

Summary
Increases in global Ca2+ in the endothelium are a crucial step in releasing relaxing factors to modulate arterial tone. In the present study we investigated spontaneous Ca2+ events in endothelial cells, and the contribution of smooth muscle cells to these Ca2+ events, in pressurized rat mesenteric resistance arteries. Spontaneous Ca2+ events were observed under resting conditions in 34% of cells. These Ca2+ events were absent in arteries preincubated with either cyclopiazonic acid or U-73122, but were unaffected by ryanodine or nicotinamide. Stimulation of smooth muscle cell depolarization and contraction with either phenylephrine or high concentrations of KCl significantly increased the frequency of endothelial cell Ca2+ events. The putative gap junction uncouplers carbenoxolone and 18· -glycyrrhetinic acid each inhibited spontaneous and evoked Ca2+ events, and the movement of calcein from endothelial to smooth muscle cells. In addition, spontaneous Ca2+ events were diminished by nifedipine, lowering extracellular Ca2+ levels, or by blockers of non-selective Ca2+ influx pathways. These findings suggest that in pressurized rat mesenteric arteries, spontaneous Ca2+ events in the endothelial cells appear to originate from endoplasmic reticulum IP3 receptors, and are subject to regulation by surrounding smooth muscle cells via myoendothelial gap junctions, even under basal conditions.
Keywords: Endothelial cells, Rat mesenteric arteries, Gap junctions, Spontaneous Ca2+ events
Introduction
Endothelial cell Ca2+ has a crucial role in controlling vascular tone and homeostasis by releasing nitric oxide (NO), prostacyclin, and endothelium-derived hyperpolarizing factor (EDHF) [1-6] and affecting gene expression, angiogenesis, cell growth, and leukocyte migration [7,8], respectively. The level of cytoplasmic [Ca2+]i can be modulated not only by the direct action of agonists and haemodynamic forces on the endothelial cells, but also indirectly by communication from surrounding cells. One important pathway for intercellular communication within resistance arteries and arterioles is via direct cell—cell coupling through myoendothelial gap junctions [9-14]. In addition to current passing between these cells through myoendothelial connections, there is also evidence for Ca2+ signalling following elevations in smooth muscle cell Ca2+ by agonists such as phenylephrine (PE) and KCl. The consequent secondary rise in endothelial Ca2+ can enhance the production of both NO and EDHF [15-18].
In intact vessels in situ, spontaneous Ca2+ events in endothelial cells have been reported in rat lung capillaries [19], rat ureter arterioles [20] and mouse cremaster arterioles [21,22]. Interestingly there are no reports of spontaneous Ca2+ events in cultured endothelial cells, although they do exhibit oscillating Ca2+ events in response to agonists [23] or flow of superfusate [24]. In cultured cells, oscillating Ca2+ events are reportedly associated with spontaneous transient outward currents (STOCs), reflecting activation of charybdotoxin-sensitive Ca2+-activated K+ (KCa) channels [24]. These various lines of evidence suggest that spontaneous Ca2+ events in the endothelium might contribute to the regulation of vascular tone.
It is clear that many Ca2+ signals occur within cellular microdomains, and may not manifest as global Ca2+ changes [25-27]. Further to this, it is also clear that at the same time as discrete, localized Ca2+ events at the cell membrane, global changes in Ca2+ can occur in parallel. For example in vascular smooth muscle cells, although global increase in [Ca2+]i (via voltage-gated Ca2+ channels) contribute to increases in arterial tone, localized spontaneous Ca2+ events from the sarcoplasmic reticulum activate large conductance KCa channels (BKCa), and thereby modulate arterial tone [28,29]. However, despite their fundamental importance spontaneous Ca2+ events in endothelial cells remain poorly understood, especially in arteries under physiological conditions. Furthermore, the mechanisms responsible for these Ca2+ events, including their source and the possibility that they are influenced by the surrounding smooth muscle cells are not known.
In the present study, we demonstrate that spontaneous endothelial cell Ca2+ events occur under resting conditions in isolated and pressurized mesenteric resistance arteries. We also show that these events were influenced by the activity of the surrounding smooth muscle cells.
Methods
Preparation of arteries for pressure myography
Male Wistar rats (200-250 g) were killed by cervical dislocation and exsanguination (Schedule 1 procedure; UK, Animals (Scientific Procedures) Act 1986). The mesenteric arcade was removed and placed in chilled MOPS buffer (4 °C) containing (mM): 145 NaCl, 4.7 KCl, 2.0 CaCl2, 1.17 MgSO4, 2.0 MOPS, 1.2 NaH2PO4, 5.0 glucose, 2.0 pyruvate, 0.02 EDTA, 2.75 NaOH (the pH of the solution was adjusted to 7.40 · 0.02 at 37 °C). A third order branch of the superior mesenteric artery was then carefully dissected free of adherent tissue. A segment of mesenteric artery (internal diameter circa 200-250 · m) was cut and cannulated at each end with glass pipettes (external diameter 150 · m), and then positioned near the base of a 1.5 ml temperature-regulated chamber (Warner Instruments) on the stage of an inverted microscope (IX 70, Olympus, UK). To avoid luminal flow, the upstream and downstream pressures were equal throughout experiments. After equilibration at 37 °C for 20 min, arteries were longitudinally stretched with a micrometer during maximal inflation (at 80 mmHg), and then maintained at 50 mmHg for the remainder of the experiments. This protocol was found to optimize the vasomotor responses to agonists [30,31]. Brightfield images were acquired using a · 10 objective and captured with an iXon 887 EMCCD camera (Andor Technology, UK) coupled to a Nipkow spinning disk confocal head (CSU22, Yokogawa, Japan), mounted on the trinocular head of the microscope, and recorded using Andor iQ software (Andor, UK). The nitric-oxide synthase inhibitor NG-nitro-l-arginine methyl ester (100 · M) was present throughout all experiments. After 30 min superfusion at 37 °C, reactivity was assessed by contraction to 2 · M PE followed by endothelium-dependent relaxation to 1 · M ACh. Only vessels relaxing · 90%, reflecting undamaged endothelium, were subsequently used. Outer diameter was measured using edge detection software (Metamorph Version 6.1; Universal Imaging).
Selective measurement of endothelial cell [Ca2+]i change in pressurized arteries
Following equilibration of pressurized arteries, filtered (0.22 · m pore) MOPS solution containing a cell-permeant acetoxymethyl ester form of Oregon Green 488 1,2-bis (2-aminophenoxy) ethane-N,N,N·,N·-tetraacetate (Oregon Green 488 BAPTA-1 AM, 10 · M) and 0.02% Pluronic F-127 was perfused through the artery lumen for 30 min to enable the selective loading of the dye into endothelial cells. The dye was then washed out by luminal perfusion with MOPS buffer, and allowed to equilibrate for at least 30 min (to allow de-esterification of the dye). After excitation at 488 nm, the fluorescence emission intensity at 515 nm from endothelial cells at the bottom of the artery were acquired using a · 40 water immersion objective (numerical aperture, 0.9; Olympus, Japan), and recorded at between 10 and 20 Hz. The full chip 512 · 512 images were cropped to enable these high acquisition rates, and enabled visualization of a field of at least 10 cells (Fig. 1A, online video 1). This optical configuration resulted in a spatial resolution of 2.6 pixel/· m, and from each artery 2 different fields were scanned for 60 s. To determine the frequency of Ca2+ activity, baseline fluorescence (F0) was determined by averaging 20 images without activity. Local fractional fluorescence increase (F/F0) was determined by dividing the fluorescence of an area 3.80 · m · 3.80 · m (10 · 10 pixels) in each image (F) by baseline average F0. Ca2+ events were defined as local F/F0 · 1.2, a value that could be distinguished above background noise. Cells that showed spontaneous Ca2+ events were further analyzed offline, by obtaining the fluorescence intensity of lines drawn over the regions of cells where waves were observed to propagate, and measured over time. This enabled multiple ‘line-scans’ to be obtained within a single-saved acquisition run. Further, the same cell and line could be used before and after treatments. In situations where changes in arterial diameter occurred, the line was re-positioned over the same cell once the focus was corrected.
Figure 1.
