

Abstract
The cochaperone CDC37 promotes association of HSP90 with the protein kinase subset of client proteins to maintain their stability and signalling functions. HSP90 inhibitors induce depletion of clients, which include several oncogenic kinases. We hypothesised that the targeting of CDC37 using siRNAs would compromise the maturation of these clients and increase the sensitivity of cancer cells to HSP90 inhibitors. Here we show that silencing of CDC37 in human colon cancer cells diminished association of kinase clients with HSP90 and reduced levels of the clients ERBB2, CRAF, CDK4 and CDK6, as well as phosphorylated AKT. CDC37 silencing promoted the proteasome-mediated degradation of kinase clients, suggesting a degradation pathway independent from HSP90 binding. Decreased cell signalling through kinase clients was also demonstrated by reduced phosphorylation of downstream substrates and colon cancer cell proliferation was subsequently reduced by inhibition of the G1/S-phase transition. Furthermore, combining CDC37 silencing with the HSP90 inhibitor 17-AAG induced more extensive and sustained depletion of kinase clients and potentiated cell cycle arrest and apoptosis. These results support an essential role for CDC37 in concert with HSP90 in maintaining oncogenic protein kinase clients and endorse the therapeutic potential of targeting CDC37 in cancer.
Keywords: CDC37, cochaperone, HSP90, client, 17-AAG
Introduction
The molecular chaperone HSP90 directs the stabilisation and activatable state of an array of signalling proteins, comprising many oncogenic protein kinases, transcription factors and hormone receptors (Maloney and Workman 2002) that regulate the hallmark traits of malignancy (Workman 2004). HSP90 activity is driven by binding and hydrolysis of ATP (Obermann et al. 1998), in a cycle which governs the conformational changes necessary for its chaperoning function (Pearl and Prodromou 2006). Pharmacological HSP90 inhibitors such as geldanamycin and radicicol act as nucleotide mimetics (Roe et al. 1999) and disrupt the stabilisation of multiple client proteins including ERBB2, CDK4 and CRAF, causing their proteasomal degradation. Chemical derivatives of these natural products and other small molecule HSP90 inhibitors have been developed as anticancer drugs (McDonald et al. 2006; Smith and Workman 2007), including 17-AAG, the first to enter into clinical trials (Pacey et al. 2006). Importantly, clinical activity was indicated in melanoma patients with NRAS or BRAF mutations (Banerji et al. 2005; Banerji et al. 2008) and trastuzumab-refractory ERBB2-positive breast cancer patients (Modi et al. 2007).
HSP90 functions within a multimeric chaperone complex (Pearl et al. 2008) assisted by a cohort of cochaperones, including AHA1 (Panaretou et al. 2002), HOP (Chen and Smith 1998), p23 (Fang et al. 1998) and CDC37 (Stepanova et al. 1997). Each seemingly has its own distinct function in regulating HSP90 activity. One of these cochaperones, CDC37, interacts predominantly with protein kinases (MacLean and Picard 2003; Pearl 2005).
CDC37 inhibits the ATPase activity of HSP90 (Siligardi et al. 2002), a property shared by HOP and p23 (Siligardi et al. 2004). By blocking closure of the N-terminal HSP90 ATP binding site, CDC37 is proposed to assist loading of kinase client proteins on to the chaperone machinery (Roe et al. 2004; Siligardi et al. 2002).
Protein kinases are fundamental in regulating signal transduction networks (Manning et al. 2002) and hence require stringent regulation. Frequent mutation and overexpression of protein kinases in cancer is critical for tumorigenesis. Known CDC37-regulated kinases include AKT (Basso et al. 2002), ERBB2 (Ghatak et al. 2005), CDK4 (Stepanova et al. 1996) and CRAF (Grammatikakis et al. 1999). Oncogenic kinase mutants such as BRAFV600E (da Rocha et al. 2005; Grbovic et al. 2006) and EGFRvIII (Lavictoire et al. 2003) also associate with HSP90/CDC37. The nature of the HSP90/CDC37 requirement is client-dependent and can affect activation, formation of mature complexes or binding partner interactions (Caplan et al. 2007).
There is growing interest in CDC37 in the context of malignancy (Pearl 2005; Gray et al 2008). CDC37 overexpression in mice induces tumour formation in the mammary gland, and CDC37 collaborates in transformation by c-myc and cyclin D1 in many tissues (Stepanova et al. 2000a). Targeted overexpression in mouse prostate increases proliferation (Stepanova et al. 2000b) and CDC37 may contribute to prostate cancer development (Schwarze et al. 2003). CDC37 overexpression is seen in pre-malignant and malignant prostate epithelia (Stepanova et al. 2000b). Together, these results support the targeting of CDC37 for cancer therapy.
Since CDC37 regulates multiple oncogenic kinase clients, we hypothesised that siRNA-mediated CDC37 silencing would compromise the stability and activity of these clients in cancer cells. The role of CDC37 was investigated in the human colon carcinoma cell lines HCT116 and HT29, used to study other cochaperones (Holmes et al. 2008; Powers et al. 2008) and HSP90 inhibitors (Clarke et al. 2000; Hostein et al. 2001). During the completion of our studies, Gray et al (2007) demonstrated that shRNA-mediated CDC37 silencing in prostate cancer cells reduced client protein activity and was synergistic with 17-AAG. Similarly, we show that silencing CDC37 in colon carcinoma cells decreases signalling through kinase clients, which inhibits cell proliferation. We also demonstrate for the first time that silencing CDC37 impairs kinase client association with HSP90 and induces proteasome-dependent degradation of these clients. Furthermore, we show that CDC37 silencing sensitises colon cancer cells to HSP90 inhibitors by potentiating kinase client depletion, cell cycle arrest, and induction of apoptosis. Depletion of HSP90 kinase clients and sensitisation to HSP90 inhibition was confirmed in breast and prostate cancer cells. This study further validates the potential for therapeutic targeting of CDC37, either in combination with current HSP90 inhibitors or as a more client-selective alternative to HSP90 blockade.
Results
CDC37 silencing reduces kinase client expression and signalling
siRNAs were designed which decreased CDC37 protein expression for up to 6 days (Fig 1a-d). Control siRNAs, which matched the targeted siRNA sequences except for an inversion of 4 central base pairs, did not affect CDC37 expression. Expression and activity of kinase client proteins were unchanged by CDC37 silencing up to 72h and 96h in HCT116 (Supplementary Fig S1) and HT29 human colon cancer cells (data not shown), respectively. However, CDC37 silencing-induced effects on client proteins were clearly seen at later times. In HCT116 cells a reproducible reduction in ERBB2, CRAF, CDK6, and CDK4 expression was observed on days 4 and 5 (Fig 1a, Supplementary Fig S2). As expected, CDC37 silencing did not affect expression of the glucocorticoid receptor, a non-kinase HSP90 client. Reduced expression of kinase clients ERBB2, CRAF, CDK6 and CDK4 was also induced by CDC37 silencing in HT29 cells at day 5 and 6 (Fig 1b, Supplementary Fig S2). No change in expression of survivin, another non-kinase HSP90 client not known to associate with CDC37, was observed in HT29 cells. CDC37 silencing did not affect expression of HSP90, HSP70 (HSC70, constitutive or HSP72, inducible) or cochaperones HOP, AHA1 or p23 (Supplementary Fig S3), confirming that the siRNAs were specific to CDC37 and did not alter the regulation of other chaperone components.
Figure 1. Silencing of CDC37 depletes kinase client proteins.
