

Abstract
Context:
There are at least twenty-four missense, non-conservative mutations found in the ACTH receptor (Melanocortin 2 receptor, MC2R) which have been associated with the autosomal recessive disease Familial Glucocorticoid Deficiency (FGD) type 1. The characterization of these mutations has been hindered by difficulties in establishing a functional heterologous cell transfection system for MC2R. Recently the melanocortin 2 receptor accessory protein (MRAP) was identified as essential for trafficking of MC2R to the cell surface; therefore a functional characterization of MC2R mutations is now possible.
Objective:
To elucidate the molecular mechanisms responsible for defective MC2R function in FGD.
Methods:
Stable cell lines expressing human MRAPα were established and transiently transfected with wild-type or mutant MC2R. Functional characterization of mutant MC2R was performed using a cell surface expression assay, a cAMP reporter assay, confocal microscopy and co-immunoprecipitation of MRAPα.
Results:
Two thirds of all MC2R mutations had a significant reduction in cell surface trafficking even though MRAPα interacted with all mutants. Analysis of those mutant receptors that reached the cell surface indicated that 4/6 failed to signal, following stimulation with ACTH.
Conclusion:
The majority of MC2R mutations found in FGD fail to function because they fail to traffic to the cell surface.
Keywords: ACTH, G protein-coupled receptors, melanocortin 2 receptors, Familial Glucocorticoid Deficiency
Introduction
Familial Glucocorticoid Deficiency (FGD) is a rare autosomal recessive disease which is characterized by isolated glucocorticoid deficiency and ACTH resistance; presenting features include severe neonatal hypoglycaemia, frequent childhood infection and excessive skin pigmentation (1-3). This is a genetically heterogeneous disorder with mutations in the ACTH receptor (Melanocortin 2 receptor, MC2R) accounting for approximately 25% of cases of FGD (4-7) and this is referred to as FGD type 1 (OMIM 202200).
MC2R is one of five melanocortin receptors (8). It is a 297 amino acid seven transmembrane receptor, the smallest of all G protein-coupled receptors (GPCRs); it is expressed predominantly in adrenocortical cells and is highly specific for ACTH, which stimulates the production of glucocorticoids. Earlier studies on MC2R were hampered by significant difficulties in expressing this GPCR in heterologous cells. The MC2R cDNA is transcribed and translated from transfected expression plasmids, but the protein product fails to traffic from the endoplasmic reticulum (ER) (9). The observation that human MC2R could be expressed in cells of adrenal origin but which lack endogenous melanocortin receptors (the mouse Y6 and OS3 cell lines) (10-12), has lead to the hypothesis that a cofactor for expression is required. Identification of the genetic cause of a second form of FGD (FGD type 2) lead to the identification of a novel gene encoding a single transmembrane domain protein, named Melanocortin 2 receptor accessory protein (MRAP) which appears to fulfil this role. MRAP interacts with MC2R and facilitates the trafficking of the receptor from ER to the cell surface (13;14).
The discovery of MRAP now provides the opportunity to characterise fully the MC2R in readily transfected and maintained cell lines, such as Chinese hamster ovary (CHO) and human embryonic kidney 293 (HEK293) cells, both of which are known not to endogenously express melanocortin receptor (MCR) and to be unresponsive to ACTH and NDP-MSH (15;16). This MCR-free system allows the complete characterization of naturally occurring mutations enabling the analysis of cell surface expression and signal transduction.
Previous studies have shown that some of these mutations have impaired ligand binding and cAMP signaling, but the mechanisms for this defect were not investigated. This comprehensive analysis of MC2R mutations is the first study to examine the effect of the clinically identified mutations on both receptor trafficking and signal transduction.
Materials and Methods
CHO and HEK293 cell lines expressing human MRAPα
Human MRAPα amplified from human adrenal cDNA (Ambion) was subcloned into pcDNA3.1(+) vector (Invitrogen). CHO cells were grown in DMEM/Ham's F12 (1:1) with 10% foetal bovine serum and penicillin/streptomycin. HEK293 cells were grown in DMEM with 10% foetal bovine serum and penicillin/streptomycin. Cells were transfected with the appropriate pcDNA3.1 vector, either empty vector or pcDNA3.1-MRAPα, using Lipofectamine ™2000 (Invitrogen) and selected with Geneticin (G418) at 700μg/ml for CHO and 1500μg/ml for HEK293 cells. Monoclonal stable cells were selected by the dilutional method plating in 96 well plates, at less than 1 cell per well. Expression of MRAPα mRNA in the selected stable cell lines was confirmed using RT-PCR. In addition, the cell surface assay with wild-type hemagglutinin (HA) epitope tagged at the N-terminus MC2R (Wt-HA-MC2R) was performed to ensure cell surface expression of MC2R.
