

Abstract
The positive association between body iron stores and risk of coronary heart disease (CHD) initially observed among a Finnish male population has not been corroborated by studies conducted in other populations. The soluble transferrin receptor (sTfR):ferritin ratio has been suggested to be a better index than ferritin to measure body iron stores. Because sTfR is sensitive to iron deficiency, this ratio can distinguish individuals with similar ferritin levels with respect to their iron status. To evaluate this novel index in relation to CHD risk, we prospectively identified and confirmed 242 incident CHD cases and randomly selected 483 controls matched for age, smoking, and fasting status among women that provided blood samples in the Nurses' Health Study during 9 y of follow-up. In both crude and multivariate analyses, neither the sTfR:ferritin ratio nor ferritin was significantly associated with an elevated risk of CHD. After multivariate adjustment for established and potential CHD risk factors, compared with women in the lowest quartile of the sTfR:ferritin ratio, women in the 2nd to 4th quartiles had relative risks (RR) (95% CI) of 1.39 (0.82, 2.36), 1.12 (0.66, 1.91), and 1.13 (0.65, 1.97; P-trend = 0.61), respectively. The multivariate RR (95% CI) for ferritin were 1.05 (0.62, 1.77), 1.19 (0.69, 2.03), and 1.05 (0.60, 1.85; P-trend = 0.90) across quartiles. Our data do not support the hypothesis that excessive body iron stores are associated with risk of CHD.
Introduction
Although animal studies have shown that iron overload exacerbates the myocardial damage caused by reperfusion and anoxia while administration of iron chelators could prevent or alleviate such damage (1), results from human studies examining associations between body iron stores and risk of coronary heart disease (CHD)9 were inconclusive (2). Evidence supporting a positive association of excessive body iron stores with CHD was primarily from studies conducted in a Finnish population (3,4), whereas studies conducted in other populations failed to corroborate such an observation (5–12). Although dietary or genetic factors may explain this discrepancy (13,14), studies are needed to shed light on this iron-heart hypothesis.
In most of these studies, plasma ferritin, transferrin saturation, total iron-binding capacity, or serum iron were used as objective markers for body iron stores. Of these biomarkers or indices, ferritin is considered the best single indicator of total body iron (15). However, body iron status is not the only determinant of plasma ferritin concentrations; acute or chronic inflammation may stimulate the production of plasma ferritin (16). Circulating soluble transferrin receptor (sTfR) is a truncated form of the tissue transferrin receptor. In contrast to ferritin, which is sensitive to excessive body iron stores and inflammation, sTfR is sensitive to iron deficiency and is believed to be free of influence by acute or chronic inflammation. Because of this complementary relationship between these 2 indices with respect to measurements of body iron stores, the sTfR:ferritin ratio was suggested to be a better marker than ferritin to measure a wide range of iron status, because this ratio has been demonstrated to distinguish between subjects with similarly high ferritin levels (17). Thus far, few prospective studies have investigated the associations of the sTfR:ferritin ratio with CHD (4) and no studies have evaluated this index among women. Therefore, we conducted a prospective nested case-control study to examine the associations of the plasma sTfR:ferritin ratio and ferritin concentrations with risk of CHD in women.
Methods
Study population.
Over 121,700 female registered nurses aged 30–55 y were enrolled in the Nurses' Health Study in 1976 and completed baseline questionnaires about their lifestyle and medical history. Blood samples were collected from 32,826 nurses in 1989 and 1990. Ninety-seven percent of the blood samples were received within 26 h of the blood draw. Immediately upon arrival, the samples were centrifuged at 1200 × g; 15 min at room temperature and divided into aliquots of plasma, erythrocytes, and buffy-coat fractions, which were then placed in liquid nitrogen freezers at −130°C or colder until analysis.
During 9 y of follow-up, of the participants who provided blood samples and were free of cardiovascular diseases and cancer at phlebotomy, 248 incident cases of CHD were identified and confirmed. By using risk-set sampling, we randomly selected 2 controls for each case and matched controls with cases for age (±1 y), smoking status (never, past, and current), and fasting status (fasting for 8 h or not). After excluding the participants with missing sTfR:ferritin ratio data, 242 CHD cases and 483 controls were available for the current analysis.
All participants gave written informed consent. The study protocol was approved by the institutional review board of the Brigham and Women's Hospital and the Human Subjects Committee Review Board of Harvard School of Public Health.
Ascertainment of CHD.