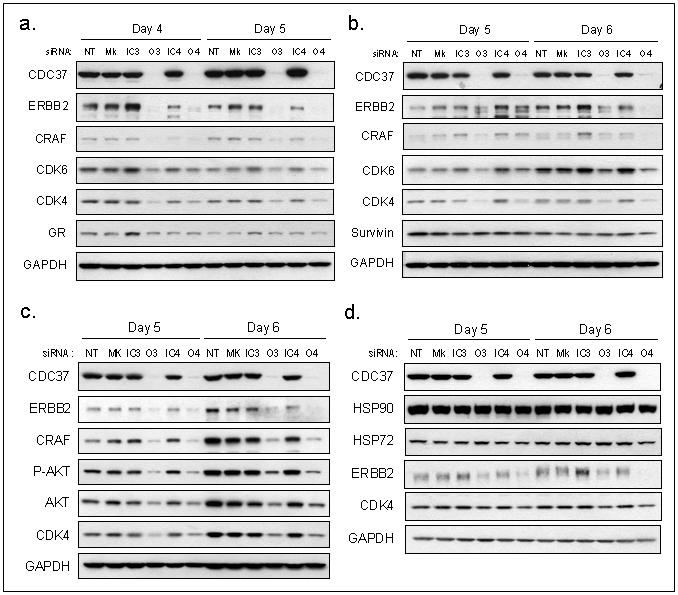
Silencing of CDC37 expression using siRNAs O3 and O4 in comparison with inactive siRNAs IC3 and IC4, no transfection (NT) and mock transfection (Mk) conditions. (a) Expression of client proteins ERBB2, CRAF, CDK6, CDK4 and GR in HCT116 human colon cancer cells at 4 and 5 days. (b) Expression of client proteins ERBB2, CRAF, CDK6, CDK4 and survivin in HT29 human colon cancer cells at 5 and 6 days. (c) Expression of ERBB2, CRAF, phosphorylated-AKT, AKT and CDK4 in SkBR3 human breast cancer cells at 5 and 6 days. (d) Expression of HSP90, HSP72, ERBB2 and CDK4 in PC3 human prostate cancer cells at 5 and 6 days. The changes in protein expression in this figure were quantitated by densitometry, as shown in Supplementary Figure S2.
CDC37 silencing also induced depletion of kinase clients in SkBr3 breast cancer cells (Fig 1c) and PC3 prostate cancer cells (Fig 1d, Supplementary Fig S2). The CDK4 decrease in PC3 cells is in contrast with results of Gray et al (2007). Also contrary to their results, we found that HSP72 and HSP90 expression were unchanged, indicating no involvement of CDC37 in heat shock regulation in any of the cell lines studied here (Fig 1d, Supplementary Fig S3).
A decrease in kinase client activity was suggested by the reduced level of their phosphorylated substrates. CDC37 silencing that depleted kinase clients at 5-6 days in HT29 cells also decreased phosphorylation of AKT (Ser473) as well as total AKT expression (Fig 2a, Supplementary Fig S4a). Interestingly, phosphorylated AKT was reduced more than total AKT levels by silencing CDC37 in HCT116 cells (Fig 2b, Supplementary Fig S4b). Decreases in phosphorylated S6 ribosomal protein (Ser235/236) and GSK3β (Ser9), without any changes in the total protein levels in HCT116 cells indicated diminished AKT signalling; decreased signalling through RAF was also demonstrated by a reduction in phosphorylated MEK1/2 (Ser217/221). Furthermore, ERK1/2 (Thr202/Tyr204) phosphorylation was decreased in HCT116 and HT29 cells while ERK2 expression remained constant. Reduced Rb phosphorylation at Ser780 and Ser795, regulated by CDK4, was also observed in HT29 cells (Fig 2a, Supplementary Fig S4a).
Figure 2. CDC37 silencing reduces kinase client cell signalling.
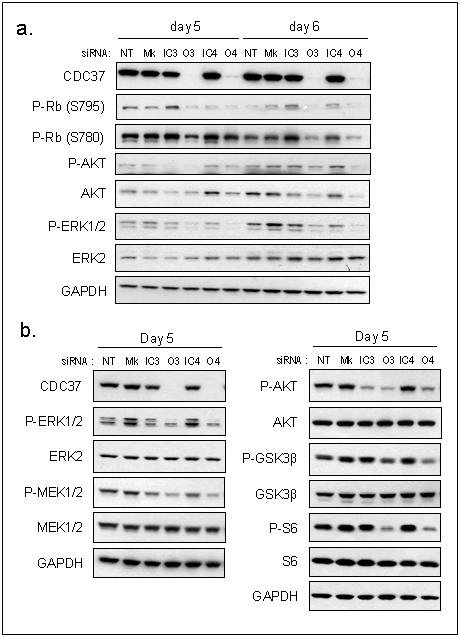
(a) Activation of pathways downstream of kinase clients as determined by phosphorylated AKT, AKT, phosphorylated ERK1/2, ERK2 and Rb in HT29 human colon cancer cells at 5 and 6 days. (b) Activation of signalling pathways as determined by phosphorylated ERK1/2, ERK2, phosphorylated MEK1/2, MEK1/2, phophorylated AKT, AKT, phosphorylated GSK3β, GSK3β, phosphorylated S6, and S6 in HCT116 human colon cancer cells at day 5. The changes in protein expression in this figure were quantitated by densitometry, as shown in Supplementary Figure S4.
Kinase client association with HSP90 requires CDC37
Consistent with the view that CDC37 promotes kinase client association with HSP90, significantly less CDC37 and HSP90 co-immunoprecipitated with CDK4 when CDC37 was silenced for 3 days in HCT116 cells (Fig 3a).
Figure 3. The association of kinase client proteins with HSP90 is inhibited by CDC37 silencing.
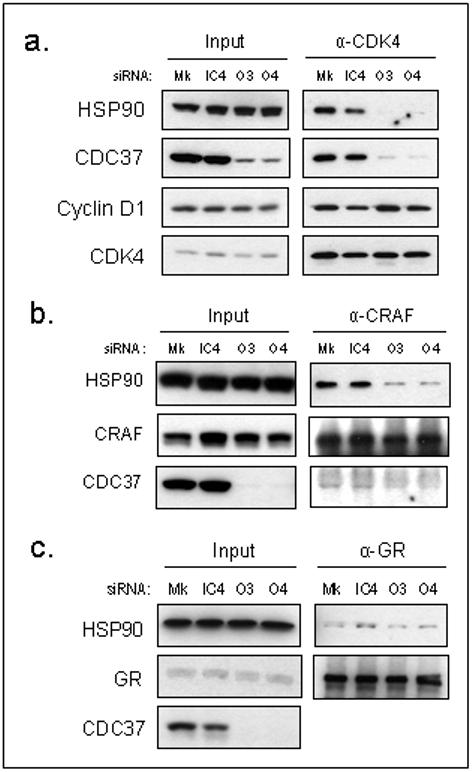
(a) Immunoprecipitation of CDK4 72h following siRNA or mock transfection and expression of HSP90, CDC37 and cyclin D1 in HCT116 human colon cancer cells. Immunoprecipitation of (b) CRAF and (c) GR, 72h following siRNA or mock transfection showing HSP90 and CDC37 co-immunoprecipitation in HCT116 human colon cancer cells.
However, in contrast to biochemical studies showing that CDC37 enhanced assembly of purified CDK4 and cyclin D1 (Lamphere et al. 1997), the association of CDK4 with cyclin D1 was not disrupted by CDC37 silencing in this p16 null cell line (Fig 3a). Furthermore after 5 days, when CDK4 levels were reduced, the association of the remaining CDK4 with cyclin D1 and p21 was increased (Supplementary Fig S5), suggesting that CDK4/cyclin D1 complexes may be stable independently of HSP90/CDC37.
In addition to CDK4, CDC37 silencing also reduced the association of CRAF with HSP90 (Fig 3b). In contrast, interaction of the non-kinase client GR with HSP90 was unaffected (Fig 3c), consistent with the client selectivity of CDC37/HSP90.
CDC37 silencing inhibits cell proliferation
Since many CDC37-regulated kinases are associated with cell cycle progression and survival, we next investigated whether cell proliferation was impeded by CDC37 silencing. Immediate effects on cell growth rate were not apparent (data not shown); however, sustained silencing by repeating the transfection at 4 days resulted in a reduction in proliferation compared with transfection and siRNA controls in HCT116 (Fig 4a) and particularly HT29 cells (Fig 4b). A trend towards reduced proliferation was observed after 6 days, and was significant with both CDC37 siRNAs at day 8 (p<0.05), consistent with the described delay in client depletion.