Site directed mutagenesis
Mutant MC2R expression constructs were generated by site-directed mutagenesis (QuickChange®II XL) using human Wt-HA-MC2R (Missouri S&T cDNA Resource Center) as template. Primer sequences are available on request. The entire mutant MC2R constructs were then sequenced to confirm the presence of specific mutations and ensure the absence of unwanted errors.
Cell surface assay
CHO cells stably expressing MRAPα (CHOα1) were seeded in 24 well plates and transfected with Wt-HA-MC2R or mutated HA-MC2R. 24 hours post transfection, cells were washed with PBS and fixed in 4% formaldehyde for 10minutes. Cells from one plate were then permeabilised with 0.025% Triton. Both permeabilised and unpermeabilsed plates were blocked with Odyssey® Blocking buffer (LI-COR) for 1 hour, followed by incubation with anti-HA antibody (1:1000) for 1 hour. Infrared-labelled secondary goat anti-mouse antibody was then added at 1:1000 (Odyssey® Goat anti-mouse IR Dye @800CW) for one hour. Thorough washes with PBS were crucial after each step. The plates were then analysed with the LI-COR Odyssey® plate reader (800nm). The relative values of cell surface expression of the receptors were calculated by the ratio of unpermeabilised and permeabilised cells which was then normalised to wild type expression in each experiment.
Immunoblotting and co-immunoprecipitation
Interaction of mutated MC2R with MRAPα was assessed by co-immunoprecipitation (Co-IP) and immunoblotting as previously described (13;14) CHO cells were transiently transfected with MRAPα-Flag, and mutant HA-MC2R constructs and twenty four hours after transfection, cells were lysed with 0.1% D-dodecyl-β-maltoside in PBS in the presence of protease inhibitors. Anti-HA agarose conjugate beads (Sigma) were added to the cell lysate and incubated overnight, at 4°C. The beads were collected, washed thoroughly and resuspended in SDS sample buffer before heating at 95°C for 10 minutes. The samples were then subjected to immunoblotting using anti-Flag (Sigma) antibodies at a dilution of 1:2000, followed by secondary antibody incubation (IRDye 800CW Goat Anti-Mouse IgG, LI-COR) at 1:10000 for 1 hour and imaged with LI-COR's Odyssey Infrared Image System.
Confocal microscopy
CHOα1 cells were seeded into 12 well plates containing sterile cover slips and transfected with either Wt-HA-MC2R, empty vector or mutated HA epitope-tagged MC2Rs for 24 hours. After fixing, the cells were permeabilised with 0.025% Triton and incubated with blocking buffer (3% BSA, 10% donkey serum in PBS) to block non-specific binding for 1 hour. The cells were then incubated with mouse anti-HA antibody (1:200 dilution in buffer A) for 90 minutes. This was followed by incubation with Cy™3-conjugated Donkey anti-mouse antibody 1:100 (Jackson ImmunoResearch, PA, US) for 1 hour. For co-localization with the ER, fixed and permeabilised cells were incubated with rabbit anti-calnexin antibodies 1:300 (Sigma) and anti-HA antibody 1:200 in blocking buffer for 90 minutes. Secondary antibody incubation was performed with Cy™3 (mouse) at 1:100 and Cy™2 (rabbit) at 1:50 (Jackson ImmunoResearch, PA, US) for 1 hour. Nuclei were stained with PBS containing 2μg/ml DAPI, 4′,6-diamidino-2-phenylindole. The cover slips were then carefully placed onto a clean slide and mounted with drops of fluorescent mounting media. Fluorescent images were taken using a Zeiss LSM 510 confocal microscope.
cAMP reporter assay
Stable HEK293 MRAPα (HEKα7) cells grown to 70-80% confluence in 6 well plates were transfected with 1000ng of receptor expression plasmids along with 900ng αGSU-846 luciferase and 100ng pRL-CMV Renilla luciferase plasmid constructs (14). 24-48 hours after transfection, cells were stimulated with ACTH (10−7M) for 6 hours. Cell lysates were collected and assayed using the Dual luciferase reporter assay system (Promega). Luciferase activity was measured using a multiplate reader (Lumistar Omega BMG Labtech), and values were normalised to the Renilla luciferase activity.
Statistical analysis
The data reported are the mean ± SEM of at least three independent experiments, performed in duplicates. Statistical comparison was performed using unpaired two-tailed Student's t-test, and P values are indicated as *, P<0.05; **, P<0.01; ***, P<0.0001.
