
Preparation of Nuclear Extracts.
Islet and αTC1 nuclear extracts were prepared as described (22). Nuclear extracts of RINm5F cells were prepared according to Dignam et al. (23). Protein concentrations were determined by the Bradford method (24).
Gel Electrophoretic Mobility-Shift Assay (EMSA).
DNA-binding reactions were performed as previously described (22). Synthetic double-stranded oligodeoxynucleotides spanning human insulin sequences from (−206 to −227) or the mutant form carrying an A → G mutation at position −211 were end-labeled by a fill-in reaction.
Purification of GSF Protein.
Nuclear extracts from 1011 RINm5F cells were precipitated with 60% ammonium sulfate, and proteins were recovered by centrifugation, resuspended in buffer C [25 mM Hepes (pH 7.9)/20% (vol/vol) glycerol/0.1 M KCl/0.2 mM EDTA/0.5 mM DTT/1 mM phenylmethylsulfonyl fluoride (PMSF)/1 mM benzamidine/1 mM sodium vanadate/1 mM NaF/5 mM β-glycerophosphate], and dialyzed against the same buffer. Insoluble proteins were removed by centrifugation, and the remaining material was chromatographed over a 100-ml heparin–agarose column (Pharmacia). The column was first washed with 0.2 M KCl in buffer C, and proteins eluted sequentially with buffer C containing 0.3 M, 0.6 M, and 1 M KCl. The 0.3 M fractions containing GSF activity were mixed with a DNA-affinity resin prepared by coupling biotinylated DNA corresponding to the GSF binding site of the human insulin promoter (−227 5′-GATCCTGGTTAAGACTCTAATGACCA-3′ −206) to 1 ml of streptavidin–agarose (Sigma). After binding (2–3 h at 4°C), the affinity matrix was washed sequentially with buffer C, buffer C containing the mutated form of the A3 element (5′-GATCCTGGTTAAGACTCTAGTGACCA-3′) at 0.5 mg/ml and with 0.2 M KCl in buffer C. Proteins were eluted with 0.3 M and 0.6 M KCl in buffer C. The 0.3 M fractions, containing GSF, were reloaded on a second DNA-affinity column and subjected to the same washes, and the bound proteins were eluted with 0.6 M KCl in buffer C. Purified proteins were precipitated with trichloroacetic acid and sodium deoxycholate to a final concentration of 25% and 0.05%, respectively. Following 10 min of incubation on ice, the proteins were recovered by centrifugation, and the pellet was dried with acetone and resuspended in sample buffer. The purified proteins were then subjected to SDS gel electrophoresis and transferred to a polyvinylidene difluoride (PVDF) membrane (Millipore). The membrane was stained with Coomassie blue R-250 in 40% methanol and 0.1% acetic acid, then destained with 50% methanol and 10% acetic acid. Two major proteins were detected and digested in situ with Lys-C (25). Peptides were separated by capillary HPLC and sequenced on an Applied Biosystems 494A sequencer. The following sequence was obtained from one of the peptides originating from the 45-kDa band: RTXTAYXRAQLXELEKEFLFN.
Elution and Renaturation of Proteins.
The PVDF membrane-bound proteins were eluted in 3 volumes of buffer containing 1% Triton X-100, 20 mM Hepes (pH 7.6), 1 mM EDTA, 100 mM NaCl, 5 mg of BSA per ml, 2 mM DTT, 1 mM PMSF, and 0.1% aprotinin, then incubated for 3 h at 37°C. Each fraction (5 μl) was tested by EMSA.
Cloning of the Human GSF/IPF-1 cDNA.
The human islet λgt11 cDNA library was screened with a labeled fragment derived from the rat somatostatin transcription factor 1 (STF-1) cDNA clone (26). Three subsequent rounds of screening resulted in the isolation of three clones, one containing part of the N terminus and two containing part of the C terminus and the 3′-untranslated regions homologous to STF-1. The missing part was obtained by reverse transcription-PCR performed on human islet RNA using primers corresponding to the flanking sequences of the isolated human cDNA clones 5′-GCCTTTCCCATGGATGAAGTCTAC-3′ and 5′-GTCCCGCCGCCGCGCTTCTTGTC-3′. The entire hGSF/IPF-1 cDNA clone was reconstructed by subsequent subcloning of the different fragments into the expression vector pcDNA3.