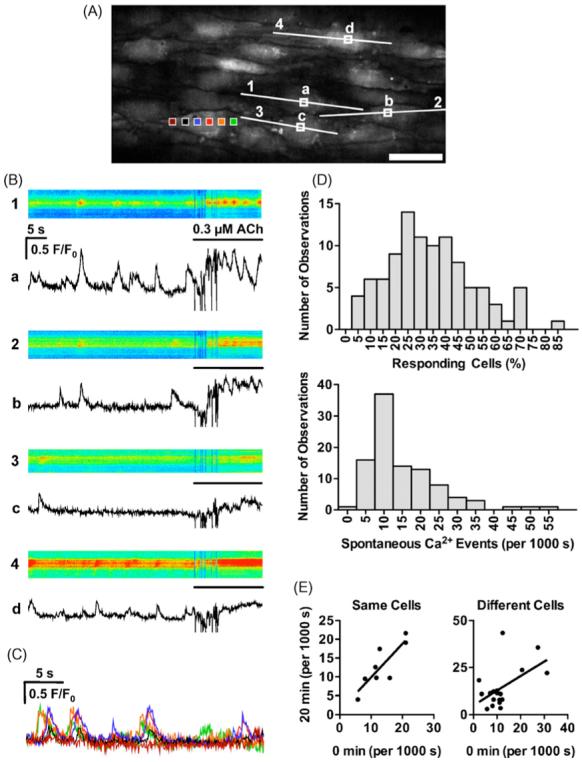
Spontaneous Ca2+ events on endothelial cells of pressurized arteries. (A) Confocal fluorescence image of arterial endothelial cells loaded with Oregon Green 488 BAPTA-1. Scale bar is 30 · m. (B) Time course of fluorescence intensity changes within individual cells shown as ‘line-scans’ in top panels (corresponding to numbered lines shown in A; pseudocolour: blue lowest to red highest) and subcellular regions of interest in bottom panels (F/F0 in the regions corresponding to the lettered boxes shown in A). ACh (0.3 · M) was added towards the end of the acquisition period, whereupon mixing created an artefact (within the first 10 s). (C) Time course of average fluorescence intensity changes within one cell to illustrate the wave-like nature of the spontaneous event. Coloured boxes in A correspond to the changes in F/F0. A movie file corresponding to these data is available online (video 1). (D) The distribution of cells displaying spontaneous Ca2+ events (Responding cells, top) and the frequency of events in these cells (Hz, bottom) under control condition (n = 99). (E) Reproducibility of Ca2+ events (at 0 and 20 min) in the same cell (left) and then at 20 min in another randomly selected cell (right).
Simultaneous measurement of endothelial cell and smooth muscle cell [Ca2+]i changes in pressurized arteries
In a separate group of experiments, Oregon Green BAPTA-1 (10 · M, prepared as above) was loaded into arteries to simultaneously monitor changes in Ca2+ levels in smooth muscle cells (bath application for 120 min) and endothelial cells (luminal application for 15 min). The dye was washed out and allowed to de-esterify for at least 30 min. This procedure ensured adequate loading of smooth muscle cells without compromising the ability to observe the spontaneous events in each cell type. Interesting and noteworthy, if the endothelial cells were loaded for longer periods, no spontaneous activity was observed. Two sets of data were obtained for each loaded artery. First, in focus smooth muscle cells (observed by lowering the plane of focus) were used to analyze Ca2+ events, and second, smooth muscle cells together with in focus endothelial cells were used to analyze endothelial cell Ca2+ events (with the ability to see small regions of smooth muscle cells above and below the endothelial cells, as in online video 2). In these experiments, the effects of carbenoxolone (100 · M, added to the bath for 90 min) and U-73122 (1 · M, luminal perfusion only) were assessed on endothelial and smooth muscle cell Ca2+ events. Note that preliminary experiments showed that extended periods of exposure to carbenoxolone (>120 min) inhibited Ca2+ events in smooth muscle cells.
Assessment of heterocellular dye transfer
In separate experiments, filtered (0.22 · m pore) MOPS solution containing calcein AM (3 · M) and 0.02% Pluronic F-127 was perfused through the artery lumen for 9 min to enable the selective loading of dye into endothelial cells. The lumen was flushed with MOPS buffer containing 0.5 · M Alexa Fluor 633 hydrazide to optimize visualization of the internal elastic lamina. The time of calcein dye loading and the incubation time before acquiring images (90 min to allow de-esterification and diffusion) were kept constant in all experiments. When used, carbenoxolone was added to the bath for 40 min prior to loading calcein, and remained present during the loading and incubation periods. The experimental setup was identical to the Ca2+ imaging experiments, except the stage was set up on an Olympus IX71 microscope with a laser scanning confocal head (FV500, Olympus, Japan). Arteries were excited at 488 and 633 nm, the fluorescence emitted at 515 and 660 nm from endothelial cells and the smooth muscle cells at the bottom of the artery were acquired through a · 40 water immersion objective (numerical aperture, 0.9; Olympus, Japan, 1024 · 650 pixels) using the same laser, pinhole, and photomultiplier tube settings in all experiments. z-stacks through the wall of the artery were obtained in 0.5 · m increments (FluoView software Version 5.0, Olympus) and reconstructed in 3-D using Imaris software (Version 5.5, Bitplane). For an estimate of cellular staining, 10 regions of interest (5 pixel diameter) were placed either (1) on the centre of endothelial cells (EC); (2) the edge of endothelial cells (where fluorescence was low) corresponding to overlying smooth muscle cells (SMC); or (3) the bright spots termed endothelial cell projections (ECP). The values for the peak intensity through the z-stack were averaged for each artery. The corresponding staining of the internal elastic lamina (IEL) was used to confirm the separation of the EC and SMC values.
Solutions and drugs
Oregon Green 488 BAPTA-1 AM (O6807), Pluronic F-127 (P3000MP) and calcein AM (C3100MP) were obtained from Molecular Probes. KB-R 7943 was purchased from Tocris. U-73122 and U-73343 were from Biomol. Cyclopiazonic acid (CPA) was from Calbiochem. All other drugs were from Sigma—Aldrich. Nifedipine was dissolved in ethanol. U-73122 and U-73343 were dissolved in chloroform, evaporated, and then dissolved in DMSO. 18· -Glycyrrhetinic acid (18· -GA) and SKF-96365 were dissolved in DMSO and then diluted in physiological buffer for experimentation. Preliminary experiments indicated that the DMSO vehicle control had no effect [30]. All other stock solutions were prepared using distilled water. When used, inhibitors were added to the incubation solution and arteries equilibrated for at least 20 min prior obtaining responses if not mentioned specially.
Data analysis
Results are summarized as means · S.E.M. of n replicates. Statistical comparisons were made using paired Student’s t-test or one-way ANOVA with Turkeys post hoc test using Prism software (Graphpad, USA). A level of P < 0.05 was considered statistically significant.
Results
Characteristics of spontaneous Ca2+ events in endothelial cells
Endothelial cells were selectively loaded following luminal perfusion of Oregon Green 488 BAPTA-1 AM in pressurized rat mesenteric arteries (Fig. 1A). In each field, more than 10 endothelial cells could be observed simultaneously. Spontaneous transient Ca2+ events were observed without agonist or mechanical stimulation in approximately one third of the analyzed endothelial cells (average 33.7 · 1.7% ranging from 5.9 to 83.3%) (Fig. 1D). The distribution of the frequency of Ca2+ events in endothelial cells in each artery ranged between 0.002 and 0.057 Hz/cell (average 0.015 · 0.001 Hz/cell) (Fig. 1D). The spontaneous Ca2+ events in endothelial cells were of lower amplitude and frequency than ACh-stimulated rises in endothelial cell Ca2+ (Fig. 1B). In most cases each event appeared to radiate within the cell (Fig. 1B, C and online video 1). The Ca2+ events did not appear to synchronize between cells and each Ca2+ event appeared localized within a subcellular region of the endothelial cell. To test the reproducibility of Ca2+ events, they were recorded at 20 min intervals. A stronger correlation was seen between the two observations from the same arterial field (r = 0.84, P < 0.01, n = 8), compared with randomly selected regions (r = 0.53, P < 0.05, n = 18) (Fig. 1E), and there was no difference in the average frequencies between the two observations (0 min: 0.013 · 0.003 Hz, 20 min: 0.013 · 0.003 Hz at the same arterial segment; 0 min: 0.012 · 0.002 Hz, 20 min: 0.014 · 0.003 Hz at a randomly selected different region; each the average of 10 cells). Additionally, because of the widely distributed frequencies of Ca2+ events between arteries, we analyzed changes in the ratio to the control of each treatment, using the same arterial region in pairs. In general, the average basal fluorescence intensity of the Oregon Green BAPTA-1 dye stayed relatively constant within the endothelial cells for periods in excess of 2 h, suggesting that cellular loss was minimal. This was a distinct advantage over the use of fluo-4 AM to monitor changes in Ca2+ levels, as fluo-4 fluorescence intensity decreased rapidly over time, such that within 30 min the dye was almost not visible within the endothelial cells, partly due to passage into smooth muscle cells (data not shown).