Results
Cell surface expression of MC2R mutations
A clonal CHO cell line stably expressing human MRAPα (CHOα1) was established as a host for transient transfection of N-terminal tagged HA MC2R and all 24 MC2R missense mutations. A cell culture plate based assay was used to monitor cell surface expression of epitope-tagged MC2Rs to the plasma membrane by detection of fluorescent signals (Figure 1). Eighteen out of 24 MC2R mutations demonstrated varying degrees of intracellular retention when compared with wild type. These are subsequently referred to as the trafficking-defective mutations. To confirm the result of cell surface expression assay, confocal analysis of CHOα1 cells expressing HA-MC2R mutants was performed (Figure 2). Wild type MC2R displayed efficient cell surface expression in permeabilised cells. Similarly D103N and R128C showed substantial plasma membrane expression, as did the other mutants which reached the cell surface, (D20N, I44M, D107N and H170L-data not shown). The trafficking-defective mutants such as R146H, T159K and Y254C (Figure 2D, E and F) exhibit marked intracellular retention consistent with the results from the cell surface assay. These mutants demonstrate ER retention, an example shown with T159K which co-localises with calnexin (Figure 2J, K and L). The other trafficking-defective mutants also displayed similar findings (data not shown). The majority of these mutations lie within the transmembrane domain (10/18) and second intracellular loop (5/18) (Figure 3A). The S74I, I130N (Figure 2G and H) and T152K demonstrated partial trafficking impairment, which did not reach a statistically significant difference on cell surface assay.
Figure 1.

Majority of MC2R mutations cause trafficking defects. The cell surface assay was used to quantify cell surface expression comparing Wt-HA-MC2R with all 24 MC2R missense mutations. The quantified relative values of cell surface expression are seen (n=3). *,P<0.05; **, P<0.01; ***, P<0.0001, relative to Wt-HA-MC2R. The majority of MC2R mutations had significant reductions in cell surface expression. There is partial cell surface expression in S74I, I130N and T152K, as there is no significance difference when compared with wild type.
Figure 2.
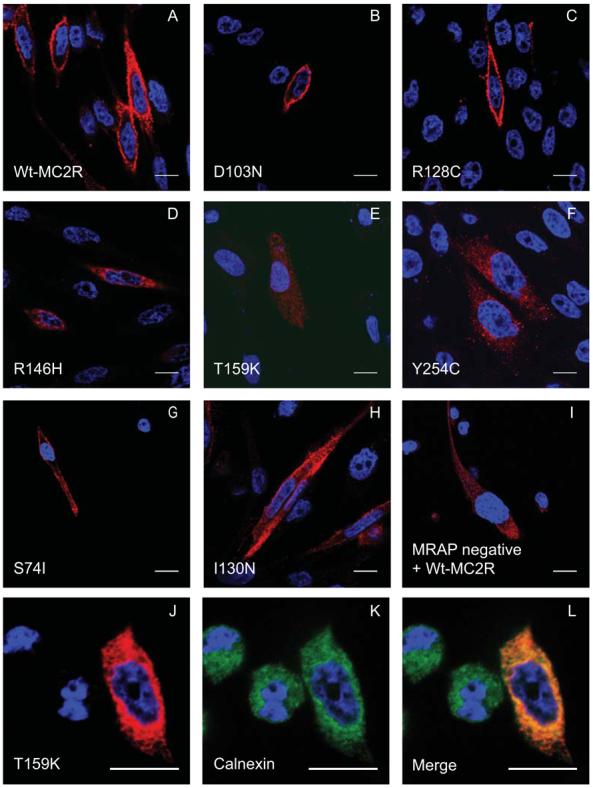
Confocal analysis of MC2R mutants in permeabilised stable CHOα1 confirms results from the cell surface assay. CHOα1 cells were transiently transfected with A: Wt-HA-MC2R (positive control); B: D103N; C: R128C all of which show good cell surface expression. D: R146H; E: T159K and F: Y254C fail to reach the plasma membrane and are retained intracellularly. G: S74I and H: I130N have partial expression at the cell surface. I: Wt-MC2R in MRAP negative cells was used as the negative control. J, K and L: MC2R harbouring mutation T159K (red) co-localise with ER marker, calnexin (green). Cells were incubated with anti-HA antibody (red) and nucleus stained with DAPI, 4′,6-diamidino-2-phenylindole (blue). The scale bar represents 10μm.
Figure 3.