Reverse Transcription–PCR.
RNA from islets or cell lines was extracted using RNAzol B (Tel-Test, Friendswood, TX). Total RNA (5 μg) was reverse-transcribed using (dT)17-adaptor as described (27). The resulting cDNAs were amplified using PCR with 100 pmol of oligonucleotides complementary to sequences in the 5′ and 3′ regions of hGSF (5′-CGGATCCTGCCTCTCATCGTGA-3′ and 5′-CGGAATTCCCGCAGCCATGAACGGC-3′) and of the control ribosomal gene coding for the L19 protein (5′-GGACAGAGTCTTGATGATCTC-3′ and 5′-CGTAAGGTCAAAGGGAATGTG-3′). Reactions were performed in 50-μl solution containing 2.5 units of Taq DNA polymerase (Promega) using the MJ Research Thermocycler (Watertown, MA). After an initial denaturation step of 5 min at 94°C, the samples were incubated for 28 cycles at 94°C, 50°C, and 72°C for 1 min each, followed by an extension at 72°C for 5 min. PCR products were analyzed by electrophoresis on 1.2% agarose.
Plasmid Construction.
The 5′-flanking region of the human insulin gene from −364 to +40 was isolated from plasmid E11 kindly provided by A. Permutt (Washington University, St. Louis). The fragment was subcloned into pGL2-Basic luciferase vector (Promega) creating the wild-type insulin–luciferase chimeric gene. The A → G mutation at position −211 was introduced by site-directed mutagenesis creating the hybrid construct. The human GSF/IPF-1 was inserted into the CMV expression vector pcDNA3 in sense (sGSF) and antisense (aGSF) orientations.
RESULTS
Characterization of GSF Binding Site in the Human Insulin Promoter.
Sequence comparison of human and rat I promoters from −206 to −227 is shown in Fig. 1 and the identical nucleotides are boxed (boxes labeled A and B). A double mutation in region B at nucleotides −219 and −220 (AA → CC) did not interfere with binding of GSF to the human sequence (Fig. 1, lane 10). In contrast, the single mutation in region A at nucleotide −211 (A → G) abolished GSF binding (lanes 5 and 11). Competition experiments showed, as expected from the above, that GSF binding was reduced by excess mutant B (lanes 4 and 9) but not mutant A (lanes 3 and 8). These results indicate that GSF binds to the A3 motif of the human insulin promoter.
Figure 1.
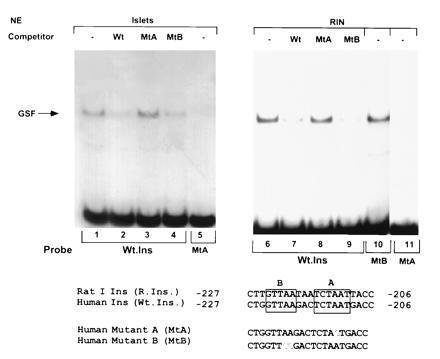
Binding of islet and RINm5F nuclear extracts to human insulin DNA sequences. EMSA was performed with nuclear extracts (NE), using as probes the wild-type human insulin sequence (Wt.Ins) and its mutated forms A (lanes 5 and 11) and B (lane 10). Competition with 50-fold excess unlabeled double-stranded oligonucleotides of wild-type (Wt; lanes 2 and 7), mutant A (MtA; lanes 3 and 8), and mutant B (MtB) sequences (lanes 4 and 9).