Participants who reported coronary events were requested to provide medical records for endpoint confirmation. Study physicians who were not aware of the exposure status of nurses reviewed available medical records. Nonfatal myocardial infarction was confirmed if the WHO criteria were met, which require typical symptoms plus either diagnostic electrocardiographic findings or elevated cardiac enzyme levels. For those whose medical records were unavailable, the diagnosis was considered probable if supported by telephone interviews or other supplemental information. We identified deaths among the Nurses' Health Study participants through reports from next of kin or postal authorities, or by searching the National Death Index. CHD deaths were identified if CHD was listed as the cause of death in hospital records, autopsy reports, or death certificates. CHD deaths were then confirmed by a previous report of CHD and if there was no other more apparent or plausible cause of death. All CHD cases in the current analysis were confirmed (91% confirmed by medical record).
Laboratory procedures.
Each case-control triplet was shipped in the same batch and analyzed in the same run. Within each triplet, samples were assayed by the same technicians in a random sequence under identical conditions.
For case-control triplets selected in 1990–1996 (n = 161), sTfR was measured by an ELISA (R & D Systems). For case-control triplets selected after 1996 (n = 81), sTfR was measured by a particle-enhanced immunoturbidimetric assay using the Hitachi 917 analyzer and Roche Diagnostics reagents. We found potential between-assay variation for sTfR measurements. Among controls, the mean ± SD was 1.55 ± 0.48 mg/L for the ELISA, whereas the mean ± SD was 2.98 ± 0.86 mg/L for the immunoturbidimetric assay. We therefore created run- and assay-specific quartiles of sTfR and used appropriate statistical methods to better control potential between-run laboratory variation (see the “Statistical methods” section). Ferritin concentrations were measured with the use of a sandwich immunoassay method (Heterogenous Sandwich Magnetic Separation Assay; Bayer) on the Technicon Immuno 1 system (Bayer).
Laboratory control samples were analyzed along with the case-control samples. Within-run CV percent (CV%) was assessed by analyzing quality-control samples placed in the same plates repeatedly. The mean CV% was 11.1% for sTfR and 6.9% for ferritin.
Assessment of diet and other covariates.
Medical history, food consumption, and lifestyle risk factors were assessed using follow-up questionnaires (including a validated semiquantitative FFQ) in 1990 when most blood samples were collected. Estimated dietary intake of iron (including iron intake from supplements) was calculated based on responses to the FFQ and the estimated nutrient contents from the Harvard Food Composition Database.
Plasma concentrations of E-selectin, interleukin-6 (IL-6), intercellular adhesion molecule-1 (ICAM-1), and vascular cell adhesion molecule-1 (VCAM-1) were measured using commercial ELISA (R&D Systems). C-reactive protein (CRP) levels were measured with a high-sensitivity, latex-enhanced immunonephelometric assay (Dade Behring).
Statistical methods.
To minimize the aforementioned between-assay laboratory variation or measurement errors, we created assay- and run-specific quartiles of body iron store markers according to the distribution among controls. We used conditional logistic regressions to estimate relative risks (RR) of CHD associated with these biomarkers. In nested case-control studies using risk-set sampling, odds ratios derived from conditional logistic regressions are unbiased estimates of RR that take into account the matching factors (18). In addition, because conditional logistic regressions estimate the RR by stratifying on matching case-control triplets (18) and because case-control triplets were assayed in the same batch and by using the same method, the impact of potential between-run laboratory variations should be minimized. In multivariate models, we adjusted for physical activity (in tertiles), alcohol intake (0, 1–4, 5–14, or ≥15 g/d), parental history of myocardial infarction before age 65 y (yes, no), postmenopausal status (yes, no), postmenopausal hormone use (never, past, current), intake of red meat (g/d), BMI (<25, 25–29, ≥30 kg/m2), history of hypertension (presence, absence), history of hypercholesterolemia (presence, absence), and history of diabetes (presence, absence). P-values for linear trend were calculated by entering a continuous score based on the median value in each quartile of iron markers into the models.
We assessed the correlation of body iron store markers with self-reported consumption of red meat (g/d) and iron (g/d) and plasma concentrations of inflammatory markers among controls. Spearman partial rank-r were calculated, adjusted for energy intake (MJ), age at phlebotomy (y), smoking status (never smoked, past smoker, and currently smoke 1–14 cigarettes/d, 15–24 cigarettes/d, or ≥25 cigarettes/d), BMI (kg/m2), fasting status (yes, no), postmenopausal status (yes, no), postmenopausal hormone use (never, past, and current), and assay wave.
All P-values were 2-sided (P < 0.05). The 95% CI were calculated for RR. Data were analyzed with the SAS software package, version 9.1 (SAS Institute).
Results
As expected, women who subsequently developed CHD had a higher BMI, drank less alcohol, were more likely to have a history of diabetes, hypertension, hypercholesterolemia, or a parental history of CHD, and had higher concentrations of plasma inflammatory markers than controls at baseline (Table 1). The cases also tended to eat more red meat than controls (P = 0.07), although their intakes of heme or nonheme iron were similar. CHD cases had significantly higher sTfR levels than did controls (P = 0.02). Other body iron store markers did not differ significantly between the groups.