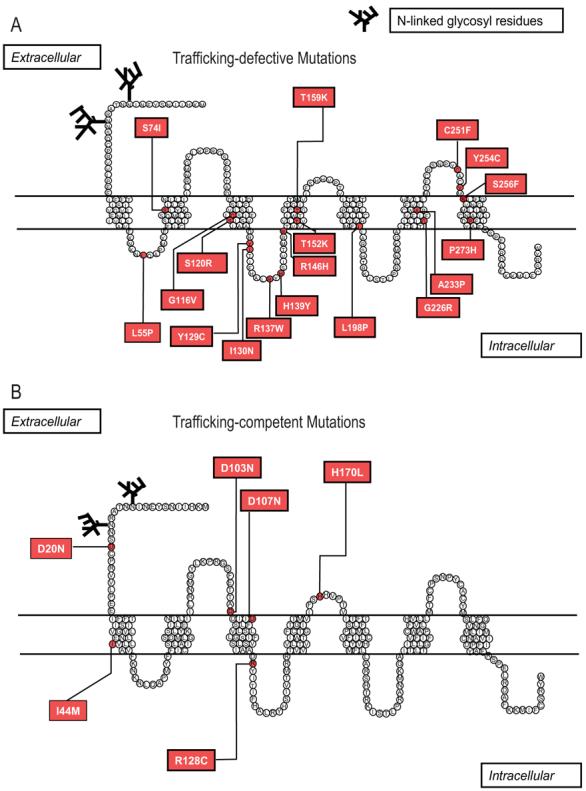
Pseudostructural plot of the MC2R mapping the locations of the 18 trafficking-defective mutations in 2A and the 6 trafficking-competent mutations in 2B.
Of the six mutants with intact trafficking function, three are located in the extracellular domain of MC2R (Figure 3B) consistent with the hypothesis that these mutations may lead to failure of ligand binding. Interestingly, one of the trafficking-competent mutation located within the 2nd intracellular loop of the MC2R is the R128 substitution (R128C), which lies within the highly conserved DRY (Asparagine-Arginine-Tyrosine) motif, which is required for G protein coupling and activation (17;18). The schematic diagram in Figure 3 maps out the distribution of the trafficking-defective and the trafficking-competent mutants.
Co-immunoprecipitation with MRAP
Considering the importance of MRAPα in cell surface expression and receptor function, we investigated whether any mutation in MC2R leads to failure of interaction with MRAPα. Wild type CHO cells were transiently transfected with MRAPα-Flag and either Wt-HA-MC2R or mutant HA-MC2R. Immunoprecipitation of MC2R followed by immunoblotting with anti-Flag antibody provides a qualitative indication of the interaction of Wt-MC2R and MRAPα (Figure 4a). Interestingly, all of the MC2R mutants examined also interacted with MRAPα. Figure 4b demonstrates interaction with MRAPα for L55P, I130N, R137W and G226R.
Figure 4.
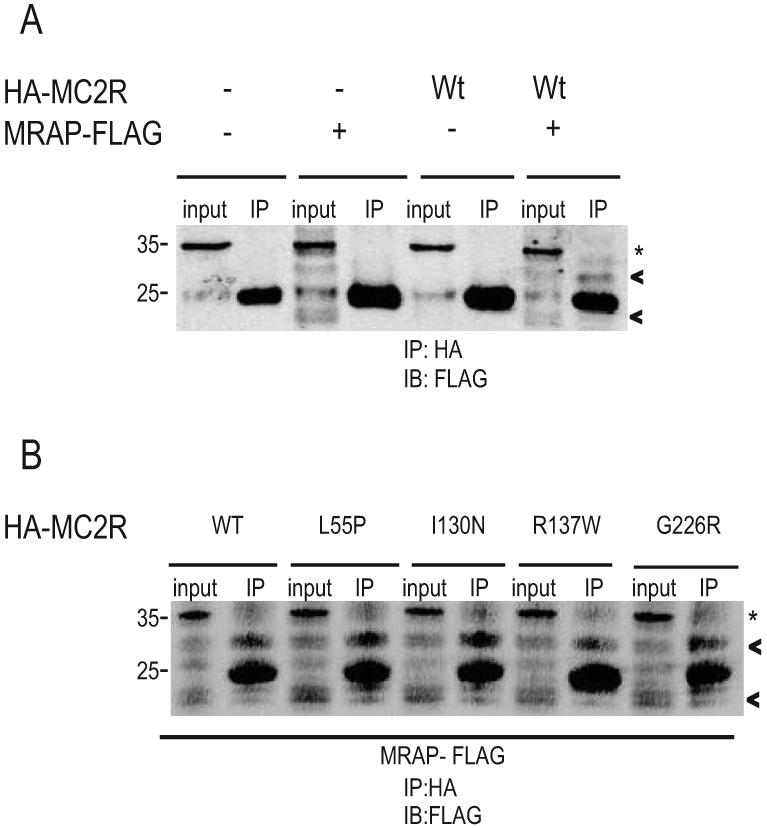
MC2R mutations do not affect interaction with MRAPα. CHO cells transiently transfected with empty vector, MRAPα-Flag, Wt-HA-MC2R or MRAPα-Flag + Wt-HA-MC2R are immunoblotted with anti-Flag antibody (4A). Cell lysates were immunoprecipitated with anti-HA and washed prior to SDS-PAGE and immunoblotting with anti-Flag antibody. Co-immunoprecipitation study demonstrates that Wt-MC2R interacts with MRAPα, the major isoforms are identified as shown by the arrow heads. IP = immunoprecipitation. * represents light chain. CHO cells were transfected with Wt-MC2R, L55P, I130N, R137W and G226R mutants and MRAPα-Flag (4B). Cell lysates were immunoprecipitated with anti-HA and washed prior to immunoblotting. Co-immunoprecipitation studies of these and all 20 additional missense mutations (data not shown) show interaction with MRAPα.