To investigate the importance of this specific site in the overall transcriptional response to glucose, we introduced the A → G mutation at position −211 to the human insulin–luciferase chimeric construct. Transfected rat islets and βTC1 cells were incubated in low or high glucose, and luciferase activities were measured. Islets transfected with the wild-type promoter showed ≈16-fold increase in luciferase activity when incubated in high vs. low glucose; in sharp contrast, the single mutation in the GSF binding site completely abolished the stimulatory effect of high glucose (Fig. 2). These results demonstrate that the A3 motif is crucial in glucose sensing of the human insulin gene. However, in βTC1 cells, the transfected mutated human insulin promoter was as highly active as the wild-type gene (Fig. 2), indicating that major differences exist between normal and transformed β cells in the control of insulin gene expression.
Figure 2.
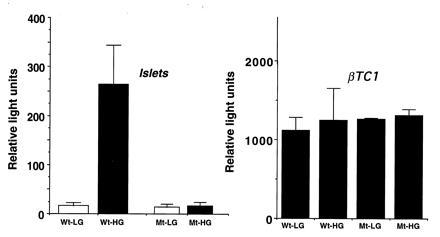
Effect of glucose on luciferase activity in transiently transfected normal islets and βTC1 cells. Wt contains the wild-type human insulin promoter linked to the luciferase reporter gene. Mt is the promoter carrying the A → G mutation (see Fig. 1). Cells were cultured in medium supplemented with 2 mM (LG) or 20 mM (HG) glucose for 48 h, extracted, and assayed for luciferase activity. Results represent the mean of five (Wt) and four (Mt) independent experiments in cultured islets and three in βTC1 cells.
Purification of the Protein Corresponding to GSF from the RINm5F Cell Line.
GSF could not be purified from normal islets due to the scarcity of tissue. We therefore chose a rat insulinoma cell line, RINm5F, and characterized the binding properties of GSF. Several lines of evidence suggest that the binding protein in RINm5F cells is similar to islet GSF (Fig. 1). Thus, complexes formed between the wild-type human insulin sequence and nuclear proteins from both normal islets and RINm5F cells showed similar migration on native gels. In addition, the binding affinities to the wild-type (Fig. 1, lanes 1 vs. 6) vs. the mutant forms of the human insulin sequences (lanes 5 vs. 11), as well as the ability of GSF to be displaced by the wild-type and the mutant forms of the insulin sequence in competition experiments, were comparable (Fig. 1, lanes 2 vs. 7, 3 vs. 8, and 4 vs. 9).
To characterize the factor(s) involved in GSF binding activity, we undertook the biochemical purification of the protein(s) from RINm5F nuclei. Using the human insulin promoter region −206 to −227 containing the A3 motif as a probe, gel mobility-shift assays were performed throughout the purification procedure to monitor the recovery of the DNA-binding activity as described in Materials and Methods. The active material, which was eluted from the final step, separated through a SDS/polyacrylamide gel, and transferred onto a PVDF membrane, revealed two major bands (Fig. 3A) at 45 and 26 kDa. Chromatography in the presence of excess nonspecific DNA and the mutated GSF binding site did not resolve the proteins, suggesting that each protein contacts directly the DNA (A3 motif). The lane containing the final eluted fraction was cut into six slices, and the proteins were eluted, renatured, and assayed for GSF binding activity by EMSA. The protein eluted from each band yielded a distinct complex in the gel-shift assay, mobility correlating with the apparent molecular mass as depicted in Fig. 3. Renatured slice number 4, which mainly included the 45-kDa band, corresponded to GSF activity, while the 26-kDa renatured polypeptide corresponded to the lower binding activity detected by EMSA (Fig. 3B). The refolded gel-purified fractions were mixed in various combinations and assayed for GSF activity; no increased binding was obtained with any combination compared with the activity of the individual components (data not shown). This suggests that GSF activity is present as a single polypeptide and indicates that the presence of other copurified proteins is not required for binding to the A3 element in the human insulin promoter.
Figure 3.