TABLE 1.
Baseline characteristics of CHD cases and controls in the Nurses' Health Study1
| Characteristics2 | Cases | Controls | P-value3 |
|---|---|---|---|
| n | 242 | 483 | |
| Demography and lifestyle | |||
| Age, y | 60.3 ± 6.5 | 60.3 ± 6.5 | 0.96 |
| BMI, kg/m2 | 26.8 ± 5.9 | 25.4 ± 4.4 | 0.001 |
| Physical activity, MET-h | 0.15 | ||
| Median | 10.3 | 11.9 | |
| Interquartile range | 4.6–21.5 | 5.8–21.8 | |
| Alcohol intake, g/d | 4.2 ± 9.4 | 6.1 ± 10.5 | 0.01 |
| Total iron intake, mg/d | 17.7 ± 12.2 | 18.0 ± 12.3 | 0.77 |
| Heme iron intake, mg/d | 1.1 ± 0.5 | 1.1 ± 0.6 | 0.49 |
| Red meat intake, g/d | 0.07 | ||
| Median | 52.2 | 46.2 | |
| Interquartile range | 32.0–86.9 | 29.2–76.0 | |
| Aspirin use, % | 62.8 | 65.8 | 0.42 |
| Smoking status, % | 0.95 | ||
| Current smoker | 31.4 | 31.7 | |
| Former smoker | 33.5 | 34.4 | |
| Never smoked | 35.1 | 34.0 | |
| Medical history, % | |||
| Diabetes | 20.3 | 6.6 | <0.0001 |
| Hypertension | 57.9 | 29.0 | <0.0001 |
| Hypercholesterolemia | 53.7 | 39.8 | 0.0004 |
| Parental MI before age 65 y | 30.2 | 16.6 | <0.0001 |
| Fasting status | 69.8 | 67.1 | 0.45 |
| Postmenopausal status | 0.18 | ||
| Premenopausal | 9.5 | 11.4 | |
| Current PMH user | 22.3 | 17.4 | |
| Past PMH user | 30.6 | 36.9 | |
| Never used PMH | 37.6 | 34.4 | |
| Biomarker4 | |||
| sTfR:ferritin ratio | 0.07 | 0.06 | 0.53 |
| Ferritin, μg/L | 105.4 ± 144.6 | 88.0 ± 77.8 | 0.08 |
| sTfR,5 mg/L | 2.20 ± 1.05 | 2.02 ± 0.92 | 0.02 |
| CRP,6mg/L | 5.67 ± 6.45 | 3.93 ± 4.94 | 0.0003 |
| ICAM-1,6μg/L | 291.2 ± 75.5 | 278.0 ± 105.0 | 0.05 |
| VCAM-1,6μg/L | 699.9 ± 169.5 | 670.4 ± 155.6 | 0.02 |
| E-selectin,6μg/L | 52.6 ± 25.0 | 46.6 ± 22.5 | 0.001 |
| IL-6,6ng/L | 2.8 ± 3.4 | 2.4 ± 3.1 | 0.12 |
Values are means ± SD or % unless indicated. Percentages are based on nonmissing data.
MET-h, metabolic equivalent-hours; MI, myocardial infarction; PMH, postmenopausal hormone use.
P-value estimates are based on Student's t test for variables expressed as 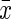 ± SD, Wilcoxon's rank-sum test for variables expressed as medians or Pearson χ2 test for variables expressed as percentages.
± SD, Wilcoxon's rank-sum test for variables expressed as medians or Pearson χ2 test for variables expressed as percentages.
All Biomarkers were measured in plasma.
Concentrations were 1.67 ± 0.49 and 1.54 ± 0.48 for cases and controls selected in 1990–1996, respectively (P = 0.007) and 3.26 ± 1.05 and 2.98 ± 0.86 for cases and controls selected in 1997–1998, respectively (P = 0.03).
Controls, n = 473 for CRP, 477 for ICAM-1, 475 for VCAM-1, 477 for E-selectin, and 461 for IL-6; cases, n = 237 for CRP, 241 for ICAM-1, 240 for VCAM-1, 241 for E-selectin, and 230 for IL-6.