Regulation of endothelial Ca2+ events by intracellular Ca2+ release pathways
CPA (10 · M), a SERCA inhibitor, or intraluminal incubation with the phospholipase C inhibitor U-73122 (1 · M), abolished spontaneous Ca2+ events in the endothelial cells (Fig. 2). These agents also reduced the rises in endothelial cell Ca2+ in response to ACh (Table 1). In contrast, neither the inactive analogue of U-73122, U-73343 (1 · M), 20 · M ryanodine nor 6 mM nicotinamide, an inhibitor of cyclic ADP-ribose synthesis, reduced the frequency of spontaneous Ca2+ events (Fig. 2). In separate experiments we showed that luminal perfusion with U-73122 did not inhibit the contraction to PE at the concentrations used to study Ca2+ events.
Figure 2.
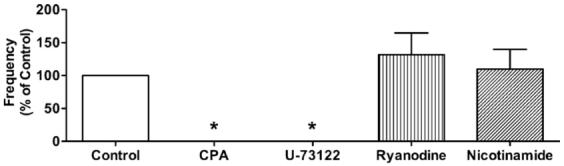
Effects of cyclopiazonic acid (CPA), U-73122, ryanodine, and nicotinamide on Ca2+ events in endothelial cells of pressurized arteries. CPA (10 · M, n = 3) and U-73122 (1 · M, n = 5) abolished the Ca2+ events. Neither ryanodine (20 · M, n = 5) nor nicotinamide (6 mM, n = 3) had a significant effect on spontaneous Ca2+ events. Values are means · S.E.M. *P < 0.05, significantly different from control.
Table 1.
Effect of agents on the peak rise in endothelial cell Ca2+ in response to 0.3 · M ACh
| Control | Treatment | n | |
|---|---|---|---|
| Cyclopiazonic acid (10 · M) | 1.51 · 0.12 | 1.25 · 0.07* | 3 |
| U-73122 (1 · M) | 1.60 · 0.18 | 1.29 · 0.18* | 3 |
| U-73433 (1 · M) | 1.47 · 0.07 | 1.55 · 0.05 | 3 |
| 1mM Extracellular Ca2+ | 1.54 · 0.10 | 1.56 · 0.09 | 3 |
| Nifedipine (1 · M) | 1.47 · 0.10 | 1.40 · 0.03 | 4 |
| SKF-96365 (50 · M) | 1.61 · 0.13 | 1.38 · 0.16* | 6 |
| NiCl2 (500 · M) | 1.54 · 0.08 | 1.39 · 0.08* | 4 |
| KB-R 7943 (3 · M) | 1.62 · 0.10 | 1.37 · 0.08* | 4 |
| Carbenoxolone (100 · M) | 1.48 · 0.03 | 1.35 · 0.11 | 4 |
| 18· -Glycyrrhetinic acid (100 · M) | 1.61 · 0.03 | 1.69 · 0.09 | 5 |
ACh was added to the bath before and after treating the arteries with the agents listed. Values are means · S.E.M.
P < 0.05, significantly different from control, paired t-test.
Role of smooth muscle cells in modulation of endothelial cell Ca2+ events
PE or KCl was added to the bath to establish any influence on the spontaneous endothelial cell Ca2+ events (Fig. 3A and B). With each agent, the concentration used stimulated vasoconstriction to the maximum level where endothelial cells were still clearly visible. The average concentration of PE and KCl used was 0.9 · 0.2 · M and 34 · 2 mM, respectively. In both cases, this translated to approximately 40% of the maximum contraction in the pressurized arteries. When arterial contraction reached a steady level, the plane of focus was adjusted, and Ca2+ events in endothelial cells were recorded. When the pressurized arteries were contracted with PE or KCl, the frequency of Ca2+ events were significantly increased (PE: 0.022 · 0.004 Hz/cell, P < 0.05, n = 11; KCl: 0.028 · 0.006 Hz/cell, P < 0.05, n = 11) (Fig. 3A and B).
Figure 3.
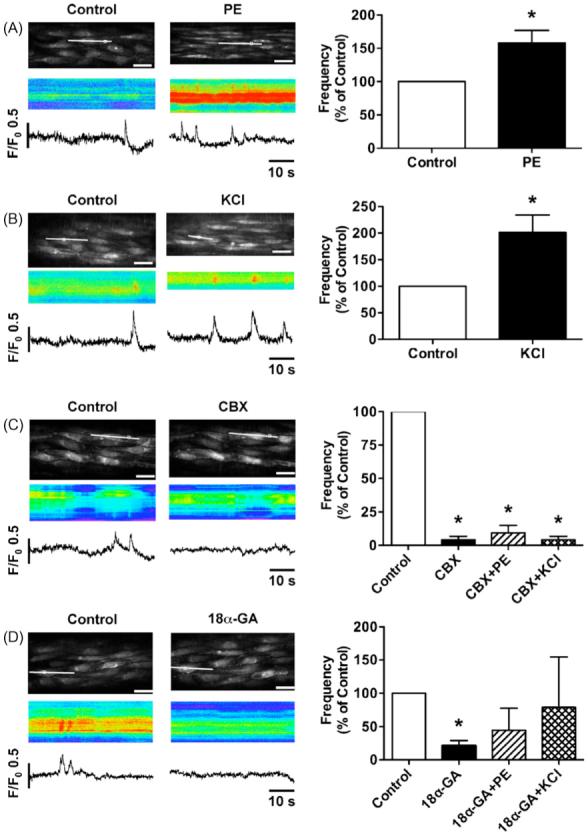
Effects of smooth muscle cells on Ca2+ events in endothelial cells. (A and B) Stimulation of smooth muscle cells with phenylephrine (PE, 0.9 · 0.2 · M, n = 11) or KCl (34 · 2 mM, n = 11) increased Ca2+ events in endothelial cells. (C and D) Application of carbenoxolone (CBX, 100 · M, n = 4) or 18· -glycyrrhetinic acid (18· -GA, 100 · M, n = 4) substantially decreased Ca2+ events. The inhibitory effects of CBX (n = 4) continued even in the presence of PE or KCl, but 18· -GA was less effective (n = 4). Values are means · S.E.M. *P < 0.05, significantly different from control.
Incubation with gap junction uncouplers, carbenoxolone (100 · M) and 18· -GA (100 · M) inhibited the frequency of endothelial Ca2+ events under resting conditions (Fig. 3C and D) without affecting the response to ACh (0.3 · M) (Table 1). To examine the inhibitory effects of these gap junction inhibitors on PE- or KCl-induced increase of Ca2+ events, the arteries were stimulated in the presence of carbenoxolone or 18· -GA (Fig. 3C and D). Preincubation with 100 · M carbenoxolone significantly prevented the increase in Ca2+ events stimulated by PE or KCl (Fig. 3C). Interestingly, the inhibitory effect of 100 · M 18· -GA on PE or KCl-induced increase in endothelial Ca2+ events was not significant (Fig. 3D), although the trend was to reduce these events.