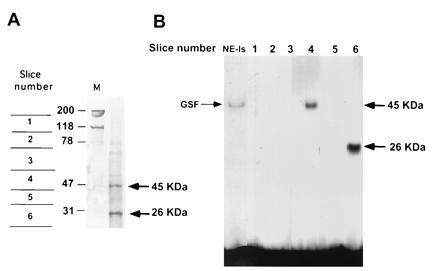
Purification of the GSF. Proteins were extracted from RINm5F nuclei and chromatographed over heparin–agarose followed by DNA affinity columns. (A) Coomassie blue staining of the final purified fraction subjected to SDS gel electrophoresis and transferred onto a PVDF membrane. Two major bands, of 45 kDa and 26 kDa, were detected. (B) EMSA of protein fractions eluted from sliced PVFD membrane containing the purified material shown in A. The 45-kDa polypeptide corresponds to GSF. NE-Is, islet nuclear extracts.
The 45-kDa and 26-kDa proteins were subjected to tryptic cleavage and resulting peptides purified by reversed-phase HPLC. The sequence of one of the peptides from the 45-kDa band matched the sequence of the previously identified IPF-1 (8). Sequence analysis of two Lys-C peptides derived from in situ digestion of the copurified 26-kDa band corresponded to the S3 ribosomal protein (28).
Sequence of hGSF/IPF-1 cDNA.
cDNA clones coding for portions of the N- and C-terminal regions of human GSF were obtained by screening a human islet cDNA library using the rat STF-1 sequence (26) homologous to mouse IPF-1. The missing cDNA was obtained by PCR using human islet RNA and the complete sequence of 1.5-kb insert determined (GenBank accession no. X99894X99894). The sequence shows an open reading frame of 283 aa; sequence comparison between human GSF, mouse IPF-1 (8), and the rat homologue STF-1 (26) or IDX-1 (29) showed a high degree of conservation (Fig. 4). The homeodomain is identical among the different species.
Figure 4.

Sequence comparison of the human GSF, mouse IPF-1, and rat STF-1/IDX-1. hGSF consists of 283 aa. The homeodomain (underlined) shares 100% amino acid identity.
Tissue Distribution of hGSF/IPF-1 mRNA.
Poly(A)-enriched mRNA from various human tissues, and β-cell lines were probed by nucleic acid hybridization for hGSF/IPF-1 mRNA. A prominent band of 1.8 kb was observed only in insulin-producing cells, and a larger mRNA species of 4.8 kb was faintly detected in most tissues (data not shown). Due to the low abundance of hGSF/IPF-1 in cells, reverse-transcribed RNAs from rat and human islets and from various cell lines were analyzed by PCR. An amplified fragment corresponding to the coding region of hGSF/IPF-1 was detected only in insulin-producing cells (Fig. 5) but not in other pancreatic cell lines like rat exocrine AR42J or mouse glucagon-producing αTC1 or in Hela cells.
Figure 5.
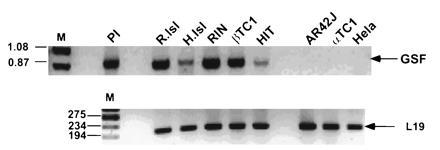
Tissue distribution of hGSF/IPF-1 mRNA. Total RNA from rat (R.Isl) and human islets (H.Isl), β-cell lines RIN, βTC1, and HIT, AR42J exocrine cells, glucagon-producing αTC1 cells, and Hela cells were reverse-transcribed. cDNAs were amplified by PCR using human primers complementary to the 5′ and 3′ ends of the hGSF/IPF-1 mRNA. A 0.88-kb fragment corresponding to the hGSF/IPF-1 coding region is shown (Upper). Primers complementary to 194 bp of the ribosomal L19 gene were used as control (Lower). Pl, hGSF/IPF-1 plasmid.
Transactivation of Human Insulin Gene by hGSF/IPF-1.