In crude and multivariate analyses, the plasma sTfR:ferritin ratio and ferritin were not significantly associated with the risk of CHD (Table 2). The multivariate RR for women in the highest quartile of the sTfR:ferritin ratio or ferritin were 1.13 (95% CI: 0.65, 1.97; P-trend = 0.61) and 1.05 (95% CI: 0.60, 1.85; P-trend = 0.90), respectively. Although sTfR was associated with an increased risk of CHD in model 1 that adjusted for matching factors only, after multivariate adjustment, especially history of chronic diseases, the associations were dramatically attenuated and became nonsignificant. After further adjustment for inflammatory markers, these associations were either further attenuated to a small extent or unchanged (data not shown). Restricting the analysis to postmenopausal or nondiabetic women did not materially change the results (data not shown). We did not detect significant effect modification by age, smoking, intake of aspirin and antioxidant vitamins, or other covariates. We stratified our analysis according to the methods of sTfR measurements and found similar results. For example, the RR (95% CI) across quartiles of sTfR levels measured by ELISA were 1.0, 1.67 (0.61, 4.57), 1.15 (0.40, 3.29), and 2.12 (0.69, 6.55), respectively. For sTfR levels measured by immunoturbidimetric assay, the corresponding RR (95% CI) were 1.0, 1.70 (0.79, 3.62), 1.82 (0.85, 3.89), and 1.66 (0.78, 3.52), respectively.
TABLE 2.
RR (95% CI) of CHD associated with plasma markers of body iron stores among U.S. women1
| Quartile of body iron store biomarker |
|||||
|---|---|---|---|---|---|
| Biomarker2 | 1 (lowest) | 2 | 3 | 4 (highest) | P-trend3 |
| sTfR:ferritin ratio | |||||
| Cases/control, n/n | 48/120 | 75/121 | 57/121 | 62/121 | |
| Mean (range) | 0.01 (0.001–0.02) | 0.02 (0.01–0.04) | 0.04 (0.02–0.08) | 0.17 (0.04–2.48) | |
| Model 1 (matching factors) | 1.0 | 1.54 (0.99, 2.41) | 1.19 (0.75, 1.90) | 1.27 (0.80, 2.02) | 0.95 |
| Model 2 (multivariate) | 1.0 | 1.42 (0.89, 2.28) | 1.14 (0.70, 1.86) | 1.15 (0.70, 1.90) | 0.72 |
| Model 3 (multivariate) | 1.0 | 1.39 (0.82, 2.36) | 1.12 (0.66, 1.91) | 1.13 (0.65, 1.97) | 0.61 |
| Ferritin, μg/L | |||||
| Cases/control, n/n | 57/120 | 53/119 | 70/123 | 62/121 | |
| Mean (range) | 21.1 (3.0–41.3) | 49.8 (34.4–68.1) | 90.5 (64.3–120.2) | 189.4 (118.4–683.0) | |
| Model 1 (matching factors) | 1.0 | 0.94 (0.60, 1.47) | 1.19 (0.77, 1.84) | 1.09 (0.69, 1.73) | 0.58 |
| Model 2 (multivariate) | 1.0 | 1.01 (0.63, 1.63) | 1.21 (0.75, 1.94) | 1.14 (0.69, 1.88) | 0.55 |
| Model 3 (multivariate) | 1.0 | 1.05 (0.62, 1.77) | 1.19 (0.69, 2.03) | 1.05 (0.60, 1.85) | 0.90 |
| sTfR, mg/L | |||||
| Cases/control, n/n | 33/121 | 61/120 | 59/122 | 89/120 | |
| Mean (range) | 1.41 (0.53–2.37) | 1.76 (1.21–2.86) | 2.11 (1.44–3.39) | 2.80 (1.73–7.45) | |
| Model 1 (matching factors) | 1.0 | 1.94 (1.17, 3.21) | 1.81 (1.10, 2.97) | 2.79 (1.70, 4.59) | 0.0004 |
| Model 2 (multivariate) | 1.0 | 1.91 (1.11, 3.26) | 1.56 (0.92, 2.63) | 2.39 (1.40, 4.07) | 0.01 |
| Model 3 (multivariate) | 1.0 | 1.74 (0.98, 3.10) | 1.40 (0.79, 2.47) | 1.54 (0.85, 2.77) | 0.36 |
The lowest quartile is the reference group. Mean (range) of each quartile is based on the distributions among controls.
Model 1 is controlled for matching factors, i.e. age at blood draw (y), smoking status (never, past, current), and fasting status (yes, no). Multivariate model 2 is further controlled for postmenopausal status (yes, no), postmenopausal hormone use (never, past, current), physical activity (in tertiles), alcohol intake (0, 0–4, 5–14, ≥15 g/d), intake of red meat (g/d), parental history of MI before age 65 y (yes, no), and BMI (<25, 25–29, ≥30 kg/m2). Based on model 2, model 3 is further adjusted for history of hypertension (presence, absence), history of hypercholesterolemia (presence, absence), and history of diabetes (presence, absence).
Linear scores derived from the medians of quartiles of biomarker levels among controls were used to estimate P-values for trend. Estimates of P-value for linear trend are all based on t tests.