To evaluate whether in addition to inhibiting spontaneous and PE-evoked endothelial cell Ca2+ events, carbenoxolone exerted a direct action to inhibit smooth muscle cells Ca2+ events, smooth muscle and endothelial cells were loaded with Oregon Green BAPTA-1 dye. Under control conditions, spontaneous Ca2+ events were observed in approximately half of the smooth muscle cells (56.2 · 12.1%, n = 5) with a frequency of 0.050 · 0.005 Hz/cell (n = 5). Virtually all of these smooth muscle cells showed at least one propagating cell-wide Ca2+ wave (CWW) (0.034 · 0.002 Hz/cell, n = 5). More localized Ca2+ activity (wavelets) was observed in 40.7 · 12.9% of smooth muscle cells (0.015 · 0.0052 Hz/cell, n = 5). The CWWs were asynchronous between cells, and 6-10 smooth muscle cells crossed each endothelial cell, making it impossible to link events between the two cell types (Fig. 4A and B and online video 2). The addition of PE significantly increased the frequency of CWWs to 0.060 · 0.007 Hz/cell (n = 3, P < 0.05) and prevented the observation of wavelets (0.001 · 0.0001 Hz/cell, n = 3, P < 0.05). In the presence of carbenoxolone, although endothelial cell Ca2+ events were inhibited (Fig. 4C), the frequency of smooth muscle CWWs and wavelets was not significantly changed either under resting conditions or in the presence of PE (Fig. 4D). The effect of carbenoxolone on endothelial cell Ca2+ events was fully reversible following 30 min of washout. Similarly, at a time when U-73122 inhibited spontaneous endothelial cell Ca2+ events, it did not alter those observed in smooth muscle cells (Fig. 4D).
Figure 4.
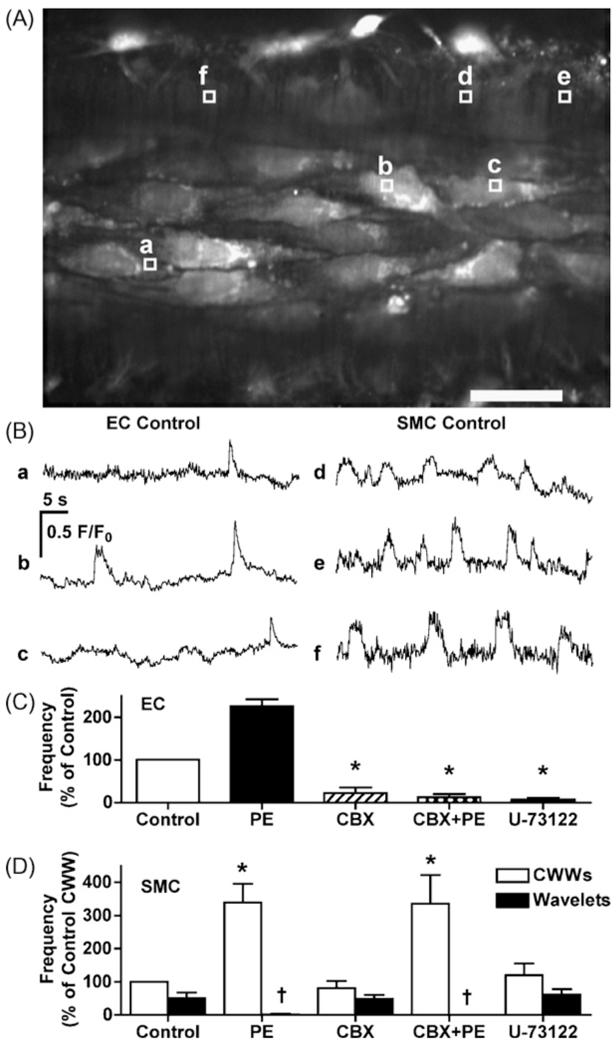
Simultaneous measurement of Ca2+ events in endothelial and smooth muscle cells in rat mesenteric arteries. (A) Confocal fluorescence image of arterial endothelial and smooth muscle cells loaded with Oregon Green 488 BAPTA-1. Scale bar is 30 · m. (B) Time course of fluorescence intensity changes within individual cells shown as subcellular regions of interest (F/F0 in the regions corresponding to the lettered boxes shown in A). A movie file corresponding to these data is available online (video 2). In these arteries, phenylephrine (PE) significantly increased Ca2+ events in both endothelial cells (C) and CWWs in smooth muscle cells (D). Either carbenoxolone (CBX, 100 · M) or U-73122 (1 · M) inhibited Ca2+ events in endothelial cells (C) without affecting smooth muscle cells events (D). Values are means · S.E.M. n = 3 for each. *P < 0.05, significantly different from control. †P < 0.05, significantly different from CBX without PE.
Since it was clear that carbenoxolone was modifying the ability of PE and KCl to evoke endothelial cell Ca2+ events, the ability of this gap junction uncoupler to prevent heterocellular dye transfer was assessed. Under control conditions, luminal perfusion of calcein AM loaded the endothelial cells uniformly, and after a few additional minutes the smooth muscle cells also became visible (Fig. 5A). In the presence of carbenoxolone (100 · M) the endothelial cell fluorescence intensity was higher (control: 1699 · 465 intensity units, n = 4; carbenoxolone: 2501 · 47 intensity units, n = 3), and there was less staining of smooth muscle cells (control: 690 · 62, n = 4; carbenoxolone: 282 · 12 intensity units, n = 3). In all images, intense fluorescence was observed in discrete spots over the endothelial cells. These spots correspond to holes through the internal elastic lamina (Fig. 5A and B), and likely represent fluorescence within the ECPs to the smooth muscle cells, which pass through these holes. It is important to note that in preliminary experiments, if higher concentrations of calcein AM were loaded, both endothelial and smooth muscle cells fluoresced, even in the presence of carbenoxolone. This may reflect direct loading of the smooth muscle cells by the AM form of calcein, or an inability of carbenoxolone to prevent this transfer of dye in high concentrations. Likewise, we cannot assess whether the low levels of smooth muscle cell staining in the presence of carbenoxolone reflects residual dye coupling, or direct loading. Yet it is also interesting to note that the dye staining within the holes was brighter in the presence of carbenoxolone (control: 1303 · 293 intensity units, n = 4; carbenoxolone: 2000 · 261 intensity units, n = 3), again consistent with less dye transfer to smooth muscle cells.
Figure 5.
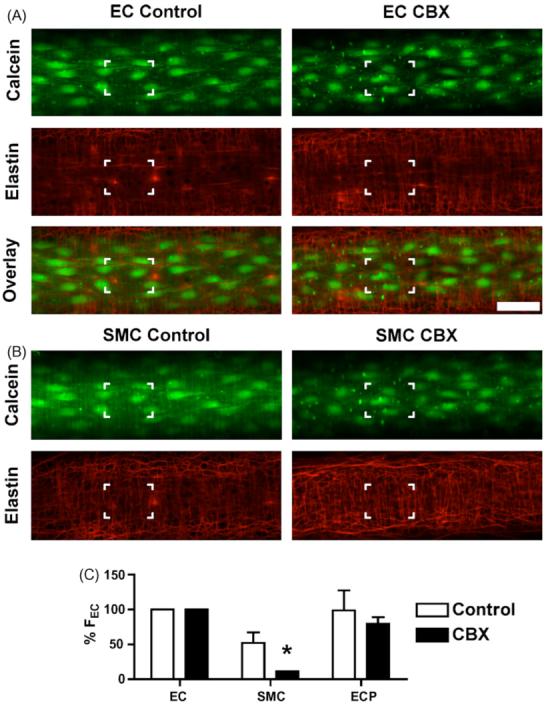
Effect of carbenoxolone on the loading of calcein into the artery wall. Image z-stacks were captured following selective endothelial cell loading of calcein AM. Confocal images at the level of the endothelial cells (A) and in the same arteries (x- and y-positions unchanged, z-position 4.5 · m down towards the outer wall) at the level of the smooth muscle cells (B). The IEL was stained with Alexa Fluor 633 hydrazide (red). Calcein (green) clearly stained the endothelial cells, with the smooth muscle cells (vertical orientation) faintly visible in the control arteries but not visible in arteries incubated with carbenoxolone. Note that nearly all the bright spots correspond to holes through the internal elastic lamina, see highlighted boxes for comparison. Bar = 50 · m. (C) The average peak fluorescence intensity in endothelial cells (ECs), smooth muscle cells (SMCs), and endothelial cell projections (ECPs) compared to the fluorescence intensity of ECs (% FEC) in control arteries (n = 4) and those treated with CBX (n = 3). See Section ‘Materials’ for details of analysis. Values are means · S.E.M. *P < 0.05, significantly different from control.
Regulation of endothelial Ca2+ events by extracellular Ca2+
When arteries were incubated with a low concentration of Ca2+ (1 mM) or the voltage-gated Ca2+ channel inhibitor nifedipine (1 · M) for 5 min, the frequency of Ca2+ events in endothelial cells was significantly decreased (Fig. 6), yet the response to ACh (0.3 · M) remained unaffected (Table 1). Luminal application of the non-selective inhibitors of Ca2+ influx, SKF-96365 (50 · M) and NiCl2 (500 · M) significantly inhibited the Ca2+ events in endothelial cells, and intraluminal incubation with the reverse-mode Na+/Ca2+ exchanger (NCX) inhibitor KB-R 7943 (3 · M) also decreased Ca2+ events in endothelial cells (Fig. 6). These agents also affected the response to ACh (0.3 · M) (Table 1).