Figure 4. Silencing of CDC37 reduces cell proliferation.

Proliferation of (a) HCT116 or (b) HT29 human colon cancer cells measured by SRB assay following no transfection, mock or siRNA transfection. Transfections were repeated at day 4 to sustain the silencing of CDC37. Mean from at least 3 independent repeats ± SE, *p<0.05 by paired students t-test in comparison with each control. (c) Representative cell cycle profile in HCT116 cells at 5 days following siRNA or mock transfection. (d) Mean cell cycle distribution of HCT116 cells at 5 days from 3 independent repeats ± SE, *p<0.05, **p<0.01 by non-paired students t-test in comparison with mock and IC4 controls.
Analysis of HCT116 cell cycle distribution after 5 days showed an increase in the proportion of cells in the G1/G0-phase, from 26.5% (mock) and 26.7% (IC4) to 35.3% (O3) and 36.6% (O4) (p<0.05; Fig 4c,d). There was also a reduction in the proportion of cells in the S-phase (p<0.05) from 56.9% (mock) and 53.7% (IC4) to 40.8% (O3) and 40.2% (O4). Therefore, decreased cell proliferation by CDC37 silencing is likely due to impaired G1/S-phase transition.
CDC37 silencing sensitises cells to HSP90 inhibitors, promoting cell cycle arrest and apoptosis
As CDC37 is required for HSP90-mediated chaperoning of many clients, we next investigated whether silencing CDC37 in combination with pharmacological HSP90 inhibition was synergistic. Silencing of CDC37 in HCT116 cells caused a significant 3-fold sensitisation to 17-AAG as determined by GI50 (p<0.05; Fig 5a, Table 1a), consistent with results reported in prostate cancer cells (Gray, Jr. et al. 2007). A similar difference in GI50 was observed using the chemically distinct HSP90 inhibitor VER49009 (Fig 5b, Table 1a). Sensitisation to 17-AAG, albeit to a lesser extent, was also observed in the RV22 prostate and MCF7 breast cancer cell lines (Table 1b), indicating that CDC37 can potentiate the anti-proliferative effects of 17-AAG in at least 3 different tumour cell types.
Figure 5. Silencing of CDC37 sensitises cancer cells to HSP90 inhibitors and potentiates 17-AAG-induced cell cycle arrest and apoptosis.
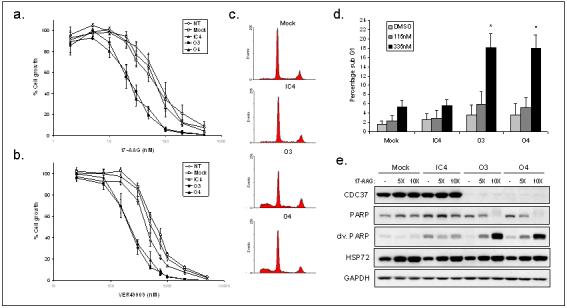
(a) 17-AAG, or (b) VER49009 induced growth inhibition of HCT116 human colon cancer cells by SRB assay from at least 3 independent repeats ± SE. Inhibitors were added 48h after transfection. (c) Representative cell cycle profile from total cells showing sub-G1/G0 events after 24h 335nM 17-AAG treatment following 72h siRNA or mock transfection. (d) Mean percentage sub-G1/G0 events from 3 independent repeats of experiment in (c) ± SE, *p<0.05 by non-paired students t-test in comparison with mock and IC4 controls. (e) 5X or 10X sensitised 17-AAG GI50 added at 72h after siRNA or mock transfection, for 48h. Total cells were analysed by western blotting and cleaved and total PARP measured.
Table 1. Effect of CDC37 silencing on sensitivity to HSP90 inhibitors.
| a. | |||||
|---|---|---|---|---|---|
| Inhibitor | NT | Mock | IC4 | O3 | O4 |
| 17-AAG | 59 ±11 | 67 ±10 | 67 ±13 | *23 ±4 | *23 ±3 |
| VER49009 | 520 ±61 | 660 ±76 | 425 ±14 | *203 ±12 | *193 ±32 |
| b. | |||||
|---|---|---|---|---|---|
| Cell line | NT | Mock | IC4 | O3 | O4 |
| RV22 | 273 ±16 | 289 ±34 | 169 ±41 | 113 ±16 | *83 ±26 |
| MCF7 | 81 ±5 | 50 ±7 | 43 ±7 | 33 ±4 | 25 ±2 |
(a) GI50 values (nM) for 17-AAG and VER49009 were determined in HCT116 human colon cancer cells by SRB assay. (b) GI50 values (nM) were determined for 17-AAG in RV22 prostate cancer cells and MCF7 breast cancer cells by SRB assay. Cells were transfected 48h prior to addition of inhibitors. Mean values are from at least 3 independent repeats ± SE.
p<0.05 by paired students t-test in comparison with each control.
17-AAG treatment at 5xGI50 concentrations for the mock/IC4 controls resulted in a similar G1/S-phase block in both control conditions and also with CDC37 silencing (Fig 5c). Following treatment at the lower concentration of 5xGI50 17-AAG for O3/O4 transfected cells, the cell cycle distribution of control transfected cells was not significantly changed compared with vehicle treated controls (p>0.05; Table 2); however, in CDC37 silenced cells, the same 17-AAG concentration induced a significant G1/S cell cycle block (p<0.01 for G1/G0 and S-phase populations).
Table 2. Cell cycle distribution following 17-AAG treatment and CDC37 silencing.
| DMSO treated | 17-AAG treated | |||||
|---|---|---|---|---|---|---|
| G1/G0 | S | G2 | G1/G0 | S | G2 | |
| Mock | 27.2 ±3.6 | 55.4 ±4.4 | 17.4 ±1.4 | 38.8 ±3.4 | 45.1 ±3.1 | 16.1 ±1.4 |
| IC4 | 32.1 ±3.1 | 51.5 ±3.8 | 16.4 ±0.9 | 38.5 ±3.9 | 49.0 ±3.4 | 14.6 ±2.3 |
| O3 | 33.0 ±2.0 | 49.0 ±1.5 | 18.1 ±1.1 | **63.5 ±5. | **22.2 ±3.6 | 14.3 ±2.5 |
| O4 | 34.9 ±2.1 | 46.2 ±2.2 | 18.9 ±0.1 | **67.2 ±1.2 | **18.5 ±3.7 | 14.3 ±3.8 |
HCT116 human colon cancer cells were treated for 24h with DMSO or 17-AAG (115nM) 72h after siRNA or mock transfection and the cell cycle distribution analysed. Mean values from 3 independent experiments are shown ± SE.
p<0.01 by non-paired students t-test in comparison with DMSO treated.
We hypothesised that apoptosis may also be promoted by combining 17-AAG and CDC37 silencing. 17-AAG treatment caused a small increase in sub-G1/G0 apoptotic cells, and CDC37 silencing for 4 days increased this significantly (p<0.05; Fig 5c,d) to 18.0% (O3 and O4), compared with 5.2% (mock) and 5.5% (IC4) using 335nM 17-AAG. CDC37 silencing alone did not cause significant apoptosis (Fig 5d). Cleaved PARP expression confirmed that apoptosis was potentiated when CDC37 silencing was combined with 17-AAG (Fig 5e).
CDC37 silencing potentiates 17-AAG-induced kinase client depletion
To investigate the mechanism of sensitisation to HSP90 inhibition, client expression was measured in CDC37 silenced cells exposed to 17-AAG. HCT116 cells were treated at 5xGI50 for each condition: 335nM, 333nM or 115nM 17-AAG for mock transfection, IC4 and O4 respectively. Using these equivalent growth inhibitory concentrations, kinase clients were depleted to a similar extent when CDC37 was silenced, despite the lower concentration of 17-AAG used (Fig 6a). Partial recovery of CRAF expression after 17-AAG exposure was evident in mock and IC4 controls from 48-72h, but absent with CDC37 silencing. There was also less recovery of AKT and CDK4 after 48h compared with mock and IC4 controls. Similar results were obtained comparing IC3 and O3 (Supplementary Fig S6), confirming prolonged kinase client depletion with silencing CDC37 in combination with 17-AAG treatment.