We examined the relationship of iron biomarkers with dietary factors as well as inflammatory markers (Table 3). Ferritin was significantly correlated with intake of red meat (r = 0.16), heme iron (r = 0.12), and total iron intake (r = 0.10). The sTfR:ferritin ratio was inversely correlated with red meat and heme iron intake, probably because of the positive correlations of ferritin with these intakes. The sTfR:ferritin ratio was not significantly correlated with the inflammatory markers, except VCAM-1. Ferritin was significantly correlated with only E-selectin. In contrast, sTfR was associated with most of the inflammatory markers, but the correlations were all weak.
TABLE 3.
Spearman partial correlation coefficients between biomarkers of body iron stores and inflammatory markers and dietary intake of iron among US women1
| Variable | sTfR:ferritin ratio | Ferritin | sTfR |
|---|---|---|---|
| Dietary intake | |||
| Total iron intake, mg/d | −0.05 | 0.10* | 0.11* |
| Heme iron intake, mg/d | −0.10* | 0.12* | 0.01 |
| Red meat intake, g/d | −0.16* | 0.16* | −0.05 |
| Biomarker2 | |||
| CRP, mg/L | 0.01 | 0.01 | 0.08 |
| ICAM-1, μg/L | 0.02 | 0.02 | 0.16* |
| VCAM-1, μg/L | 0.12* | −0.08 | 0.20* |
| IL-6, ng/L | 0.05 | −0.01 | 0.12* |
| E-selectin, μg/L | −0.08 | 0.14* | 0.14* |
| sTfR:ferritin ratio | 1.0 | — | — |
| Ferritin, μg/L | −0.95* | 1.0 | — |
| sTfR, mg/L | 0.43* | −0.17* | 1.0 |
Among controls only. n = 483 unless otherwise indicated. Spearman partial correlation coefficients are controlled for age, smoking (never, past, current) and fasting status (yes, no), postmenopausal status (yes, no), postmenopausal hormone use (never, past, current), BMI (<25, 25–29, ≥30 kg/m2), history of hypertension (presence, absence), history of hypercholesterolemia (presence, absence), and history of diabetes (presence, absence), and assay wave. For correlations of dietary iron or red meat intake, total energy intake (MJ) was further adjusted. *P < 0.05; **P < 0.01.
n = 473 for CRP, 477 for ICAM-1, 475 for VCAM-1, 459 for IL-6, and 477 for E-selectin.
Discussion
In this prospective case-control study, markers of body iron stores, including the sTfR:ferritin ratio, were not associated with risk of CHD among U.S. women after controlling for established and potential confounders.
Based on the observation that after menopause, women's incidence of CHD increases along with the accumulation of body iron stores, Sullivan (19) raised the iron-heart hypothesis, suggesting excessive body iron stores are a risk factor of CHD. Although this hypothesis was supported by evidence from some animal studies (20,21), most epidemiologic studies in humans have not detected significant associations between excessive body iron stores and risk of CHD (2,5–11), except studies conducted in a Finnish population (3,4). Several factors may underlie the inconsistency among these studies. In comparison to the U.S. population, Finnish populations have a higher intake of red meat (14), which has been shown to be a risk factor for CHD and a determinant for body iron stores (22). In addition, populations of northern European origin have relatively high prevalence of a HFE (hemochromatosis) gene polymorphism (Cys282Tyr), which was significantly associated with higher body iron stores and an elevated risk of CHD among the Finnish population (13).
sTfR is a truncated form of tissue transferrin receptor, which has been shown to be a sensitive marker for iron deficiency (17,23). In contrast to serum ferritin, sTfR concentrations are thought to be unaffected by inflammation (17,23). In addition, studies have indicated that sTfR and ferritin are inversely regulated at the posttranscriptional level (23). For people with similarly elevated levels (including marginally elevated levels) of ferritin, this ratio further distinguished these subjects with respect to their body iron status (17). Therefore, the sTfR:ferritin ratio has been suggested by some researchers to reflect a wider range of body iron status than ferritin or sTfR alone (17,23,24). So far, few studies have examined the association between the sTfR:ferritin ratio and risk of CHD. In a small case-control study conducted in Finnish men, a lower sTfR:ferritin ratio was associated with an elevated risk of CHD (4), but this association was not present in another retrospective case-control study (25). In our analysis, the sTfR:ferritin ratio was not an independent risk factor for CHD after multivariate adjustment.