Figure 6.
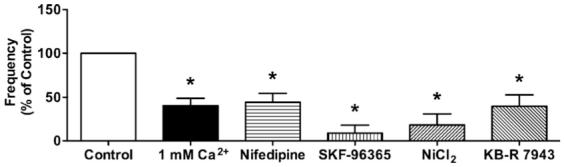
Effects of lowered extracellular Ca2+, nifedipine, SKF-96365, NiCl2, and KB-R 7943. Lower extracellular Ca2+ (1 mM) decreased the frequency of Ca2+ events compared to control (2 mM Ca2+, n = 5). To decrease Ca2+ in smooth muscle cells, the arteries were incubated with the voltage-gated Ca2+ channel inhibitor, nifedipine. Nifedipine (1 · M, n = 5) decreased Ca2+ events in endothelial cells. The frequency of Ca2+ events was less in the presence of non-selective inhibitors of Ca2+ influx SKF-96365 (n = 5), NiCl2 (500 · M, n = 3), or KB-R 7943 (3 · M, n = 4). Values are means · S.E.M. *P < 0.05, significantly different from control.
Discussion
The present study demonstrates that spontaneous endothelial Ca2+ events occur in isolated and pressurized rat mesenteric arteries, and can be regulated not only by Ca2+ influx and Ca2+ stores in these cells, but also by the adjacent smooth muscle cells. This is the first demonstration of spontaneous Ca2+ events in endothelial cells under physiological conditions, and suggests that the regulation of Ca2+ events within endothelial cells by the smooth muscle cells most probably occurs through myoendothelial gap junctions.
In cultured bovine aortic endothelial cells, Ca2+ events have been shown to be associated with STOCs, reflecting the activation of Ca2+-activated K+ channels [24]. Rat mesenteric arteries contain three type of KCa; large-conductance Ca2+-activated K+ channels (BKCa) are confined to the smooth muscle cells, whereas intermediate- and small-conductance Ca2+-activated K+ channels (IKCa and SKCa, respectively) are located in the endothelial cells [7,16,32]. In smooth muscle cells, spontaneous Ca2+ events, or Ca2+ sparks, are known to activate BKCa and as a result decrease in arterial tone [25-27]. Therefore, it is most likely that spontaneous Ca2+ events in our present study can cause STOCs originating in endothelial cells and contribute to the activation of SKCa or IKCa, resulting in hyperpolarization to regulate vascular tone. Certainly, consistent with this suggestion mice deficit in either SKCa or IKCa have a raised blood pressure [33,34].
Release of Ca2+ from endothelial cell intracellular stores
Although there are no reports of spontaneous Ca2+ events in cultured endothelial cells, there is one report of these events in lung capillary endothelial cells [19], suggesting that they can occur in the absence of a direct influence from smooth muscle cells. The other reports of spontaneous Ca2+ events within endothelial cells were in intact arterioles in situ [20-22], where the influence from adjacent smooth muscle cells cannot be excluded.
In the present study, depletion of endothelial cell intracellular Ca2+ stores with CPA (10 · M) almost fully blocked the Ca2+ events, and inhibition of phospholipase C with U-73122 (1 · M) abolished them. By perfusing these inhibitors through the artery lumen, we appeared to confine their action to endothelial cells, as U-73122 did not inhibit Ca2+ events in smooth muscle cells, and did not inhibit contraction to PE. The observation that responses to the endothelial-cell-specific agonist ACh were inhibited as well as the spontaneous events shows that the inhibitors did act on the endothelial cells. Although some studies have suggested that ryanodine receptors and cyclic ADP-ribose contribute to agonist-evoked increases in endothelial cell Ca2+ increases [35,36], neither ryanodine nor nicotinamide had a significant inhibitory effect on the spontaneous endothelial Ca2+ events in our study. This concurs with a lack of effect of ryanodine on spontaneous Ca2+ events in rat ureter endothelial cells in situ [20]. Furthermore, ryanodine in higher concentrations (100 · M) also failed to inhibit the Ca2+ events (data not shown). Therefore, our data support a central role for intracellular Ca2+ stores as the source of spontaneous Ca2+ events, and IP3, rather than ryanodine receptors as the endoplasmic reticulum Ca2+ release channels involved in the initiation and propagation of Ca2+ events within the endothelial cells of rat mesenteric arteries exposed to a physiological pressure. However, a selective and cell-permeant inhibitor for IP3 receptors will be necessary to resolve this point definitively.
Regulation of endothelial Ca2+ events via myoendothelial gap junctions
The selective · 1 agonist PE, stimulates direct smooth muscle depolarization and contraction in the rat mesenteric artery, it has no direct action on the endothelial cells [16,17,37]. So both KCl and PE would be expected to depolarize endothelial cells, either directly or indirectly via smooth muscle cells [38]. If there is a large effect of voltage on Ca2+ influx, these depolarizing effects on the endothelial cells should, if anything, act to decrease Ca2+ by reducing the driving force for Ca2+ entry into the endothelial cells [39,40]. Therefore taken together, our findings that PE and KCl each increased the frequency of endothelial cell Ca2+ events suggest this effect is unlikely to be explained by a direct action on the endothelial cells, but is more likely to be secondary to smooth muscle activation. In addition, since there is no evidence for voltage-gated Ca2+ channels in the endothelial cells [41] of this vascular bed, the inhibitory effect of nifedipine on Ca2+ events will reflect inhibition of voltage-gated Ca2+ channels located in the smooth muscle cells.
The gap junction uncouplers carbenoxolone and 18· -GA inhibited spontaneous endothelial cell Ca2+ events in unstimulated pressurized mesenteric arteries. Carbenoxolone did not affect the ability of endothelial cells to respond to ACh, neither did it alter the magnitude of Ca2+ increase, nor the frequency of oscillations. Therefore, it is unlikely that carbenoxolone is modifying the M3-receptor signalling pathways, which include stimulation of phospholipase C, the release of Ca2+ from internal stores and Ca2+ influx [30]. Therefore we propose that carbenoxolone is indeed preventing intercellular Ca2+ signalling through gap junctions, including myoendothelial gap junctions. The inhibition of cell—cell dye coupling from the endothelial cells to smooth muscle cells with carbenoxolone supports this view. The observation that calcein remained concentrated within the endothelial cells, including the endothelial cell projections which form gap junctions with the muscle, strongly suggests that the dye normally leaves the endothelium via gap junctions (and/or possibly connexin hemichannels) [42]. This technique has been used previously to show the effectiveness of another group of gap junction uncoupling agents, the connexin mimetic gap peptides [43].
Both carbenoxolone and 18· -GA reduced the ability of PE and KCl to increase Ca2+ events within endothelial cells, suggesting that a Ca2+ signal from the muscle passes through myoendothelial gap junctions to stimulate the endothelial cells. In this regard, carbenoxolone was more effective than 18· -GA, which is consistent with several studies showing 18· -GA to be less effective than carbenoxolone [44-47]. However, caution should be exercised in interpreting these findings; although these uncouplers have inhibitory effects on gap junctions, they are also known to have non-specific effects directly on endothelial cells [44,47,48]. Although carbenoxolone had little effect on endothelial cell resting membrane potential in rat mesenteric arteries which had been cut open and pinned out, it did appear to reduce hyperpolarization to ACh. Further, carbenoxolone inhibited the inter-endothelial cell passage of lucifer yellow dye. Interestingly, the effects of carbenoxolone were shown to be reversible for electrical, but not dye coupling [47], suggesting current passes more readily between cells. In the presence of an effective gap junction uncoupler, the hyperpolarization to ACh would be less influenced by adjacent cells, which may normally act to amplify and average responses throughout the tissue, including to slightly damaged cells. However, in arteries under a physiological tension, we failed to record a depression of ACh-evoked in endothelial cell hyperpolarization with carbenoxolone, at a time when the hyperpolarization to levcromakalim was blocked (unpublished observations). Note that the KATP channels activated by levcromakalim are found only in the smooth muscle, not the endothelial cells in the mesenteric artery [31,49]. Furthermore, carbenoxolone did not depress the increase in endothelial cell Ca2+ evoked with ACh in pressurized mesenteric arteries [50]. Taken together, these data indicate carbenoxolone can block gap junctions, albeit that some unrecognized actions on the artery wall may also occur.