To investigate whether quantitative changes in GSF/IPF-1 expression affect insulin gene expression, rat islets and βTC1 cells were cotransfected with the human insulin promoter–luciferase chimeric gene together with different concentrations of the plasmid expressing hGSF/IPF-1. Fig. 6A shows that, while in βTC1 cells increasing amounts of ectopic hGSF/IPF-1 did not influence the expression of the reporter luciferase gene, in normal islet cells a 2-fold increase in luciferase activity was observed with low levels of expressed protein, followed by a decline in enzymatic activity at higher concentrations, yielding a bell-shaped curve. To determine whether hGSF/IPF-1 is able to transactivate the human insulin promoter in non-β cells, glucagon-producing αTC1 cells, which lack detectable amounts of GSF/IPF-1 (data not shown), were transfected with CMV-hGSF/IPF-1 in the sense (sGSF) or antisense (aGSF) orientations together with the wild-type or mutated human insulin–luciferase constructs. In these cells, the expressed GSF transactivated the wild-type construct 7-fold, while only 2-fold stimulation was observed with the mutant promoter (Fig. 6B). Similar experiments in normal islets showed the same 2-fold transactivation of the wild-type insulin promoter activity as in Fig. 6A; however, the mutant construct failed to be transactivated. In the transformed βTC1 cells, hGSF/IPF-1 expression plasmid had no effect on either construct.
Figure 6.
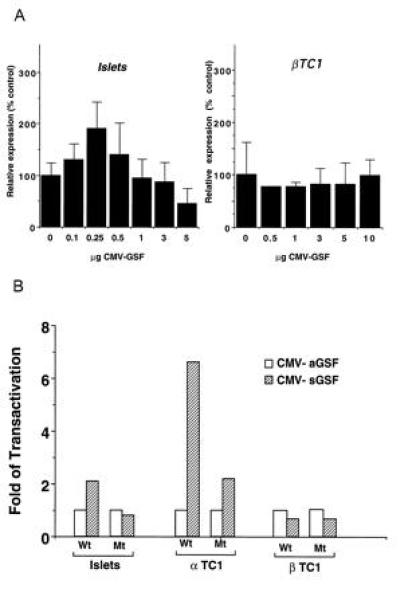
Transactivation of the human insulin promoter. (A) Indicated amounts of CMV–hGSF were cotransfected with wild-type insulin–luciferase construct in rat islets and in βTC1 cells. Expression is presented as percentage of control. Each column gives mean ± SD of three to seven independent experiments. (B) Rat islets, αTC1, and βTC1 cells were cotransfected with 0.25 μg (islets) or 5 μg (cell lines) CMV–sGSF (sense orientation, hatched bars) or CMV-aGSF (antisense, open bars) and with the wild-type human insulin (Wt) or the mutated insulin (Mt) luciferase constructs. Forty-eight hours after transfection, cell extracts were tested for luciferase activity. Results represent mean of three independent experiments. Luciferase activity was normalized to internal control β-galactosidase value.
DISCUSSION
We have previously identified an islet-specific GSF whose DNA binding to the A3 motif of the rat I and human insulin genes is modulated by extracellular glucose levels (22). Here we show that an intact A3 motif is required for GSF binding, and a single A → G mutation at −211 in the 5′-flanking region of the human insulin gene completely abolished the glucose effect in transiently transfected adult islets. However, in the non-glucose-responsive βTC1 cells the mutated human insulin promoter was as active as the wild-type sequence. The results obtained from normal adult rat islets strongly support our contention that the GSF binding site is a crucial physiological element in the glucose-regulated expression of the insulin gene. Similar results were obtained by Petersen et al. (30) in newborn rat islets stressing anew the need of using normal islets to obtain physiologically significant information.