Figure 6. Silencing of CDC37 promotes 17-AAG-induced depletion of kinase client proteins.
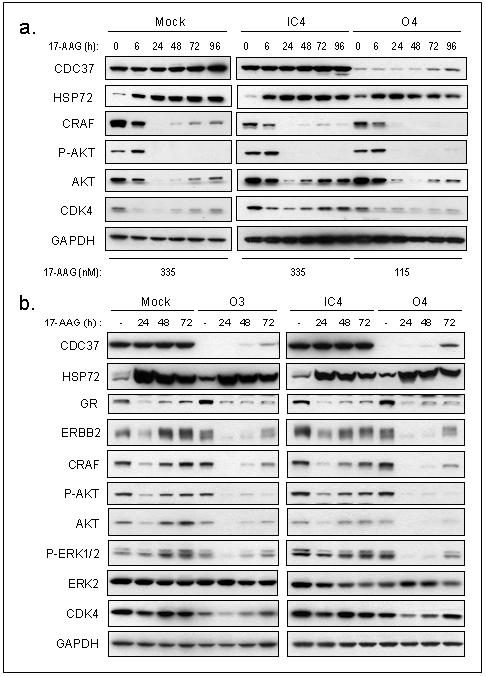
(a) Time course of HSP72, CRAF, phosphorylated AKT, AKT and CDK4 expression following 48h siRNA or mock transfection and 17-AAG treatment at 5XGI50 in HCT116 human colon cancer cells. (d) Time course of HSP72, GR, ERBB2, CRAF, phosphorylated AKT, AKT, phosphorylated ERK1/2, ERK2 and CDK4 expression following siRNA or mock transfection for 72h and 115nM 17-AAG treatment in HCT116 human colon cancer cells.
Since 5xGI50 17-AAG caused extensive depletion of kinase clients in the mock and IC4 conditions, an equimolar dose of 115nM (5xGI50 of O3/O4), equivalent to 1.7x GI50 of mock/IC4 controls, was used to compare the extent of client loss. Using 115nM 17-AAG, minimal depletion of most clients was observed at 24h in the control transfected cells, with almost complete recovery at 48h (Fig 6b). Following CDC37 silencing, however, kinase clients were fully depleted at 24h and remained so for longer than in mock and IC4 controls. Even by 72h 17-AAG treatment, expression of kinase clients had only partially recovered. Thus when CDC37 was silenced, kinases were extensively depleted using lower 17-AAG concentrations, providing an explanation for the observed sensitisation. This effect also appeared specific to CDC37-associated clients, since 17-AAG-induced depletion of GR was unchanged by CDC37 silencing (Fig 6b). HSP72 induction by 17-AAG was also equivalent in all transfection conditions, confirming that CDC37 does not influence the heat shock response.
Proteasome-dependent degradation of kinase clients by CDC37 silencing and 17-AAG
To compare the mechanism by which kinase clients are depleted, we investigated the effects of the proteasome inhibitor MG132 on 17-AAG treated or CDC37 silenced cells. As expected, inhibition of proteasomal degradation by MG132 caused an increase in the accumulation of kinase clients in an insoluble form following 17-AAG exposure (Fig 7a). Importantly, proteasome inhibition in the absence of HSP90 inhibitor also led to a greater accumulation of insoluble CDK4 and CRAF in CDC37 silenced cells compared with the non-silenced control (Fig 7a). The accumulation of insoluble CDK4 in CDC37 silenced cells was time-dependent (Supplementary Fig S7). Proteasomal targeting of CDK4 following CDC37 silencing was further indicated by increased ubiquitination of this client after MG132 treatment, as compared to the control transfected cells (Fig 7b). These results show that both 17-AAG and CDC37 silencing lead to client depletion via the ubiquitin-proteasome pathway.
Figure 7. Kinase client association with HSP90 and proteasomal degradation after silencing of CDC37 and 17-AAG.
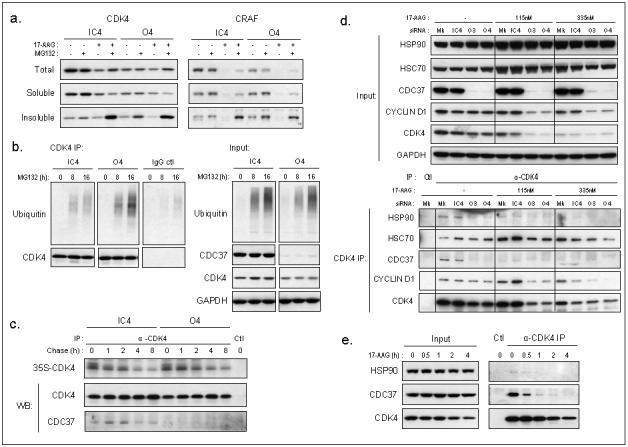
(a) Separation of proteins into total, soluble, or detergent-insoluble fractions after 72h siRNA transfection of human HCT116 colon cancer cells followed by treatment with DMSO, MG132 or 17-AAG or drug combination at 5XGI50 concentrations. (b) Left: Ubiquitin from CDK4 or goat IgG immunoprecipitation of total lysate after siRNA transfection for 72h and 0, 8 or 16h MG132 treatment at 5XGI50, right: input from total proteins. (c) CDK4 immunoprecipitates from pulse chase, labelling for 2h with 35S Met/Cys protein labelling mix after 96h siRNA transfection. Ctl is without antibody. The immunoprecipitates were also analysed by western blotting (WB). Note that the incorporation of the label is similar in CDC37 silenced versus non-silenced cells and that the turnover of newly synthesised CDK4 is comparable with both conditions (t1/2 ∼8h). (d) CDK4 immunoprecipitation following 24h 17-AAG treatment after siRNA or mock transfection for 72h. HSP90, HSC70, CDC37 and cyclin D1 association with CDK4. (e) Immunoprecipitation of CDK4 after 30min, 1h, 2h or 4h 17-AAG treatment at 5XGI50 and association with HSP90 and CDC37, Ctl IP is without antibody.
Analysis of the rate of CDK4 turnover by pulse chase showed that the stabilisation of newly synthesised CDK4 was not affected (t1/2 ∼8h) after 4 days silencing of CDC37 (Fig 7c), suggesting that the degradation of mature but unstable CDK4 was promoted. This differs from the reported shortening of CDK4 half-life, as well as other kinase clients, induced by geldanamycin (Stepanova et al. 1996; Yun and Matts 2005).
To further understand how kinase clients may be directed for proteasomal degradation, we next compared the interactions of client proteins with the HSP90 chaperone complex following 17-AAG exposure, CDC37 silencing, or the combination. Treatment with 17-AAG for 24h markedly decreased the association of CDK4 with HSP90 and CDC37 (Fig 7d). Similarly, as demonstrated earlier (Fig 3a,b), CDC37 silencing also severely restricted association of the client with HSP90. When CDC37 was silenced in combination with HSP90 inhibition, co-immunoprecipitation of CDK4 with CDC37 and HSP90 was reduced further. CDK4/cyclin D1 association remained after 17-AAG treatment and CDC37 silencing (Fig 7d), consistent with earlier results indicating that the stability of this complex is not dependent on HSP90/CDC37 (Fig 3a, Supplementary Fig S5). Notably, 17-AAG promoted the association of CDK4 with HSC70; however, CDC37 silencing alone did not appear to increase CDK4-HSC70 binding (Fig 7d). Similar results were obtained with CDK6 immunoprecipitates, where CDC37 silencing or 17-AAG treatment reduced the interaction with HSP90 and CDC37, and 17-AAG treatment marginally increased HSC70 co-immunoprecipitation with CDK6 (Supplementary Fig S8). The loss of CDK4 association with HSP90 and CDC37 occurred rapidly, as early as 1h after 17-AAG treatment and before CDK4 expression was depleted (Fig 7e). Collectively, these results suggest that CDC37 silencing and 17-AAG both cause the kinase client to dissociate from HSP90, but there appear to be differences in the involvement of HSC70 in directing client degradation (Fig 8).