By using the approach of loading endothelial cells and smooth muscle cells with Ca2+ dye, we established that carbenoxolone did not affect the spontaneous or PE-evoked Ca2+ events in smooth muscle cells. Importantly, and in contrast to another report in the same tissue [37], we used concentrations of PE that did not stimulate vasomotion, and therefore only observed asynchronous Ca2+ events that likely did not rely on cell—cell coupling, supporting a lack of non-specific actions of carbenoxolone on smooth muscle Ca2+ events. A similar imaging approach has been used previously [37], but the authors were able to carefully load a different Ca2+-sensitive dye into each cell type and enabled simultaneous Ca2+ imaging in endothelial and smooth muscle cells. That study fully supports our data that both KCl and PE increase endothelial cell Ca2+ levels, and showed that another gap junction uncoupler, palmitoleic acid, inhibited the rise in endothelial cell Ca2+ levels, and in the case of KCl, did not affect the response in smooth muscle cells [37]. Further support of an influence from the muscle layers via gap junctions comes from our use of nifedipine, which decreases [Ca2+]i by inhibiting voltage-gated Ca2+ channels in smooth muscle cells, and significantly inhibited Ca2+ events in endothelial cells. Overall, these separate lines of evidence from pressurized mesenteric arteries strongly support the suggestion that regulation of Ca2+ events within endothelial cells by the adjacent smooth muscle cells is affected through gap junctions, even under resting conditions.
Among the potential candidates for Ca2+ signalling through myoendothelial gap junctions are IP3 and Ca2+ [15,37,51,52]. The exact contribution possible by Ca2+ and IP3 passing through gap junctions remains to be defined. Generally, PE acts to increase IP3 and thus release Ca2+ from the sarcoplasmic reticulum, while KCl increases Ca2+ via depolarization and the opening of smooth muscle voltage-gated Ca2+ channels [53,54]. As PE and KCl each had a similar effect on spontaneous Ca2+ events in the endothelium, Ca2+ rather than IP3 may be expected to be the predominant influence diffusing from the smooth muscle to the endothelial cells. Ca2+ and IP3 are, however, interdependent in terms of their regenerative process [54]. Indeed, the ability of KCl to evoke rises in endothelial cell Ca2+ appears dependent on phospholipase C and IP3-induced Ca2+ release [37], supporting an important role for this signalling pathway in rat mesenteric arteries. Therefore, in order to define the relative importance of each signal, a specific and effective cell-permeant inhibitor targeted to IP3 receptors or the selective loading of specific inhibitory IP3 receptor antibodies into endothelial cells are called for. An additional consideration is that the passage of Ca2+ via myoendothelial gap junctions into the endothelium is likely to contribute to Ca2+ accumulation into the ER [37,52], which together with the likely expression of IP3 receptors within this signalling microdomain [52,55], suggests that this filling/release pathway may contribute to the increase in Ca2+ events in endothelial cells. How the influence from smooth muscle cells relates to ‘spontaneous’ activation of phospholipase C in endothelial cells has yet to be established, but this may reflect parallel pathways both required for the generation of endothelial cell Ca2+ events.
Endothelial cell Ca2+ influx through the cell membrane
Endothelial cell Ca2+ events were substantially reduced in the presence of inhibitors of non-selective cation channels (NSCC), SKF-96365 or NiCl2. Additionally, an inhibitor of the reverse mode of NCX, KB-R 7943 also significantly decreased the endothelial cell Ca2+ events. NSCCs by definition are not highly selective for Ca2+ (PCa/PNa < 10) [7] and have a higher relative membrane permeability to Na+ compared with voltage-gated Ca2+ channels (PCa/PNa > 1000) [56]. Furthermore, as Na+ (· 150 mM) is much more concentrated than Ca2+ (2 mM) in the extracellular space, NSCC opening will affect the entry of Na+ in addition to Ca2+. Na+ entry into the plasmalemmal space through NSCC, would be predicted to contribute to a reversed mode function of the NCX, resulting in Ca2+ influx into the endothelial cells. Several studies have suggested that Na+ influx through receptor-operated cation channels/store-operated Ca2+ channels is serially coupled to Ca2+ influx through NCX in vascular smooth muscle cells, and that blockade of either the NCX or NSCC can abolish Ca2+ oscillations and puffs in both vascular smooth muscle cells [57-59] and an endothelial cell line [36], respectively. These data suggest that both NSCC and NCX in the endothelial cells might contribute to the Ca2+ influx, enhancing Ca2+ levels in the endoplasmic reticulum.
In summary, spontaneous Ca2+ events occur in endothelial cells and appear to originate from opening of IP3 receptors in the endoplasmic reticulum. Release of Ca2+ via the IP3 receptor, detected as puffs, will cause a local Ca2+ increase favouring activation of neighbouring IP3 receptors [60] and simultaneous opening of store-operated NSCC. Together these events would be predicted to activate KCa channels in the endothelial cell membrane, which functionally would serve to decrease artery tone (and thus blood pressure). Na+ influx into the endothelial cells (along with Ca2+) will promote the reverse mode of NCX, contributing to Ca2+ influx. Ca2+ and/or IP3 moving from the smooth muscle cells through myoendothelial gap junctions will contribute to this series of events. So overall, it is not only Ca2+ influx across the endothelial cell membrane that is important in determining Ca2+ levels in the cytoplasm and endoplasmic reticulum, but also the adjacent smooth muscle.
Conclusion
Spontaneous Ca2+ events in endothelial cells occur to a significant extent in mesenteric arteries at physiological pressures. These localized increases in cytoplasmic Ca2+ concentration, originate from the endoplasmic reticulum, and are subject to modulation not only by ion channels in the endothelial membrane, but also by the smooth muscle cells through gap junction connections. The physiological significance of the present observations remains to be determined.
Supplementary Material
Acknowledgement
This work was supported by the Wellcome Trust, UK.
Footnotes
Appendix A. Supplementary data
Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.ceca. 2007.11.012.