To characterize the DNA-binding component of the GSF protein complex, we used DNA affinity chromatography and purified two major peptides of 45 and 26 kDa that specifically interacted with the GSF binding site. Elution and renaturation of the 45-kDa protein was sufficient to form the complex with the A3 element, with a mobility corresponding to GSF. Its amino acid sequence showed that GSF is similar to the cloned IPF-1 (8). IPF-1 was originally described as an insulin promoter A1 element-binding protein; the evidence presented here shows that it also binds the A3 motif within the human insulin promoter region, corresponding to GSF. MacFarlane et al. (31) reported that a previously described binding activity, IUF1, appears to correspond to GSF, since it also binds the A3 element and is modulated by extracellular glucose in normal islets; furthermore, antibodies against mouse IPF-1 specifically competed for IUF1 binding. IPF-1, required for pancreas development in mice, was mainly expressed in the β cells of adult islets (9, 32). Simultaneously, an identical protein, STF-1 (or IDX-1), was shown to transactivate the somatostatin gene (26, 29). It was further suggested that IPF-1/STF-1 expression was initiated in the stem cells of the duodenum and pancreas and that it is transiently expressed in non-β cells during development (32). Based on the results presented we suggest that, in addition to being involved in the determination/differentiation of pancreatic tissue and acting as a key regulator of insulin gene expression, the protein named varyingly GSF/IPF-1/STF-1/IDX-1, at the terminally differentiated stage of the β cell, acquires the role of a glucose-sensitive factor, controlling the physiological expression of the insulin gene (triple function). This type of multiple roles for a transcription factor is not unique to the β cell. As example, Pit-1 (GHF-1), a transcription factor restricted to the anterior pituitary gland, can determine cell phenotype, regulate the proliferation of differentiated cell types, and play a role in more transient regulation of gene expression (33).
GSF/IPF-1 was expressed only in insulin-producing cell lines; it was not found in pancreatic non-insulin-producing cells, such as exocrine (AR42J) or glucagon-producing (αTC1) cell lines, or any other human tissue tested. A larger mRNA species was faintly observed in most tissues; its significance is unclear.
Several groups (13, 14) have described synergism between the E2 box and the adjacent A3–A4 motifs in the stimulation of the rat insulin I promoter. Here we provide direct evidence that synergism does not occur at the DNA binding level, since no other protein copurified with GSF enhanced the binding in reconstitution experiments. This was confirmed by expressing hGSF/IPF-1 in αTC1 and Hela cells, where only one complex migrating as GSF was detected with probes containing the A3 motif (data not shown).
The second 26-kDa protein, which tightly bound to the A3 motif, corresponding to the lower complex obtained by EMSA, was not a proteolytic degradation product of the 45-kDa GSF polypeptide. Its amino acid sequence matched that of the S3 ribosomal protein (28). The drosophilia S3 contains a nuclear localization signal and binds and specifically cleaves DNA that contains an apurinic/apyrimidinic site (34); such a site is present in the GSF-binding sequence used for DNA affinity chromatography. In any event, mixture of the purified polypeptides did not modify the binding pattern of each one tested separately, negating cooperativity between the two proteins.
hGSF/IPF-1 transactivated the human insulin promoter in non-insulin-producing cell lines such as αTC1 and Hela cells (data not shown) but not in the insulin-producing transformed βTC1 cells. Interestingly, in normal islets, low levels of hGSF/IPF-1 were more efficient in mediating a slight transactivation; higher concentrations had an inhibitory effect upon gene expression. We suggest that this repression might occur by a protein–protein interaction—e.g., hGSF/IPF-1 might compete for binding to a TFIID-associated factor capable of mediating transcriptional activation or to a coactivator present only in limited amounts in normal adult islets, but not in the transformed βTC1 cells. It was reported that STF-1 induces insulin expression by acting cooperatively with the helix–loop–helix protein E47 (14). Recently, STF-1 was also found to form heterodimeric complexes with Pbx, the mammalian homologue of the drosophilia extradentile (35). These heterodimers bound the TAAT sequence of the somatostatin promoter but not the same sequence (A3 motif) of the insulin promoter, suggesting that this preference may form the basis for target site selection in developing islet cells and that the transcriptional function of the gene may be highly context-dependent. Indeed, GSF/IPF-1, like other homeodomain proteins, may not act alone. It is possible that in normal β cells, GSF/IPF-1 behaves like the octamer-binding protein Oct-2; this transcription factor binds the immunoglobulin promoter and interacts specifically with the coactivator OBF-1, which is only expressed in B lymphocytes and has no intrinsic DNA binding activity (36). Such potential interactions of GSF/IPF-1, mainly in the glucose-stimulated normal adult β cell, remain to be investigated.