Because the sTfR:ferritin ratio is a function of 2 factors, its association with risk of CHD could be driven by both or either of its 2 components. For example, in the aforementioned Finnish study (4), because ferritin was associated with the risk of CHD among the same population (3), inverse associations for the sTfR:ferritin ratio may have been driven by ferritin's effects per se. Few epidemiologic data exist regarding sTfR in relation to CHD and its risk factors. In the current analysis, unadjusted associations for sTfR were explained by established CHD risk factors. In addition, we observed unexpected correlations between sTfR and inflammatory markers. Several lines of evidence indicate that tissue transferrin receptor expression can be regulated by inflammatory cytokines and reactive oxygen species (26,27). Interestingly, in vitro and animal studies suggest that expression of tissue transferrin receptor may also be regulated by insulin (28,29), levels of which are closely related to inflammation in the setting of insulin resistance (30). Although more data are needed to elucidate the associations between sTfR and inflammation, it is possible that, like ferritin, sTfR concentrations were determined by other factors in addition to iron status. The sTfR:ferritin ratio as a marker of body iron stores was examined primarily among anemia patients or young adults receiving repeated phlebotomy (17,24). Whether this ratio measures body iron stores under a broader setting is still unknown, especially in the presence of inflammation or insulin resistance.
The free radical-generating feature of iron ion is the biological basis supporting the iron-heart hypothesis. However, because it is the free iron that can amplify the oxidative stress and the main physiological function of iron-related proteins, such as ferritin and transferrin, is to sequester and transport free iron, markers measuring body iron stores within normal range may not be relevant in terms of oxidative stress. Indeed, the current evidence regarding body iron stores and oxidative stress is inconclusive (31). More recently, researchers have suggested that nontransferrin–bound iron, a more chemically active form of iron in blood, is a more relevant marker for examining the iron-heart hypothesis (31). However, results from a prospective study did not show positive associations with CHD, but rather suggested the opposite (32). Further data are needed, however, especially because careful interpretation and standardization are needed for the nontransferrin–bound iron assay (33). So far, only 1 randomized clinical trial examined the effect of phlebotomy on risk of mortality and cardiovascular disease among patients of symptomatic peripheral arterial disease. The data indicated that reduction of body iron stores did not affect total mortality, although post hoc analysis suggested that among younger patients, phlebotomy significantly reduced the risk of a composite outcome, including death, nonfatal myocardial infarction, or stroke (12). Whether phlebotomy has any specific effect on reducing risk of CHD is unclear. Taken together, current epidemiologic and clinical trial evidence does not support the role of excessive iron stores in the etiology of CHD (34).
A potential explanation for the lack of association between body iron stores and risk of CHD is that the biomarkers currently used in epidemiological studies may not be biologically relevant. Yuan et al. (35) suggested that tissue iron, as measured by liver iron or foam cell iron content, was a more relevant exposure for the iron-heart hypothesis, because these biomarkers have been shown to be associated with advanced atherosclerosis and unstable atheroma plaques. Alternatively, subunits of ferritin may be a better marker than ferritin for assessing body iron status. Studies have demonstrated that H-ferritin subunit expression was selectively induced by tumor necrosis factor-α (36) and L-ferritin subunit was more sensitive to iron overload (37). Therefore, L-ferritin concentrations could be a more specific marker for iron overload and the H:L ratio can be used to distinguish between inflammation- and excessive iron-induced elevation of ferritin. More data are needed to examine these hypotheses.
The current study employed prospective design in which levels of biomarkers were unlikely influenced by the occurrence of disease. By using risk-set sampling, we also minimized the possibility of control selection bias. However, several limitations of this study are also worth discussion. sTfR concentrations were measured by 2 different methods in the current study. Between-assay variation of sTfR levels may bias our findings. However, we created run-specific quartiles of sTfR levels and used conditional logistic regressions to minimize the influence of such measurement errors. When estimating correlations of sTfR levels with other risk factors, we compared results within each batch and found similar results. In addition, single baseline measurements of body iron store markers will not perfectly reflect long-term values. Random variations of the measurements of the biomarkers may attenuate the true associations. Because of the observational nature of the current study, we cannot fully exclude the possibility that these null results may be explained by residual confounding caused by certain healthy lifestyle or dietary factors. Lastly, this study was conducted in women only and thus the results may not be generalized to men.
In summary, our data do not provide evidence to support the hypothesis that excessive body iron stores are associated with risk of CHD.
Supported by research grants HL60712, HL34594, CA78293, and CA87969 from the NIH. Dr. Sun is supported by a postdoctoral fellowship from the Unilever Corporate Research. Dr. Hu is a recipient of the American Heart Association Established Investigator Award.
Author disclosures: Q. Sun, J. Ma, N. Rifai, O. H. Franco, K. M. Rexrode, and F. B. Hu, no conflicts of interest.
Abbreviations used: CHD, coronary heart disease; CRP, C-reactive protein; CV%, CV percent; ICAM-1, intercellular adhesion molecule-1; IL-6, interleukin-6; RR, relative risk; sTfR, soluble transferrin receptor; VCAM-1, vascular cell adhesion molecule-1.