Figure 8. Model for the fate of kinase clients and the effects of HSP90 inhibition or targeting CDC37.
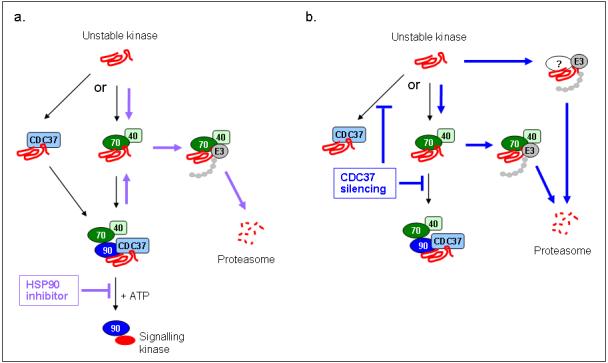
(a) Based on the results in this paper, there may be (at least) two possible routes of recruitment to HSP90: kinases either associate with HSC70/HSP40 and then form a stable complex with HSP90/CDC37, or may associate directly with CDC37 and are then recruited to HSP90. Under normal conditions the kinase is stabilised and/or rendered competent for activation by the ATP-driven HSP90 chaperone cycle, permitting kinase signalling. Treatment with an ATP-competitive HSP90 inhibitor blocks the conformational modification of kinases bound to HSP90, which in an unfolded state may return to an HSC70/HSP40 complex. The association of ubiquitinating enzymes (eg. E3) with HSC70 facilitates polyubiquitination of the unfolded kinase, which is subsequently degraded via the proteasome. (b) By silencing CDC37 expression, the binding of the unstable kinase to HSP90 is reduced. This may leave the unstable kinase prone to factors that directly target it for degradation or it may be ubiquitinated in the HSC70/HSP40 complex which is coupled to the proteasome degradation machinery. Black arrows indicate routes under normal conditions, purple arrows indicate proposed routes during HSP90 inhibition and blue arrows indicate proposed routes during CDC37 silencing. Note that the pulse chase experiment (Fig 7c) indicates that, at least in the case of CDK4, CDC37 depletion appears to result in degradation of the mature but unstable kinase, whereas there is evidence that HSP90 inhibition leads to degradation of newly synthesised client (Stepanova et al. 1996).
Discussion
HSP90 guides the stabilisation, activatable competence and degradation of client proteins, thereby regulating multiple signalling pathways. The dependence of oncogenic cell signalling on this chaperone has been exploited by HSP90 inhibitors, with several now in phase I/II clinical trials (Smith and Workman 2007). As HSP90-mediated stabilisation of signalling-competent proteins requires associating cochaperones (Pearl et al. 2008), understanding how these components orchestrate the modification of client proteins may identify alternative approaches to target the chaperone. Here we have explored the role of the cochaperone CDC37 to determine its potential for therapeutic intervention and investigated the putative beneficial effect of CDC37 silencing combined with HSP90 inhibitors.
Using siRNAs, we have demonstrated the importance of CDC37 in stabilising several kinase clients in colon, breast and prostate cancer cell lines. A consistent reduction in ERBB2, CRAF, CDK4 and CDK6 expression was evident when CDC37 was silenced in human colon cancer cells. There was differential depletion between clients, an effect also seen with pharmacological HSP90 inhibition. Interestingly, one of the highly sensitive clients to HSP90 inhibition, ERBB2, was generally the most extensively depleted by CDC37 silencing. Similarly, the expression of CDK6, which is relatively insensitive to HSP90 inhibition, was less affected by CDC37 silencing. Thus, client proteins vary in their dependency on HSP90/CDC37 to maintain stability, with some clients, such as ERBB2, being particularly prone to degradation upon disruption of either of the stabilising components.
However, client stability appears to be cell context-dependent, since in HT29 cells ERBB2 depletion following CDC37 silencing was less prominent compared with other clients.
Our results showed that HSP90 association with clients CDK4 and CRAF was greatly reduced by CDC37 silencing, consistent with CDC37 promoting kinase client loading onto HSP90 (Siligardi et al. 2002). CDC37 may act either by first binding to the client and then recruiting it to HSP90, or by its presence in the chaperone complex enabling stable interaction with kinase clients. The order of binding is unclear since CDC37-HSP90 complexes free of clients (Zhang et al. 2004; Roe et al. 2004) and CDC37-kinase complexes without HSP90 (Vaughan et al. 2006) have been isolated.
An emerging view of the HSP90 chaperone system, supported by our studies of another cochaperone, AHA1, suggests that client stabilisation and activation may be two distinct functions (Holmes et al. 2008). Taken together with our present results, we hypothesise that the intermediate HSP90 chaperone complex, to which CDC37 promotes kinase loading, predominantly controls stability by preventing degradation, whereas formation of a mature complex by AHA1-promoted ATP hydrolysis modifies the competence of the client for activation. As this view suggests that stabilisation of clients precedes activation, modulation of CDC37 is likely also to impact on signalling activity.
Consistent with this, CDC37 silencing reduced the phosphorylation of substrates downstream of CDK4, CRAF and AKT. It is difficult to definitively ascertain the effect of CDC37 silencing on client activation per se, owing to the clear decrease in client expression. In contrast to our findings, using CDC37 shRNA in prostate cancer cells Gray and colleagues (2007) demonstrated reduced client activity without significant client depletion. Our results show that both extensive and sustained CDC37 silencing is required before client expression is affected since we did not observe kinase client depletion until 4-5 days, despite substantial loss of CDC37 expression by 48h. Under these conditions we were able to demonstrate considerable ERBB2 depletion and some decrease in CDK4, even in PC3 prostate cancer cells for which Gray et al reported no change in client levels. Additionally, in HCT116 cells, we showed reduced AKT activity by a decrease in phosphorylation, without changes in total AKT. This is consistent with HSP90/CDC37 protecting AKT from dephosphorylation (Sato et al. 2000). Additionally, depletion of ERBB2 by CDC37 silencing may also promote phosphatase PP1 activity, which increases AKT dephosphorylation (Xu et al. 2003). Interestingly, we found that silencing CDC37, like 17-AAG, did not restrict association of the remaining CDK4 with its binding partner cyclin D1 or its inhibitor p21; rather, these interactions were augmented after 5 days suggesting that CDK4 bound to cyclin D1/p21 is less prone to degradation when HSP90/CDC37 chaperoning is disrupted.
Pharmacological HSP90 inhibitors induce pleiotropic molecular changes. Thus isolating those fundamental to the antitumour activity is challenging; however, selectively targeting client subsets could reduce redundant or even detrimental effects. We demonstrate here that destabilisation of the CDC37-regulated subset of clients is sufficient to inhibit proliferation of human colon carcinoma cells. Our data suggest that cell proliferation was reduced by restricted S-phase entry, consistent with the observed depletion of CDK4/6 and reduced Rb phosphorylation.
In addition, we found that CDC37 silencing sensitised HCT116 colon cancer cells to HSP90 inhibitors VER49009 and 17-AAG by a factor of 2- to 3-fold respectively, as measured by the SRB assay, and also potentiated both cell cycle arrest and apoptosis by 17-AAG. A similar trend was seen in the RV22 prostate cancer and MCF7 breast cancer cell lines, albeit to a lesser extent. The sensitisation to HSP90 inhibition in HCT116 cells was likely attributable to the more extensive and prolonged depletion of kinase client proteins when CDC37 was silenced before addition of pharmacological inhibitors. Since CDC37 silencing restricted kinase association with HSP90, and there is evidence that HSP90 is involved in the proteasomal degradation of clients through association with ubiquitin ligases (Connell et al 2001; Xu et al, 2002), it may appear contradictory that kinase clients were subsequently degraded more effectively by HSP90 inhibition after CDC37 silencing. In fact, we show here that both CDC37 silencing and 17-AAG cause a marked decrease in association of the kinase clients studied with HSP90/CDC37, and the combined treatment reduces the interaction further. Similarly, geldanamycin disrupts CRAF binding to CDC37 and HSP90 (Grammatikakis et al. 1999), even though it does not displace CDC37 from HSP90 (Siligardi et al. 2002). It should be noted, however, that salt-labile kinase client-bound HSP90 complexes have been isolated upon exposure to geldanamycin (Hartson et al 2000), which may be responsible for proteasome targeting.