References
- [1].Chen GF, Suzuki H. Calcium dependency of the endothelium-dependent hyperpolarization in smooth muscle cells of the rabbit carotid artery. J. Physiol. 1990;421:521–534. doi: 10.1113/jphysiol.1990.sp017959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Domeier TL, Segal SS. Electromechanical and pharmacomechanical signalling pathways for conducted vasodilatation along endothelium of hamster feed arteries. J. Physiol. 2007;579:175–186. doi: 10.1113/jphysiol.2006.124529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Edwards G, Dora KA, Gardener MJ, Garland CJ, Weston AH. K+ is an endothelium-derived hyperpolarizing factor in rat arteries. Nature. 1998;396:269–272. doi: 10.1038/24388. [DOI] [PubMed] [Google Scholar]
- [4].Feletou M, Vanhoutte PM. Endothelium-derived hyperpolarizing factor: where are we now? Arterioscler. Thromb. Vasc. Biol. 2006;26:1215–1225. doi: 10.1161/01.ATV.0000217611.81085.c5. [DOI] [PubMed] [Google Scholar]
- [5].Marchenko SM, Sage SO. Effects of shear stress on [Ca2+]i and membrane potential of vascular endothelium of intact rat blood vessels. Exp. Physiol. 2000;85:43–48. [PubMed] [Google Scholar]
- [6].Parkington HC, Coleman HA, Tare M. Prostacyclin and endothelium-dependent hyperpolarization. Pharmacol. Res. 2004;49:509–514. doi: 10.1016/j.phrs.2003.11.012. [DOI] [PubMed] [Google Scholar]
- [7].Nilius B, Droogmans G. Ion channels and their functional role in vascular endothelium. Physiol. Rev. 2001;81:1415–1459. doi: 10.1152/physrev.2001.81.4.1415. [DOI] [PubMed] [Google Scholar]
- [8].Paria BC, Bair AM, Xue J, Yu Y, Malik AB, Tiruppathi C. Ca2+ influx induced by protease-activated receptor-1 activates a feed-forward mechanism of TRPC1 expression via nuclear factor-kappaB activation in endothelial cells. J. Biol. Chem. 2006;281:20715–20727. doi: 10.1074/jbc.M600722200. [DOI] [PubMed] [Google Scholar]
- [9].Beny JL, Pacicca C. Bidirectional electrical communication between smooth muscle and endothelial cells in the pig coronary artery. Am. J. Physiol. 1994;266:H1465–H1472. doi: 10.1152/ajpheart.1994.266.4.H1465. [DOI] [PubMed] [Google Scholar]
- [10].De Wit C. Connexins pave the way for vascular communication. News Physiol. Sci. 2004;19:148–153. doi: 10.1152/nips.01520.2004. [DOI] [PubMed] [Google Scholar]
- [11].Xia J, Little TL, Duling BR. Cellular pathways of the conducted electrical response in arterioles of hamster cheek pouch in vitro. Am. J. Physiol. Heart Circ. Physiol. 1995;269:H2031–H2038. doi: 10.1152/ajpheart.1995.269.6.H2031. [DOI] [PubMed] [Google Scholar]
- [12].Little TL, Xia J, Duling BR. Dye tracers define differential endothelial and smooth muscle coupling patterns within the arteriolar wall. Circ. Res. 1995;76:498–504. doi: 10.1161/01.res.76.3.498. [DOI] [PubMed] [Google Scholar]
- [13].Yamamoto Y, Imaeda K, Suzuki H. Endothelium-dependent hyperpolarization and intercellular electrical coupling in guinea-pig mesenteric arterioles. J. Physiol. 1999;514:505–513. doi: 10.1111/j.1469-7793.1999.505ae.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Sandow SL, Tare M, Coleman HA, Hill CE, Parkington HC. Involvement of myoendothelial gap junctions in the actions of endothelium-derived hyperpolarizing factor. Circ. Res. 2002;90:1108–1113. doi: 10.1161/01.res.0000019756.88731.83. [DOI] [PubMed] [Google Scholar]
- [15].Dora KA, Doyle MP, Duling BR. Elevation of intracellular calcium in smooth muscle causes endothelial cell generation of NO in arterioles. Proc. Natl. Acad. Sci. U.S.A. 1997;94:6529–6534. doi: 10.1073/pnas.94.12.6529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Dora KA, Hinton JM, Walker SD, Garland CJ. An indirect influence of phenylephrine on the release of endothelium-derived vasodilators in rat small mesenteric artery. Br. J. Pharmacol. 2000;129:381–387. doi: 10.1038/sj.bjp.0703052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Oishi H, Budel S, Schuster A, Stergiopulos N, Meister JJ, Beny JL. Cytosolic-free calcium in smooth-muscle and endothelial cells in an intact arterial wall from rat mesenteric artery in vitro. Cell Calcium. 2001;30:261–267. doi: 10.1054/ceca.2001.0233. [DOI] [PubMed] [Google Scholar]
- [18].Yashiro Y, Duling BR. Integrated Ca2+ signaling between smooth muscle and endothelium of resistance vessels. Circ. Res. 2000;87:1048–1054. doi: 10.1161/01.res.87.11.1048. [DOI] [PubMed] [Google Scholar]
- [19].Ying XY, Minamiya Y, Fu CZ, Bhattacharya J. Ca2+ waves in lung capillary endothelium. Circ. Res. 1996;79:898–908. doi: 10.1161/01.res.79.4.898. [DOI] [PubMed] [Google Scholar]
- [20].Burdyga T, Shmygol A, Eisner DA, Wray S. A new technique for simultaneous and in situ measurements of Ca2+ signals in arteriolar smooth muscle and endothelial cells. Cell Calcium. 2003;34:27–33. doi: 10.1016/s0143-4160(03)00019-8. [DOI] [PubMed] [Google Scholar]
- [21].Duza T, Sarelius IH. Conducted dilations initiated by purines in arterioles are endothelium dependent and require endothelial Ca2+ Am. J. Physiol. Heart Circ. Physiol. 2003;285:H26–H37. doi: 10.1152/ajpheart.00788.2002. [DOI] [PubMed] [Google Scholar]
- [22].Duza T, Sarelius IH. Increase in endothelial cell Ca2+ in response to mouse cremaster muscle contraction. J. Physiol. 2004;555:459–469. doi: 10.1113/jphysiol.2003.051029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Huser J, Blatter LA. Elementary events of agonist-induced Ca2+ release in vascular endothelial cells. Am. J. Physiol. Cell Physiol. 1997;273:C1775–C1782. doi: 10.1152/ajpcell.1997.273.5.C1775. [DOI] [PubMed] [Google Scholar]
- [24].Hoyer J, Kohler R, Distler A. Mechanosensitive Ca2+ oscillations and STOC activation in endothelial cells. FASEB J. 1998;12:359–366. doi: 10.1096/fasebj.12.3.359. [DOI] [PubMed] [Google Scholar]
- [25].Jaggar JH. Intravascular pressure regulates local and global Ca2+ signaling in cerebral artery smooth muscle cells. Am. J. Physiol. Cell Physiol. 2001;281:C439–C448. doi: 10.1152/ajpcell.2001.281.2.C439. [DOI] [PubMed] [Google Scholar]
- [26].Nelson MT, Cheng H, Rubart M, Santana LF, Bonev AD, Knot HJ, Lederer WJ. Relaxation of arterial smooth muscle by calcium sparks. Science. 1995;270:633–637. doi: 10.1126/science.270.5236.633. [DOI] [PubMed] [Google Scholar]
- [27].Nelson MT, Quayle JM. Physiological roles and properties of potassium channels in arterial smooth muscle. Am. J. Physiol. Cell Physiol. 1995;268:C799–C822. doi: 10.1152/ajpcell.1995.268.4.C799. [DOI] [PubMed] [Google Scholar]
- [28].Bolton TB, Imaizumi Y. Spontaneous transient outward currents in smooth muscle cells. Cell Calcium. 1996;20:141–152. doi: 10.1016/s0143-4160(96)90103-7. [DOI] [PubMed] [Google Scholar]
- [29].Earley S, Heppner TJ, Nelson MT, Brayden JE. TRPV4 forms a novel Ca2+ signaling complex with ryanodine receptors and BKCa channels. Circ. Res. 2005;97:1270–1279. doi: 10.1161/01.RES.0000194321.60300.d6. [DOI] [PubMed] [Google Scholar]
- [30].McSherry IN, Spitaler MM, Takano H, Dora KA. Endothelial cell Ca2+ increases are independent of membrane potential in pressurized rat mesenteric arteries. Cell Calcium. 2005;38:23–33. doi: 10.1016/j.ceca.2005.03.007. [DOI] [PubMed] [Google Scholar]
- [31].Takano H, Dora KA, Spitaler MM, Garland CJ. Spreading dilatation in rat mesenteric arteries associated with calcium-independent endothelial cell hyperpolarization. J. Physiol. 2004;556:887–903. doi: 10.1113/jphysiol.2003.060343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Walker SD, Dora KA, Ings NT, Crane GJ, Garland CJ. Activation of endothelial cell IKCa with 1-ethyl-2-benzimidazolinone evokes smooth muscle hyperpolarization in rat isolated mesenteric artery. Br. J. Pharmacol. 2001;134:1548–1554. doi: 10.1038/sj.bjp.0704415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Taylor MS, Bonev AD, Gross TP, Eckman DM, Brayden JE, Bond CT, Adelman JP, Nelson MT. Altered expression of small-conductance Ca2+-activated K+ (SK3) channels modulates arterial tone and blood pressure. Circ. Res. 2003;93:124–131. doi: 10.1161/01.RES.0000081980.63146.69. [DOI] [PubMed] [Google Scholar]