References
- 1.McCord JM. Is iron sufficiency a risk factor in ischemic heart disease? Circulation. 1991;83:1112–4. [DOI] [PubMed] [Google Scholar]
- 2.Danesh J, Appleby P. Coronary heart disease and iron status: meta-analyses of prospective studies. Circulation. 1999;99:852–4. [DOI] [PubMed] [Google Scholar]
- 3.Salonen JT, Nyyssonen K, Korpela H, Tuomilehto J, Seppanen R, Salonen R. High stored iron levels are associated with excess risk of myocardial infarction in eastern Finnish men. Circulation. 1992;86:803–11. [DOI] [PubMed] [Google Scholar]
- 4.Tuomainen TP, Punnonen K, Nyyssonen K, Salonen JT. Association between body iron stores and the risk of acute myocardial infarction in men. Circulation. 1998;97:1461–6. [DOI] [PubMed] [Google Scholar]
- 5.Liao Y, Cooper RS, McGee DL. Iron status and coronary heart disease: negative findings from the NHANES I epidemiologic follow-up study. Am J Epidemiol. 1994;139:704–12. [DOI] [PubMed] [Google Scholar]
- 6.Sempos CT, Looker AC, Gillum RF, Makuc DM. Body iron stores and the risk of coronary heart disease. N Engl J Med. 1994;330:1119–24. [DOI] [PubMed] [Google Scholar]
- 7.Sempos CT, Looker AC, Gillum RE, McGee DL, Vuong CV, Johnson CL. Serum ferritin and death from all causes and cardiovascular disease: the NHANES II Mortality Study. National Health and Nutrition Examination Study. Ann Epidemiol. 2000;10:441–8. [DOI] [PubMed] [Google Scholar]
- 8.Baer DM, Tekawa IS, Hurley LB. Iron stores are not associated with acute myocardial infarction. Circulation. 1994;89:2915–8. [DOI] [PubMed] [Google Scholar]
- 9.Magnusson MK, Sigfusson N, Sigvaldason H, Johannesson GM, Magnusson S, Thorgeirsson G. Low iron-binding capacity as a risk factor for myocardial infarction. Circulation. 1994;89:102–8. [DOI] [PubMed] [Google Scholar]
- 10.Manttari M, Manninen V, Huttunen JK, Palosuo T, Ehnholm C, Heinonen OP, Frick MH. Serum ferritin and ceruloplasmin as coronary risk factors. Eur Heart J. 1994;15:1599–603. [DOI] [PubMed] [Google Scholar]
- 11.Klipstein-Grobusch K, Koster JF, Grobbee DE, Lindemans J, Boeing H, Hofman A, Witteman JC. Serum ferritin and risk of myocardial infarction in the elderly: the Rotterdam Study. Am J Clin Nutr. 1999;69:1231–6. [DOI] [PubMed] [Google Scholar]
- 12.Zacharski LR, Chow BK, Howes PS, Shamayeva G, Baron JA, Dalman RL, Malenka DJ, Ozaki CK, Lavori PW. Reduction of iron stores and cardiovascular outcomes in patients with peripheral arterial disease: a randomized controlled trial. JAMA. 2007;297:603–10. [DOI] [PubMed] [Google Scholar]
- 13.Tuomainen TP, Kontula K, Nyyssonen K, Lakka TA, Helio T, Salonen JT. Increased risk of acute myocardial infarction in carriers of the hemochromatosis gene Cys282Tyr mutation: a prospective cohort study in men in eastern Finland. Circulation. 1999;100:1274–9. [DOI] [PubMed] [Google Scholar]
- 14.Ascherio A, Willett WC. Are body iron stores related to the risk of coronary heart disease? N Engl J Med. 1994;330:1152–4. [DOI] [PubMed] [Google Scholar]
- 15.Cook JD, Lipschitz DA, Miles LE, Finch CA. Serum ferritin as a measure of iron stores in normal subjects. Am J Clin Nutr. 1974;27:681–7. [DOI] [PubMed] [Google Scholar]
- 16.Gabay C, Kushner I. Acute-phase proteins and other systemic responses to inflammation. N Engl J Med. 1999;340:448–54. [DOI] [PubMed] [Google Scholar]
- 17.Skikne BS, Flowers CH, Cook JD. Serum transferrin receptor: a quantitative measure of tissue iron deficiency. Blood. 1990;75:1870–6. [PubMed] [Google Scholar]
- 18.Rothman KJ, Greenland S. Modern epidemiology. 2nd ed. Philadelphia: Lippincott Williams & Wilkins; 1998.