We demonstrated that, again like 17-AAG treatment, CDC37 silencing induced the proteasome-dependent degradation of kinase clients. Thus, at least in some cases, unchaperoned protein kinase clients may be inherently susceptible to degradation and HSP90/CDC37 may serve to protect these proteins against targeting to the proteasome. Consistent with this view, C/EBPα-mediated disruption of CDK4/CDC37/HSP90 complexes similarly induced CDK4 ubiquitination and proteasomal degradation (Wang et al. 2002). Studies of Cdc37 in yeast also support a role in protecting kinase clients from proteasomal degradation (Mandal et al. 2007).
Interestingly, 17-AAG caused an increase in client association with HSC70, indicating that clients may be shifting from HSP90 to HSC70 prior to proteasomal degradation. In contrast, we found that CDC37 silencing did not appear to affect kinase association with HSC70. Therefore, binding to HSC70 may not be important in directing kinase clients for degradation following CDC37 silencing. Of note, combinatorial silencing of HSC70 and HSP72 was similarly found to induce the proteasomal degradation of kinase clients (Powers et al. 2008). Blocking both client recruitment to HSP90 and substrate maturation by combinatorial CDC37 silencing and HSP90 inhibition may consequently drive the degradation of kinase clients via pathways that are either independent of, or dependent on, HSC70. This may provide an explanation for the combinatorial client depletion we observed. Based on our results, a speculative model is presented in Fig 8.
An alternative mechanism by which CDC37 silencing might sensitise to HSP90 inhibitors could apply if CDC37 itself was able to protect kinase clients to some extent during pharmacological HSP90 inhibition. Yeast Cdc37 mutants deficient in Hsp90 binding exhibited only limited ability to stabilise clients (Lee et al. 2002). Kinase clients can also be depleted to non-detectable levels by HSP90 inhibitors and there is no conclusive evidence that CDC37 has its own chaperoning activity in mammalian cells. Additionally, only a small fraction of Cdc37 binds Hsp90 in yeast (Turnbull et al. 2005), suggesting that Hsp90-independent functions may be more likely than in mammalian cells where HSP90-bound CDC37 is abundant. We therefore favour the involvement of alternative routes to proteasomal degradation as the mechanism for sensitisation.
We have demonstrated that, in contrast to HSP90 inhibitors, CDC37 silencing did not induce HSP72 in any of the cell lines studied, and similarly did not affect expression of other HSF1-regulated genes such as AHA1 or HSP90. Furthermore, 17-AAG-mediated HSP72 induction was unchanged by CDC37 silencing. Thus, in contrast to Gray et al (2007), we did not find evidence that CDC37 influences the heat shock response by HSF1 activation. HSP70 induction limits the efficacy of HSP90 inhibitors through its anti-apoptotic role (Guo et al. 2005). Thus the targeting of CDC37 could be therapeutically advantageous compared with HSP90 inhibitors by avoiding this. Although we did not observe significant apoptosis by silencing CDC37 alone, we measured considerable apoptosis when 17-AAG and CDC37 silencing were combined. This further supports the potential therapeutic value of combinatorial targeting of CDC37 and HSP90.
Chemically targeting the CDC37-HSP90 interaction is becoming potentially more feasible with the progressive structural characterisation of HSP90, cochaperone and client interactions (Vaughan et al. 2006; Pearl et al. 2008). Recently, celastrol, which demonstrates anticancer activity, was reported to inhibit CDC37 and HSP90 association (Zhang et al. 2008). Celastrol exhibits some similarities to HSP90 inhibitors (Hieronymus et al. 2006), although it is unlikely to act through inhibition of CDC37 alone, since it causes heat shock induction (Westerheide et al. 2004) and proteasome inhibition (Yang et al. 2006). It is nonetheless clear from our studies reported here that CDC37 has considerable potential as a more client-selective alternative for targeting the HSP90 chaperone system, as well as enhancing the anti-proliferative and pro-apoptotic effects of HSP90 inhibitors. The increased activity and predominant existence of HSP90 in cochaperone-bound complexes in tumour cells (Kamal et al. 2003) provides a basis for therapeutic selectivity, as with HSP90 inhibitors (Workman et al. 2007). Additionally, the heightened dependence of overexpressed or mutated kinase client proteins on chaperone stabilisation further suggests the potential for increased susceptibility to CDC37 inhibition in malignant versus normal cells.
Materials and Methods
Cell culture
Human cancer cell lines were obtained from ATCC. All cells were cultured in DMEM (Sigma Aldrich, UK), except RV22, which were cultured in RPMI (Invitrogen, UK), and supplemented with 10% FCS (PAA Laboratories, UK), 2mM L-glutamine and non-essential amino acids. Cells were maintained at 37°C in a humidified incubator with 5% CO2.
siRNA transfection
CDC37 siRNAs were synthesised by Dharmacon with the following target sequences ACACAAGACCUUCGUGGAA (O3) and CGGCAGUUCUUCACUAAGA (O4). Inverted control siRNA sequences with a 4bp inversion in the centre were ACACAAGAUUCCCGUGGAA (IC3) and CGGCAGUUCACUUCUAAGA (IC4). Transfection of 20nM siRNAs in HCT116, RV22, PC3, SkBr3 and MCF7 cells was carried out using Oligofectamine (Invitrogen) according to the manufacturer’s protocol. 100nM siRNAs were transfected in HT29 cells using Dharmafect4 (Dharmacon) according to the manufacturer’s instructions.
Western blotting and immunoprecipitation
These are detailed in supplementary methods.
Cellular growth inhibition assay
Cells were seeded (3-5 ×103 cells/ml) into a 96-well plate 24h before transfection. Sulforhodamine B assay was carried out as described (Holford et al. 1998), adding 17-AAG or VER49009 (Sharp et al. 2007) 48h after transfection.
Cell cycle analysis
Cells were harvested, washed in PBS and fixed overnight at 4°C in 70% ethanol, then prepared as described (Raynaud et al. 2007). Samples were analysed using a BD LSR II flow cytometer. WinMDI and Cylchred software were used for cell cycle phase distribution analysis.
Pulse chase
This is detailed in supplementary methods
Supplementary Material
Acknowledgements
We thank our colleagues in the Signal Transduction and Molecular Pharmacology Team and Chaperone Project Team for helpful discussions. The authors’ work is supported by Cancer Research UK [CUK] Programme grant number C309/A8274. JS is the recipient of a studentship from The Institute of Cancer Research. PW is a Cancer Research UK Life Fellow.