Acknowledgments
We are indebted to Drs. D. Goeddel, S. McKnight, and T. Hoey (Tularik, San Francisco) for substantial help, guidance, and encouragement throughout the purification procedure. We thank Drs. T. Henkel and J. Hou (Tularik) for helpful suggestions. We are grateful to Dr. W. Henzel (Genentech, South San Francisco, CA) for peptide sequencing. We thank Dr. R. Stein (Vanderbilt University, Nashville, TN) for the gift of rat STF-1, and Dr. A. M. Permutt (Washington University) for the human islet cDNA library. Our thanks to Drs. Y. Bergman and Y. Ben-Neriah (Hebrew University Hadassah Medical School, Jerusalem) for critically reading the manuscript. This work was supported by the Juvenile Diabetes Foundation International and the Israel Science Foundation (D.M.).
Footnotes
Abbreviations: GSF, glucose-sensitive factor; IPF-1, insulin promoter factor 1; STF-1, somatostatin transcription factor 1; CMV, cytomegalovirus; EMSA, electrophoretic mobility-shift assay.
Data deposition: The sequence reported in this paper has been deposited in the GenBank data base (accession no. X99894X99894).
References
- 1.Stein R. Trends Endocrinol Metab. 1993;4:96–101. doi: 10.1016/1043-2760(93)90086-t. [DOI] [PubMed] [Google Scholar]
- 2.Docherty K, Clark A R. FASEB J. 1994;8:20–27. doi: 10.1096/fasebj.8.1.8299887. [DOI] [PubMed] [Google Scholar]
- 3.German M, Ashcroft S, Docherty K, Edlund H, Edlund T, et al. Diabetes. 1995;44:1002–1004. doi: 10.2337/diab.44.8.1002. [DOI] [PubMed] [Google Scholar]
- 4.Murre C, McCaw P S, Baltimore D. Cell. 1989;56:777–783. doi: 10.1016/0092-8674(89)90682-x. [DOI] [PubMed] [Google Scholar]
- 5.Naya F J, Stellrecht C M M, Tsai M-J. Genes Dev. 1995;8:1009–1019. doi: 10.1101/gad.9.8.1009. [DOI] [PubMed] [Google Scholar]
- 6.Dandoy-Dron F, Monthioux E, Jami J, Bucchini D. Nucleic Acids Res. 1991;18:4925–4930. doi: 10.1093/nar/19.18.4925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ohlsson H, Thor S, Edlund T. Mol Endocrinol. 1991;5:897–904. doi: 10.1210/mend-5-7-897. [DOI] [PubMed] [Google Scholar]
- 8.Ohlsson H, Karlsson K, Edlund T. EMBO J. 1993;12:4251–4259. doi: 10.1002/j.1460-2075.1993.tb06109.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jonsson J, Cartsson L, Edlund T, Edlund H. Nature (London) 1994;371:606–609. doi: 10.1038/371606a0. [DOI] [PubMed] [Google Scholar]
- 10.Karlsson O, Thor S, Norberg T, Ohlsson H, Edlund T. Nature (London) 1990;344:879–882. doi: 10.1038/344879a0. [DOI] [PubMed] [Google Scholar]
- 11.German M S, Wang J, Fernald A A, Espinosa R, III, Lebeau M M, Bell G I. Genomics. 1994;24:403–404. doi: 10.1006/geno.1994.1639. [DOI] [PubMed] [Google Scholar]
- 12.Emans L A, Landers D W, Moss L G. Proc Natl Acad Sci USA. 1992;89:7300–7304. doi: 10.1073/pnas.89.16.7300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.German M S, Wang J, Chadwick R B, Rutter W J. Genes Dev. 1992;6:2165–2176. doi: 10.1101/gad.6.11.2165. [DOI] [PubMed] [Google Scholar]
- 14.Peers B, Leonard J, Sharma S, Teitelman G, Montminy M R. Mol Endocrinol. 1994;8:1798–1806. doi: 10.1210/mend.8.12.7708065. [DOI] [PubMed] [Google Scholar]