- 19.Sullivan JL. Iron and the sex difference in heart disease risk. Lancet. 1981;1:1293–4. [DOI] [PubMed] [Google Scholar]
- 20.McCord JM. Iron, free radicals, and oxidative injury. Semin Hematol. 1998;35:5–12. [PubMed] [Google Scholar]
- 21.Araujo JA, Romano EL, Brito BE, Parthe V, Romano M, Bracho M, Montano RF, Cardier J. Iron overload augments the development of atherosclerotic lesions in rabbits. Arterioscler Thromb Vasc Biol. 1995;15:1172–80. [DOI] [PubMed] [Google Scholar]
- 22.Qi L, van Dam RM, Rexrode K, Hu FB. Heme iron from diet as a risk factor for coronary heart disease in women with type 2 diabetes. Diabetes Care. 2007;30:101–6. [DOI] [PubMed] [Google Scholar]
- 23.Baynes RD. Assessment of iron status. Clin Biochem. 1996;29:209–15. [DOI] [PubMed] [Google Scholar]
- 24.Punnonen K, Irjala K, Rajamaki A. Serum transferrin receptor and its ratio to serum ferritin in the diagnosis of iron deficiency. Blood. 1997;89:1052–7. [PubMed] [Google Scholar]
- 25.Braun S, Ndrepepa G, von Beckerath N, Vogt W, Schomig A, Kastrati A. Value of serum ferritin and soluble transferrin receptor for prediction of coronary artery disease and its clinical presentations. Atherosclerosis. 2004;174:105–10. [DOI] [PubMed] [Google Scholar]
- 26.Seiser C, Teixeira S, Kuhn LC. Interleukin-2-dependent transcriptional and post-transcriptional regulation of transferrin receptor mRNA. J Biol Chem. 1993;268:13074–80. [PubMed] [Google Scholar]
- 27.Tsuji Y, Miller LL, Miller SC, Torti SV, Torti FM. Tumor necrosis factor-alpha and interleukin 1-alpha regulate transferrin receptor in human diploid fibroblasts. Relationship to the induction of ferritin heavy chain. J Biol Chem. 1991;266:7257–61. [PubMed] [Google Scholar]
- 28.Davis RJ, Corvera S, Czech MP. Insulin stimulates cellular iron uptake and causes the redistribution of intracellular transferrin receptors to the plasma membrane. J Biol Chem. 1986;261:8708–11. [PubMed] [Google Scholar]
- 29.Moutafchiev DA, Sirakov LM. Effect of insulin and adrenaline on the 59Fe transferrin uptake of lactating mouse mammary gland cells. Horm Metab Res. 1992;24:420–3. [DOI] [PubMed] [Google Scholar]
- 30.Eckel RH, Grundy SM, Zimmet PZ. The metabolic syndrome. Lancet. 2005;365:1415–28. [DOI] [PubMed] [Google Scholar]
- 31.Wood RJ. The iron-heart disease connection: is it dead or just hiding? Ageing Res Rev. 2004;3:355–67. [DOI] [PubMed] [Google Scholar]
- 32.van der A DL, Marx JJ, Grobbee DE, Kamphuis MH, Georgiou NA, van Kats-Renaud JH, Breuer W, Cabantchik ZI, Roest M, et al. Non-transferrin-bound iron and risk of coronary heart disease in postmenopausal women. Circulation. 2006;113:1942–9. [DOI] [PubMed] [Google Scholar]
- 33.Jacobs EM, Hendriks JC, van Tits BL, Evans PJ, Breuer W, Liu DY, Jansen EH, Jauhiainen K, Sturm B, et al. Results of an international round robin for the quantification of serum non-transferrin-bound iron: need for defining standardization and a clinically relevant isoform. Anal Biochem. 2005;341:241–50. [DOI] [PubMed] [Google Scholar]
- 34.Hu FB. The iron-heart hypothesis: search for the ironclad evidence. JAMA. 2007;297:639–41. [DOI] [PubMed] [Google Scholar]
- 35.Yuan XM, Li W. The iron hypothesis of atherosclerosis and its clinical impact. Ann Med. 2003;35:578–91. [DOI] [PubMed] [Google Scholar]
- 36.Kwak EL, Larochelle DA, Beaumont C, Torti SV, Torti FM. Role for NF-kappa B in the regulation of ferritin H by tumor necrosis factor-alpha. J Biol Chem. 1995;270:15285–93. [DOI] [PubMed] [Google Scholar]
- 37.Leggett BA, Fletcher LM, Ramm GA, Powell LW, Halliday JW. Differential regulation of ferritin H and L subunit mRNA during inflammation and long-term iron overload. J Gastroenterol Hepatol. 1993;8:21–7. [DOI] [PubMed] [Google Scholar]