References
- Banerji U, Affolter A, Judson I, Marais R, Workman P. BRAF and NRAS mutations in melanoma: potential relationships to clinical response to HSP90 inhibitors. Mol. Cancer Ther. 2008;7:737–739. doi: 10.1158/1535-7163.MCT-08-0145. [DOI] [PubMed] [Google Scholar]
- Banerji U, et al. Phase I pharmacokinetic and pharmacodynamic study of 17-allylamino, 17-demethoxygeldanamycin in patients with advanced malignancies. J. Clin. Oncol. 2005;23:4152–4161. doi: 10.1200/JCO.2005.00.612. [DOI] [PubMed] [Google Scholar]
- Basso AD, Solit DB, Chiosis G, Giri B, Tsichlis P, Rosen N. Akt forms an intracellular complex with heat shock protein 90 (Hsp90) and Cdc37 and is destabilized by inhibitors of Hsp90 function. J. Biol. Chem. 2002;277:39858–39866. doi: 10.1074/jbc.M206322200. [DOI] [PubMed] [Google Scholar]
- Caplan AJ, Mandal AK, Theodoraki MA. Molecular chaperones and protein kinase quality control. Trends Cell Biol. 2007;17:87–92. doi: 10.1016/j.tcb.2006.12.002. [DOI] [PubMed] [Google Scholar]
- Chen S, Smith DF. Hop as an adaptor in the heat shock protein 70 (Hsp70) and hsp90 chaperone machinery. J. Biol. Chem. 1998;273:35194–35200. doi: 10.1074/jbc.273.52.35194. [DOI] [PubMed] [Google Scholar]
- Clarke PA, Hostein I, Banerji U, Stefano FD, Maloney A, Walton M, et al. Gene expression profiling of human colon cancer cells following inhibition of signal transduction by 17-allylamino-17-demethoxygeldanamycin, an inhibitor of the hsp90 molecular chaperone. Oncogene. 2000;19:4125–4133. doi: 10.1038/sj.onc.1203753. [DOI] [PubMed] [Google Scholar]
- Connell P, Ballinger CA, Jiang J, Wu Y, Thompson LJ, Hohfeld J, Patterson C. The co-chaperone CHIP regulates protein triage decisions mediated by heat-shock proteins. Nat. Cell Biol. 2001;3:93–96. doi: 10.1038/35050618. [DOI] [PubMed] [Google Scholar]
- da Rocha DS, Friedlos F, Light Y, Springer C, Workman P, Marais R. Activated B-RAF is an Hsp90 client protein that is targeted by the anticancer drug 17-allylamino-17-demethoxygeldanamycin. Cancer Res. 2005;65:10686–10691. doi: 10.1158/0008-5472.CAN-05-2632. [DOI] [PubMed] [Google Scholar]
- Fang Y, Fliss AE, Rao J, Caplan AJ. SBA1 encodes a yeast hsp90 cochaperone that is homologous to vertebrate p23 proteins. Mol. Cell Biol. 1998;18:3727–3734. doi: 10.1128/mcb.18.7.3727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghatak S, Misra S, Toole BP. Hyaluronan constitutively regulates ErbB2 phosphorylation and signaling complex formation in carcinoma cells. J. Biol. Chem. 2005;280:8875–8883. doi: 10.1074/jbc.M410882200. [DOI] [PubMed] [Google Scholar]
- Grammatikakis N, Lin JH, Grammatikakis A, Tsichlis PN, Cochran BH. p50(cdc37) acting in concert with Hsp90 is required for Raf-1 function. Mol. Cell Biol. 1999;19:1661–1672. doi: 10.1128/mcb.19.3.1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray PJ, Jr., Stevenson MA, Calderwood SK. Targeting Cdc37 inhibits multiple signaling pathways and induces growth arrest in prostate cancer cells. Cancer Res. 2007;67:11942–11950. doi: 10.1158/0008-5472.CAN-07-3162. [DOI] [PubMed] [Google Scholar]
- Gray PJ, Jr., Prince T, Cheng J, Stevenson MA, Calderwood SK. Targeting the oncogene and kinome chaperone CDC37. Nat. Rev. Cancer. 2008;8:491–495. doi: 10.1038/nrc2420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grbovic OM, Basso AD, Sawai A, Ye Q, Friedlander P, Solit D, et al. V600E B-Raf requires the Hsp90 chaperone for stability and is degraded in response to Hsp90 inhibitors. Proc. Natl. Acad. Sci. U. S. A. 2006;103:57–62. doi: 10.1073/pnas.0609973103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo F, et al. Abrogation of heat shock protein 70 induction as a strategy to increase antileukemia activity of heat shock protein 90 inhibitor 17-allylamino-demethoxy geldanamycin. Cancer Res. 2005;65:10536–10544. doi: 10.1158/0008-5472.CAN-05-1799. [DOI] [PubMed] [Google Scholar]
- Hartson SD, Irwin AD, Shao J, Scroggins BT, Volk L, Huang W, Matts RL. p50(cdc37) is a nonexclusive Hsp90 cohort which participates intimately in Hsp90-mediated folding of immature kinase molecules. Biochemistry. 2000;39:7631–7644. doi: 10.1021/bi000315r. [DOI] [PubMed] [Google Scholar]
- Hieronymus H, et al. Gene expression signature-based chemical genomic prediction identifies a novel class of HSP90 pathway modulators. Cancer Cell. 2006;10:321–330. doi: 10.1016/j.ccr.2006.09.005. [DOI] [PubMed] [Google Scholar]
- Holford J, Sharp SY, Murrer BA, Abrams M, Kelland LR. In vitro circumvention of cisplatin resistance by the novel sterically hindered platinum complex AMD473. Br. J. Cancer. 1998;77:366–373. doi: 10.1038/bjc.1998.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes JL, Sharp SY, Hobbs S, Workman P. Silencing of HSP90 cochaperone AHA1 expression decreases client protein activation and increases cellular sensitivity to the HSP90 inhibitor 17-allylamino-17-demethoxygeldanamycin. Cancer Res. 2008;68:1188–1197. doi: 10.1158/0008-5472.CAN-07-3268. [DOI] [PubMed] [Google Scholar]
- Hostein I, Robertson D, DiStefano F, Workman P, Clarke PA. Inhibition of signal transduction by the Hsp90 inhibitor 17-allylamino-17-demethoxygeldanamycin results in cytostasis and apoptosis. Cancer Res. 2001;61:4003–4009. [PubMed] [Google Scholar]
- Kamal A, Thao L, Sensintaffar J, Zhang L, Boehm MF, Fritz LC, et al. A high-affinity conformation of Hsp90 confers tumour selectivity on Hsp90 inhibitors. Nature. 2003;425:407–410. doi: 10.1038/nature01913. [DOI] [PubMed] [Google Scholar]
- Lamphere L, Fiore F, Xu X, Brizuela L, Keezer S, Sardet C, et al. Interaction between Cdc37 and Cdk4 in human cells. Oncogene. 1997;14:1999–2004. doi: 10.1038/sj.onc.1201036. [DOI] [PubMed] [Google Scholar]
- Lavictoire SJ, Parolin DA, Klimowicz AC, Kelly JF, Lorimer IA. Interaction of Hsp90 with the nascent form of the mutant epidermal growth factor receptor EGFRvIII. J. Biol. Chem. 2003;278:5292–5299. doi: 10.1074/jbc.M209494200. [DOI] [PubMed] [Google Scholar]
- Lee P, Rao J, Fliss A, Yang E, Garrett S, Caplan AJ. The Cdc37 protein kinase-binding domain is sufficient for protein kinase activity and cell viability. J. Cell Biol. 2002;159:1051–1059. doi: 10.1083/jcb.200210121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLean M, Picard D. Cdc37 goes beyond Hsp90 and kinases. Cell Stress.Chaperones. 2003;8:114–119. doi: 10.1379/1466-1268(2003)008<0114:cgbhak>2.0.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maloney A, Workman P. HSP90 as a new therapeutic target for cancer therapy: the story unfolds. Expert. Opin. Biol. Ther. 2002;2:3–24. doi: 10.1517/14712598.2.1.3. [DOI] [PubMed] [Google Scholar]
- Mandal AK, et al. Cdc37 has distinct roles in protein kinase quality control that protect nascent chains from degradation and promote posttranslational maturation. J. Cell Biol. 2007;176:319–328. doi: 10.1083/jcb.200604106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Science. 2002;298:1912–1934. doi: 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- McDonald E, Workman P, Jones K. Inhibitors of the HSP90 molecular chaperone: attacking the master regulator in cancer. Curr. Top. Med. Chem. 2006;6:1091–1107. doi: 10.2174/156802606777812004. [DOI] [PubMed] [Google Scholar]