- 15.Nielsen D A, Welsh M, Casadaban M J, Steiner D F. J Biol Chem. 1985;260:13585–13589. [PubMed] [Google Scholar]
- 16.Welsh M, Nielsen D A, MacKrell A J, Steiner D F. J Biol Chem. 1985;260:13590–13594. [PubMed] [Google Scholar]
- 17.Melloul, D. & Cerasi, E. (1994) Diabetologia 37, Suppl. 2, S3–S10. [DOI] [PubMed]
- 18.German M S, Moss L G, Rutter W J. J Biol Chem. 1990;265:22063–22066. [PubMed] [Google Scholar]
- 19.Efrat S, Surana M, Fleischer N. J Biol Chem. 1991;266:11141–11143. [PubMed] [Google Scholar]
- 20.German M S, Wang J. Mol Cell Biol. 1994;14:4067–4075. doi: 10.1128/mcb.14.6.4067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sharma A, FuscoDeMane D, Henderson E, Efrat S, Stein R. Mol Endocrinol. 1995;9:1468–1476. doi: 10.1210/mend.9.11.8584024. [DOI] [PubMed] [Google Scholar]
- 22.Melloul D, Ben Neriah Y, Cerasi E. Proc Natl Acad Sci USA. 1993;90:3865–3869. doi: 10.1073/pnas.90.9.3865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dignam J D, Lebovitz R M, Roeder R G. Nucleic Acids Res. 1983;11:1475–1489. doi: 10.1093/nar/11.5.1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bradford M M. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 25.Henzel W J, Grimley C G, Bourell J H, Billeci T M, Wong S C, Stult J T. Companion Methods Enzymol. 1994;6:239–247. [Google Scholar]
- 26.Leonard J, Peers B, Johnson T, Ferrere L, Lee S, Montminy M R. Mol Endocrinol. 1994;7:1275–1283. doi: 10.1210/mend.7.10.7505393. [DOI] [PubMed] [Google Scholar]
- 27.Frohman M A, Dush M K, Martin G R. Proc Natl Acad Sci USA. 1988;85:8998–9002. doi: 10.1073/pnas.85.23.8998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wilson D M, III, Deutsch W A, Kelley M R J. Nucleic Acids Res. 1993;21:2516. doi: 10.1093/nar/21.10.2516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Miller C P, McGhee R E, Habener J. EMBO J. 1994;13:1145–1156. doi: 10.1002/j.1460-2075.1994.tb06363.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Petersen H V, Serup P, Leonard J, Michelsen B, Madsen O. Proc Natl Acad Sci USA. 1994;91:10465–10469. doi: 10.1073/pnas.91.22.10465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.MacFarlane W M, Read M L, Gilligan M, Bujalska I, Docherty K. Biochem J. 1994;303:625–631. doi: 10.1042/bj3030625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Guz Y, Montminy M R, Stein R, Leonard J, Gamer L W, Wright C V E, Teitelman G. Development (Cambridge, UK) 1995;121:11–18. doi: 10.1242/dev.121.1.11. [DOI] [PubMed] [Google Scholar]
- 33.Andersen B, Rosenfeld M G. J Biol Chem. 1994;269:29335–29338. [PubMed] [Google Scholar]
- 34.Wilson D M, Deutsch W A, Kelley M R J. J Biol Chem. 1994;269:25359–25364. [PubMed] [Google Scholar]
- 35.Peers B, Sharma S, Johnson T, Kamps M, Montminy M R. Mol Cell Biol. 1995;15:7091–7097. doi: 10.1128/mcb.15.12.7091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Strubin M, Newell J W, Mattias P. Cell. 1995;80:497–506. doi: 10.1016/0092-8674(95)90500-6. [DOI] [PubMed] [Google Scholar]