Signaling function of the trafficking-competent mutants
To determine whether there is a loss of MC2R signaling in the mutants expressed at the cell surface, HEKα7 cells were transfected with either Wt-HA-MC2R, untagged Wt-MC2R or mutant HA-MC2R and stimulated with ACTH. The stable HEK cells were chosen in place of the CHOα1 because of their high levels of transfection efficiency, protein expression and good cAMP responsiveness. Interestingly, results showed that the untagged Wt-MC2R generated 40% more cAMP than Wt-HA-MC2R. Furthermore, both D20N and I44M demonstrated similar function when compared with Wt-HA-MC2R (Figure 5). However, all other mutants (D103N, D107N, R128C and H170L) had significant impairment of cAMP generation, as seen by the decreased luciferase activity.
Figure 5.
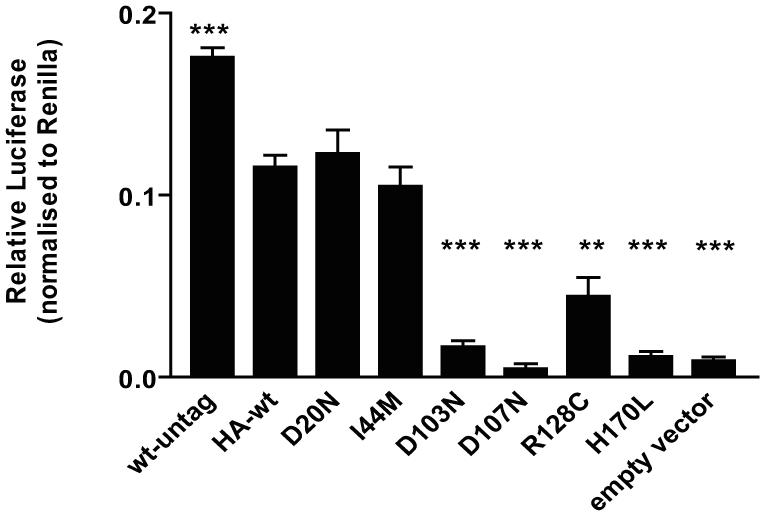
Most trafficking-competent mutants fail to signal. Luciferase assay was employed to assess the effect of MC2R mutations on cell signalling in response to ACTH (10−7M) in the HEKα7 clonal cells. All trafficking-competent mutations were transiently transfected. **,P<0.01; ***,P<0.0001 (compared with Wt-HA-MC2R). Despite good cell surface expression, 4/6 mutations did not respond to stimulation by ACTH after 6 hours treatment.
Discussion
In this study, we systematically characterized the functional defect of twenty four naturally occurring missense mutations of the MC2R found in FGD type 1. It transpires that the majority of the mutations traffic poorly to the plasma membrane despite direct interaction with MRAP and those that do traffic efficiently mainly fail to signal after stimulation with ACTH. This study provides a novel insight into the molecular mechanisms accounting for the MC2R functional defects, and confirms the problem of MC2R trafficking from the ER as that most likely to cause FGD type 1.
Four out of six trafficking-competent mutations, D103N, D107N, R128C and H170L, failed to signal despite receptor cell surface expression. Previous studies found that both D103N and D107N fail to bind ACTH (10;19).
The only receptor mutation which traffics successfully to the cell surface and is located intracelluarly is R128C. This is an interesting mutation as it lies in the Asp-Arg-Tyr (DRY) region in the second intracellular loop, which is the most conserved region of all GPCRs (20). Indeed, the arginine residue in this motif is the only amino acid conserved among all subclass 1 receptors (21). Substitution of the arginine residue within the highly conserved DRY motif will abolish or drastically reduce G protein coupling (17;18) and is proposed to represent the primary trigger for the release of GDP from the receptor-G protein complex (22). Interestingly, substitution of the next amino acid in the DRY motif - the Y129C mutant caused significant intracellular retention. Tyrosine is the least conserved of the triad sequence, and in other GPCRs this tyrosine mutation often only marginally affects receptor function, if at all (23).