- [34].Si H, Heyken WT, Wolfle SE, Tysiac M, Schubert R, Grgic I, Vilianovich L, Giebing G, Maier T, Gross V, Bader M, de Wit C, Hoyer J, Kohler R. Impaired endothelium-derived hyperpolarizing factor-mediated dilations and increased blood pressure in mice deficient of the intermediate-conductance Ca2+-activated K+ channel. Circ. Res. 2006;99:537–544. doi: 10.1161/01.RES.0000238377.08219.0c. [DOI] [PubMed] [Google Scholar]
- [35].Zhang G, Teggatz EG, Zhang AY, Koeberl MJ, Yi F, Chen L, Li PL. Cyclic ADP ribose-mediated Ca2+ signaling in mediating endothelial nitric oxide production in bovine coronary arteries. Am. J. Physiol. Heart Circ. Physiol. 2006;290:H1172–H1181. doi: 10.1152/ajpheart.00441.2005. [DOI] [PubMed] [Google Scholar]
- [36].Paltauf-Doburzynska J, Frieden M, Spitaler M, Graier WF. Histamine-induced Ca2+ oscillations in a human endothelial cell line depend on transmembrane ion flux, ryanodine receptors and endoplasmic reticulum Ca2+-ATPase. J. Physiol. 2000;524(Pt. 3):701–713. doi: 10.1111/j.1469-7793.2000.00701.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Lamboley M, Pittet P, Koenigsberger M, Sauser R, Beny JL, Meister JJ. Evidence for signaling via gap junctions from smooth muscle to endothelial cells in rat mesenteric arteries: possible implication of a second messenger. Cell Calcium. 2005;37:311–320. doi: 10.1016/j.ceca.2004.11.004. [DOI] [PubMed] [Google Scholar]
- [38].Richards GR, Weston AH, Burnham MP, Feletou M, Vanhoutte PM, Edwards G. Suppression of K+-induced hyperpolarization by phenylephrine in rat mesenteric artery: relevance to studies of endothelium-derived hyperpolarizing factor. Br. J. Pharmacol. 2001;134:1–5. doi: 10.1038/sj.bjp.0704256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Kamouchi M, Droogmans G, Nilius B. Membrane potential as a modulator of the free intracellular Ca2+ concentration in agonist-activated endothelial cells. Gen. Physiol. Biophys. 1999;18:199–208. [PubMed] [Google Scholar]
- [40].Malli R, Frieden M, Hunkova M, Trenker M, Graier WF. Ca2+ refilling of the endoplasmic reticulum is largely preserved albeit reduced Ca2+ entry in endothelial cells. Cell Calcium. 2007;41:63–76. doi: 10.1016/j.ceca.2006.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Colden-Stanfield M, Schilling WP, Ritchie AK, Eskin SG, Navarro LT, Kunze DL. Bradykinin-induced increases in cytosolic calcium and ionic currents in cultured bovine aortic endothelial cells. Circ. Res. 1987;61:632–640. doi: 10.1161/01.res.61.5.632. [DOI] [PubMed] [Google Scholar]
- [42].Evans WH, De Vuyst E, Leybaert L. The gap junction cellular internet: connexin hemichannels enter the signalling limelight. Biochem. J. 2006;397:1–14. doi: 10.1042/BJ20060175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Griffith TM, Chaytor AT, Taylor HJ, Giddings BD, Edwards DH. cAMP facilitates EDHF-type relaxations in conduit arteries by enhancing electrotonic conduction via gap junctions. Proc. Natl. Acad. Sci. U.S.A. 2002;23:23. doi: 10.1073/pnas.092089799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Coleman HA, Tare M, Parkington HC. K+ currents underlying the action of endothelium-derived hyperpolarizing factor in guinea-pig, rat and human blood vessels. J. Physiol. 2001;531:359–373. doi: 10.1111/j.1469-7793.2001.0359i.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Goto K, Fujii K, Kansui Y, Abe I, Iida M. Critical role of gap junctions in endothelium-dependent hyperpolarization in rat mesenteric arteries. Clin. Exp. Pharmacol. Physiol. 2002;29:595–602. doi: 10.1046/j.1440-1681.2002.03689.x. [DOI] [PubMed] [Google Scholar]
- [46].Schuster A, Oishi H, Beny JL, Stergiopulos N, Meister JJ. Simultaneous arterial calcium dynamics and diameter measurements: application to myoendothelial communication. Am. J. Physiol. Heart Circ. Physiol. 2001;280:H1088–H1096. doi: 10.1152/ajpheart.2001.280.3.H1088. [DOI] [PubMed] [Google Scholar]
- [47].Tare M, Coleman HA, Parkington HC. Glycyrrhetinic derivatives inhibit hyperpolarization in endothelial cells of guinea pig and rat arteries. Am. J. Physiol. Heart Circ. Physiol. 2002;282:H335–H341. doi: 10.1152/ajpheart.2002.282.1.H335. [DOI] [PubMed] [Google Scholar]
- [48].Davidson JS, Baumgarten IM. Glycyrrhetinic acid derivatives: a novel class of inhibitors of gap-junctional intercellular communication. Structure—activity relationships. J. Pharmacol. Exp. Ther. 1988;246:1104–1107. [PubMed] [Google Scholar]
- [49].White TA, Johnson S, Walseth TF, Lee HC, Graeff RM, Munshi CB, Prakash YS, Sieck GC, Kannan MS. Subcellular localization of cyclic ADP-ribosyl cyclase and cyclic ADP-ribose hydrolase activities in porcine airway smooth muscle. Biochim. Biophys. Acta. 2000;1498:64–71. doi: 10.1016/s0167-4889(00)00077-x. [DOI] [PubMed] [Google Scholar]
- [50].Mather S, Dora KA, Sandow SL, Winter P, Garland CJ. Rapid endothelial cell-selective loading of connexin 40 antibody blocks endothelium-derived hyperpolarizing factor dilation in rat small mesenteric arteries. Circ. Res. 2005;97:399–407. doi: 10.1161/01.RES.0000178008.46759.d0. [DOI] [PubMed] [Google Scholar]
- [51].Dora KA. Intercellular Ca2+ signalling: the artery wall. Semin. Cell Dev. Biol. 2001;12:27–35. doi: 10.1006/scdb.2000.0214. [DOI] [PubMed] [Google Scholar]
- [52].Isakson BE, Ramos SI, Duling BR. Ca2+ and inositol 1,4,5-trisphosphate-mediated signaling across the myoendothelial junction. Circ. Res. 2007;100:246–254. doi: 10.1161/01.RES.0000257744.23795.93. [DOI] [PubMed] [Google Scholar]
- [53].Kotecha N, Hill MA. Myogenic contraction in rat skeletal muscle arterioles: smooth muscle membrane potential and Ca2+ signaling. Am. J. Physiol. Heart Circ. Physiol. 2005;289:H1326–H1334. doi: 10.1152/ajpheart.00323.2005. [DOI] [PubMed] [Google Scholar]
- [54].Rottingen J, Iversen JG. Ruled by waves? Intracellular and intercellular calcium signalling. Acta Physiol. Scand. 2000;169:203–219. doi: 10.1046/j.1365-201x.2000.00732.x. [DOI] [PubMed] [Google Scholar]
- [55].Grayson TH, Haddock RE, Murray TP, Wojcikiewicz RJ, Hill CE. Inositol 1,4,5-trisphosphate receptor subtypes are differentially distributed between smooth muscle and endothelial layers of rat arteries. Cell Calcium. 2004;36:447–458. doi: 10.1016/j.ceca.2004.04.005. [DOI] [PubMed] [Google Scholar]
- [56].Bean BP. Classes of calcium channels in vertebrate cells. Annu. Rev. Physiol. 1989;51:367–384. doi: 10.1146/annurev.ph.51.030189.002055. [DOI] [PubMed] [Google Scholar]
- [57].Arnon A, Hamlyn JM, Blaustein MP. Na+ entry via store-operated channels modulates Ca2+ signaling in arterial myocytes. Am. J. Physiol. Cell Physiol. 2000;278:C163–C173. doi: 10.1152/ajpcell.2000.278.1.C163. [DOI] [PubMed] [Google Scholar]
- [58].Lee CH, Poburko D, Sahota P, Sandhu J, Ruehlmann DO, van Breemen C. The mechanism of phenylephrine-mediated [Ca2+]i oscillations underlying tonic contraction in the rabbit inferior vena cava. J. Physiol. 2001;534:641–650. doi: 10.1111/j.1469-7793.2001.t01-1-00641.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Lee CH, Rahimian R, Szado T, Sandhu J, Poburko D, Behra T, Chan L, van Breemen C. Sequential opening of IP3-sensitive Ca2+ channels and SOC during alpha-adrenergic activation of rabbit vena cava. Am. J. Physiol. Heart Circ. Physiol. 2002;282:H1768–H1777. doi: 10.1152/ajpheart.00637.2001. [DOI] [PubMed] [Google Scholar]
- [60].Iino M, Endo M. Calcium-dependent immediate feedback control of inositol 1,4,5-triphosphate-induced Ca2+ release. Nature. 1992;360:76–78. doi: 10.1038/360076a0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.