Both D20N and I44M were found to have efficient cell surface expression and cell signaling. I44M was described in a Finnish girl by Weber et al (6), as a compound heterozygous mutation in combination with L192fs. This frame shift results in a nonsense sequence of 54 residues followed by a premature stop codon. It is not clear how the I44M mutation alters receptor function, if at all, as this isoleucine in the first transmembrane domain is not a conserved residue and is substituted by the relatively hydrophobic methionine in the bovine ACTH receptor. No novel splice site was created by either mutation as predicted by analysis using http://www.fruitfly.org/seq_tools/splice.html. A further possibility is that both D20N and I44M are in linkage disequilibrium with a more functionally significant mutation elsewhere in the gene and outside the coding region such as the previously reported −2 substitution in the MC2R promoter initiator element (24;25). This is a relatively common polymorphism which is found in 6.5% of a healthy population (25) and normal subjects homozygous for the rarer C allele displayed higher ACTH/cortisol ratios in response to CRH testing (24). This variant has been proposed as a cause of FGD when combined as a compound heterozygote with a frameshift mutation on the other allele (25).
The majority of the mutant receptors trafficked inefficiently to the plasma membrane. Notably, the most severely affected mutations are located towards the C-terminus of the receptor. Previous functional studies performed for a variety of mutations such as G116V (26), R137W (27), R146H (6;10), T159K (10), C251F (19) and Y254C (27) all found that there was impaired receptor signaling when stimulated with ACTH and low affinity for ACTH binding. It is now apparent that this was because of impaired cell surface expression of the receptors. We investigated the hypothesis that mutations that affect trafficking do so by interfering with the interaction between MRAP and MC2R, as the latter plays an important role in facilitating trafficking of the receptor to the cell surface. No mutation was found to block this interaction, indicating that this was not the mechanism underlying trafficking failure.
Several inherited diseases are now found to result from GPCR trafficking defects. These include, rhodpsin mutations in retinitis pigmentosa (28), vasopressin 2 receptor mutations causing nephrogenic diabetes insipudus (29) and GnRHR point mutations causing hypogonadotrophic hypogonadism (30). The strict quality control mechanisms within cells ensures that improperly folded proteins are targeted for degradation via the proteosome or other pathways (31). Some low molecular weight compounds have been shown to inhibit aggregation and/or enable mutant proteins to escape the quality control system and, theoretically, this will result in the “rescue” of their function. These small molecules, named chemical chaperones, are thought to non-selectively stabilise mutant proteins and facilitate their folding (32). Receptor ligands or enzyme inhibitors, which selectively recognise the mutant proteins and rescue conformational mutants, are referred to as pharmacological chaperones, and these present promising therapeutic avenues (33;34). In principle this approach could also be applied for rescuing MC2R mutations with trafficking defects, but this therapy is not likely to be practical, as there already exists a simple and effective treatment in the form of replacement with hydrocortisone.
Acknowledgment
We thank Irina Bogdarina for her help with cloning and technical support.
Footnotes
Disclosure summary:
TTC and LFC are supported by the UK Medical Research Council (MRC) Clinical Training Fellowship programme, SNC by an MRC grant and TRW and LAM by the Wellcome Trust. AJLC, JPC and PJK have nothing to declare.
Reference List
- 1.Shepard TH, Landing BH, Mason DG. Familial Addison's disease; case reports of two sisters with corticoid deficiency unassociated with hypoaldosteronism. AMA J Dis Child. 1959;97(2):154–162. [PubMed] [Google Scholar]
- 2.Franks RC, Nance WE. Hereditary adrenocortical unresponsiveness to ACTH. Pediatrics. 1970;45(1):43–48. [PubMed] [Google Scholar]
- 3.Clark AJ, Weber A. Adrenocorticotropin insensitivity syndromes. Endocr Rev. 1998;19(6):828–843. doi: 10.1210/edrv.19.6.0351. [DOI] [PubMed] [Google Scholar]
- 4.Clark AJ, McLoughlin L, Grossman A. Familial glucocorticoid deficiency associated with point mutation in the adrenocorticotropin receptor. Lancet. 1993;341(8843):461–462. doi: 10.1016/0140-6736(93)90208-x. [DOI] [PubMed] [Google Scholar]
- 5.Tsigos C, Arai K, Hung W, Chrousos GP. Hereditary isolated glucocorticoid deficiency is associated with abnormalities of the adrenocorticotropin receptor gene. J Clin Invest. 1993;92(5):2458–2461. doi: 10.1172/JCI116853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Weber A, Toppari J, Harvey RD, Klann RC, Shaw NJ, Ricker AT, Nanto-Salonen K, Bevan JS, Clark AJ. Adrenocorticotropin receptor gene mutations in familial glucocorticoid deficiency: relationships with clinical features in four families. J Clin Endocrinol Metab. 1995;80(1):65–71. doi: 10.1210/jcem.80.1.7829641. [DOI] [PubMed] [Google Scholar]
- 7.Clark AJ, Metherell LA, Cheetham ME, Huebner A. Inherited ACTH insensitivity illuminates the mechanisms of ACTH action. Trends Endocrinol Metab. 2005;16(10):451–457. doi: 10.1016/j.tem.2005.10.006. [DOI] [PubMed] [Google Scholar]
- 8.Mountjoy KG, Robbins LS, Mortrud MT, Cone RD. The cloning of a family of genes that encode the melanocortin receptors. Science. 1992;257(5074):1248–1251. doi: 10.1126/science.1325670. [DOI] [PubMed] [Google Scholar]
- 9.Noon LA, Franklin JM, King PJ, Goulding NJ, Hunyady L, Clark AJ. Failed export of the adrenocorticotrophin receptor from the endoplasmic reticulum in non-adrenal cells: evidence in support of a requirement for a specific adrenal accessory factor. J Endocrinol. 2002;174(1):17–25. doi: 10.1677/joe.0.1740017. [DOI] [PubMed] [Google Scholar]
- 10.Elias LL, Huebner A, Pullinger GD, Mirtella A, Clark AJ. Functional characterization of naturally occurring mutations of the human adrenocorticotropin receptor: poor correlation of phenotype and genotype. J Clin Endocrinol Metab. 1999;84(8):2766–2770. doi: 10.1210/jcem.84.8.5924. [DOI] [PubMed] [Google Scholar]
- 11.Swords FM, Baig A, Malchoff DM, Malchoff CD, Thorner MO, King PJ, Hunyady L, Clark AJ. Impaired desensitization of a mutant adrenocorticotropin receptor associated with apparent constitutive activity. Mol Endocrinol. 2002;16(12):2746–2753. doi: 10.1210/me.2002-0099. [DOI] [PubMed] [Google Scholar]
- 12.Yang YK, Ollmann MM, Wilson BD, Dickinson C, Yamada T, Barsh GS, Gantz I. Effects of recombinant agouti-signaling protein on melanocortin action. Mol Endocrinol. 1997;11(3):274–280. doi: 10.1210/mend.11.3.9898. [DOI] [PubMed] [Google Scholar]
- 13.Metherell LA, Chapple JP, Cooray S, David A, Becker C, Ruschendorf F, Naville D, Bégeot M, Khoo B, Nurnberg P, Huebner A, Cheetham ME, Clark AJ. Mutations in MRAP, encoding a new interacting partner of the ACTH receptor, cause familial glucocorticoid deficiency type 2. Nat Genet. 2005;37(2):166–170. doi: 10.1038/ng1501. [DOI] [PubMed] [Google Scholar]
- 14.Cooray SN, Almiro Do Vale I, Leung KY, Webb TR, Chapple JP, Egertova M, Cheetham ME, Elphick MR, Clark AJ. The melanocortin 2 receptor accessory protein exists as a homodimer and is essential for the function of the melanocortin 2 receptor in the mouse y1 cell line. Endocrinology. 2008;149(4):1935–1941. doi: 10.1210/en.2007-1463. [DOI] [PubMed] [Google Scholar]
- 15.Blondet A, Doghman M, Rached M, Durand P, Bégeot M, Naville D. Characterization of cell lines stably expressing human normal or mutated EGFP-tagged MC4R. J Biochem. 2004;135(4):541–546. doi: 10.1093/jb/mvh064. [DOI] [PubMed] [Google Scholar]
- 16.Rached M, El Mourabit H, Buronfosse A, Blondet A, Naville D, Bégeot M, Penhoat A. Expression of the human melanocortin-2 receptor in different eukaryotic cells. Peptides. 2005;26(10):1842–1847. doi: 10.1016/j.peptides.2004.11.037. [DOI] [PubMed] [Google Scholar]
- 17.Zhu SZ, Wang SZ, Hu J, el Fakahany EE. An arginine residue conserved in most G protein-coupled receptors is essential for the function of the m1 muscarinic receptor. Mol Pharmacol. 1994;45(3):517–523. [PubMed] [Google Scholar]
- 18.Jones PG, Curtis CA, Hulme EC. The function of a highly-conserved arginine residue in activation of the muscarinic M1 receptor. Eur J Pharmacol. 1995;288(3):251–257. doi: 10.1016/0922-4106(95)90036-5. [DOI] [PubMed] [Google Scholar]
- 19.Naville D, Penhoat A, Barjhoux L, Jaillard C, Fontanay S, Saez J, Durand P, Bégeot M. Characterization of the human ACTH receptor gene and in vitro expression. Endocr Res. 1996;22(4):337–348. doi: 10.1080/07435809609043716. [DOI] [PubMed] [Google Scholar]
- 20.Oliveira L, Paiva AC, Sander C, Vriend G. A common step for signal transduction in G protein-coupled receptors. Trends Pharmacol Sci. 1994;15(6):170–172. doi: 10.1016/0165-6147(94)90137-6. [DOI] [PubMed] [Google Scholar]
- 21.Probst WC, Snyder LA, Schuster DI, Brosius J, Sealfon SC. Sequence alignment of the G-protein coupled receptor superfamily. DNA Cell Biol. 1992;11(1):1–20. doi: 10.1089/dna.1992.11.1. [DOI] [PubMed] [Google Scholar]
- 22.Acharya S, Karnik SS. Modulation of GDP release from transducin by the conserved Glu134-Arg135 sequence in rhodopsin. J Biol Chem. 1996;271(41):25406–25411. doi: 10.1074/jbc.271.41.25406. [DOI] [PubMed] [Google Scholar]
- 23.Rovati GE, Capra V, Neubig RR. The highly conserved DRY motif of class A G protein-coupled receptors: beyond the ground state. Mol Pharmacol. 2007;71(4):959–964. doi: 10.1124/mol.106.029470. [DOI] [PubMed] [Google Scholar]
- 24.Slawik M, Reisch N, Zwermann O, Maser-Gluth C, Stahl M, Klink A, Reincke M, Beuschlein F. Characterization of an adrenocorticotropin (ACTH) receptor promoter polymorphism leading to decreased adrenal responsiveness to ACTH. J Clin Endocrinol Metab. 2004;89(7):3131–3137. doi: 10.1210/jc.2003-032010. [DOI] [PubMed] [Google Scholar]
- 25.Tsiotra PC, Koukourava A, Kaltezioti V, Geffner ME, Naville D, Bégeot M, Raptis SA, Tsigos C. Compound heterozygosity of a frameshift mutation in the coding region and a single base substitution in the promoter of the ACTH receptor gene in a family with isolated glucocorticoid deficiency. J Pediatr Endocrinol Metab. 2006;19(9):1157–1166. doi: 10.1515/jpem.2006.19.9.1157. [DOI] [PubMed] [Google Scholar]
- 26.Collares CV, Antunes-Rodrigues J, Moreira AC, Franca SN, Pereira LA, Soares MM, Elias Junior J, Clark AJ, de Castro M, Elias LL. Heterogeneity in the molecular basis of ACTH resistance syndrome. Eur J Endocrinol. 2008;159(1):61–68. doi: 10.1530/EJE-08-0079. [DOI] [PubMed] [Google Scholar]
- 27.Fluck CE, Martens JW, Conte FA, Miller WL. Clinical, genetic, and functional characterization of adrenocorticotropin receptor mutations using a novel receptor assay. J Clin Endocrinol Metab. 2002;87(9):4318–4323. doi: 10.1210/jc.2002-020501. [DOI] [PubMed] [Google Scholar]
- 28.Mendes HF, van der SJ, Chapple JP, Cheetham ME. Mechanisms of cell death in rhodopsin retinitis pigmentosa: implications for therapy. Trends Mol Med. 2005;11(4):177–185. doi: 10.1016/j.molmed.2005.02.007. [DOI] [PubMed] [Google Scholar]
- 29.Bernier V, Lagace M, Bichet DG, Bouvier M. Pharmacological chaperones: potential treatment for conformational diseases. Trends Endocrinol Metab. 2004;15(5):222–228. doi: 10.1016/j.tem.2004.05.003. [DOI] [PubMed] [Google Scholar]
- 30.Janovick JA, Maya-Nunez G, Conn PM. Rescue of hypogonadotropic hypogonadism-causing and manufactured GnRH receptor mutants by a specific protein-folding template: misrouted proteins as a novel disease etiology and therapeutic target. J Clin Endocrinol Metab. 2002;87(7):3255–3262. doi: 10.1210/jcem.87.7.8582. [DOI] [PubMed] [Google Scholar]
- 31.Sitia R, Braakman I. Quality control in the endoplasmic reticulum protein factory. Nature. 2003;426(6968):891–894. doi: 10.1038/nature02262. [DOI] [PubMed] [Google Scholar]
- 32.Welch WJ, Brown CR. Influence of molecular and chemical chaperones on protein folding. Cell Stress Chaperones. 1996;1(2):109–115. doi: 10.1379/1466-1268(1996)001<0109:iomacc>2.3.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Conn PM, Leanos-Miranda A, Janovick JA. Protein origami: therapeutic rescue of misfolded gene products. Mol Interv. 2002;2(5):308–316. doi: 10.1124/mi.2.5.308. [DOI] [PubMed] [Google Scholar]
- 34.Cohen FE, Kelly JW. Therapeutic approaches to protein-misfolding diseases. Nature. 2003;426(6968):905–909. doi: 10.1038/nature02265. [DOI] [PubMed] [Google Scholar]