- Modi S, et al. Combination of trastuzumab and tanespimycin (17-AAG, KOS-953) is safe and active in trastuzumab-refractory HER-2 overexpressing breast cancer: a phase I dose-escalation study. J. Clin. Oncol. 2007;25:5410–5417. doi: 10.1200/JCO.2007.11.7960. [DOI] [PubMed] [Google Scholar]
- Obermann WM, Sondermann H, Russo AA, Pavletich NP, Hartl FU. In vivo function of Hsp90 is dependent on ATP binding and ATP hydrolysis. J. Cell Biol. 1998;143:901–910. doi: 10.1083/jcb.143.4.901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacey S, Banerji U, Judson I, Workman P. Hsp90 inhibitors in the clinic. Handb. Exp. Pharmacol. 2006:331–358. doi: 10.1007/3-540-29717-0_14. [DOI] [PubMed] [Google Scholar]
- Panaretou B, et al. Activation of the ATPase activity of hsp90 by the stress-regulated cochaperone aha1. Mol. Cell. 2002;10:1307–1318. doi: 10.1016/s1097-2765(02)00785-2. [DOI] [PubMed] [Google Scholar]
- Pearl LH. Hsp90 and Cdc37 - a chaperone cancer conspiracy. Curr. Opin. Genet. Dev. 2005;15:55–61. doi: 10.1016/j.gde.2004.12.011. [DOI] [PubMed] [Google Scholar]
- Pearl LH, Prodromou C. Structure and mechanism of the Hsp90 molecular chaperone machinery. Annu. Rev. Biochem. 2006;75:271–294. doi: 10.1146/annurev.biochem.75.103004.142738. [DOI] [PubMed] [Google Scholar]
- Pearl LH, Prodromou C, Workman P. The Hsp90 molecular chaperone: an open and shut case for treatment. Biochem. J. 2008;410:439–453. doi: 10.1042/BJ20071640. [DOI] [PubMed] [Google Scholar]
- Powers MV, Clarke PA, Workman P. Dual targeting of HSC70 and HSP72 inhibits HSP90 function and induces tumor-specific apoptosis. Cancer Cell. 2008 doi: 10.1016/j.ccr.2008.08.002. In press. [DOI] [PubMed] [Google Scholar]
- Raynaud FI, et al. Pharmacologic characterization of a potent inhibitor of class I phosphatidylinositide 3-kinases. Cancer Res. 2007;67:5840–5850. doi: 10.1158/0008-5472.CAN-06-4615. [DOI] [PubMed] [Google Scholar]
- Roe SM, Ali MM, Meyer P, Vaughan CK, Panaretou B, Piper PW, et al. The Mechanism of Hsp90 regulation by the protein kinase-specific cochaperone p50(cdc37) Cell. 2004;116:87–98. doi: 10.1016/s0092-8674(03)01027-4. [DOI] [PubMed] [Google Scholar]
- Roe SM, Prodromou C, O’Brien R, Ladbury JE, Piper PW, Pearl LH. Structural basis for inhibition of the Hsp90 molecular chaperone by the antitumor antibiotics radicicol and geldanamycin. J. Med. Chem. 1999;42:260–266. doi: 10.1021/jm980403y. [DOI] [PubMed] [Google Scholar]
- Sato S, Fujita N, Tsuruo T. Modulation of Akt kinase activity by binding to Hsp90. Proc. Natl. Acad. Sci. U. S. A. 2000;97:10832–10837. doi: 10.1073/pnas.170276797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarze SR, Fu VX, Jarrard DF. Cdc37 enhances proliferation and is necessary for normal human prostate epithelial cell survival. Cancer Res. 2003;63:4614–4619. [PubMed] [Google Scholar]
- Sharp SY, et al. Inhibition of the heat shock protein 90 molecular chaperone in vitro and in vivo by novel, synthetic, potent resorcinylic pyrazole/isoxazole amide analogues. Mol. Cancer Ther. 2007;6:1198–1211. doi: 10.1158/1535-7163.MCT-07-0149. [DOI] [PubMed] [Google Scholar]
- Siligardi G, Hu B, Panaretou B, Piper PW, Pearl LH, Prodromou C. Co-chaperone regulation of conformational switching in the Hsp90 ATPase cycle. J. Biol. Chem. 2004;279:51989–51998. doi: 10.1074/jbc.M410562200. [DOI] [PubMed] [Google Scholar]
- Siligardi G, Panaretou B, Meyer P, Singh S, Woolfson DN, Piper PW, et al. Regulation of Hsp90 ATPase activity by the co-chaperone Cdc37p/p50cdc37. J. Biol. Chem. 2002;277:20151–20159. doi: 10.1074/jbc.M201287200. [DOI] [PubMed] [Google Scholar]
- Smith JR, Workman P. Targeting the cancer chaperone HSP90. Drug Discov. Today: Ther. Strategies. 2007;4:219–227. [Google Scholar]
- Stepanova L, Finegold M, DeMayo F, Schmidt EV, Harper JW. The oncoprotein kinase chaperone CDC37 functions as an oncogene in mice and collaborates with both c-myc and cyclin D1 in transformation of multiple tissues. Mol. Cell Biol. 2000a;20:4462–4473. doi: 10.1128/mcb.20.12.4462-4473.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stepanova L, Leng X, Harper JW. Analysis of mammalian Cdc37, a protein kinase targeting subunit of heat shock protein 90. Methods Enzymol. 1997;283:220–229. doi: 10.1016/s0076-6879(97)83018-2. [DOI] [PubMed] [Google Scholar]
- Stepanova L, Leng X, Parker SB, Harper JW. Mammalian p50Cdc37 is a protein kinase-targeting subunit of Hsp90 that binds and stabilizes Cdk4. Genes Dev. 1996;10:1491–1502. doi: 10.1101/gad.10.12.1491. [DOI] [PubMed] [Google Scholar]
- Stepanova L, Yang G, DeMayo F, Wheeler TM, Finegold M, Thompson TC, et al. Induction of human Cdc37 in prostate cancer correlates with the ability of targeted Cdc37 expression to promote prostatic hyperplasia. Oncogene. 2000b;19:2186–2193. doi: 10.1038/sj.onc.1203561. [DOI] [PubMed] [Google Scholar]
- Turnbull EL, Martin IV, Fantes PA. Cdc37 maintains cellular viability in Schizosaccharomyces pombe independently of interactions with heat-shock protein 90. FEBS J. 2005;272:4129–4140. doi: 10.1111/j.1742-4658.2005.04825.x. [DOI] [PubMed] [Google Scholar]
- Vaughan CK, Gohlke U, Sobott F, Good VM, Ali MM, Prodromou C, et al. Structure of an Hsp90-Cdc37-Cdk4 complex. Mol. Cell. 2006;23:697–707. doi: 10.1016/j.molcel.2006.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Goode T, Iakova P, Albrecht JH, Timchenko NA. C/EBPalpha triggers proteasome-dependent degradation of cdk4 during growth arrest. EMBO J. 2002;21:930–941. doi: 10.1093/emboj/21.5.930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westerheide SD, Bosman JD, Mbadugha BN, Kawahara TL, Matsumoto G, Kim S, et al. Celastrols as inducers of the heat shock response and cytoprotection. J. Biol. Chem. 2004;279:56053–56060. doi: 10.1074/jbc.M409267200. [DOI] [PubMed] [Google Scholar]
- Workman P. Combinatorial attack on multistep oncogenesis by inhibiting the Hsp90 molecular chaperone. Cancer Lett. 2004;206:149–157. doi: 10.1016/j.canlet.2003.08.032. [DOI] [PubMed] [Google Scholar]
- Workman P, Burrows F, Neckers L, Rosen N. Drugging the cancer chaperone HSP90: combinatorial therapeutic exploitation of oncogene addiction and tumor stress. Ann.N.Y.Acad.Sci. 2007;1113:202–216. doi: 10.1196/annals.1391.012. [DOI] [PubMed] [Google Scholar]
- Xu W, Marcu M, Yuan X, Mimnaugh E, Patterson C, Neckers L. Chaperone-dependent E3 ubiquitin ligase CHIP mediates a degradative pathway for c-ErbB2/Neu. Proc.Natl.Acad.Sci.U.S.A. 2002;99:12847–12852. doi: 10.1073/pnas.202365899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Yuan X, Jung YJ, Yang Y, Basso A, Rosen N, et al. The heat shock protein 90 inhibitor geldanamycin and the ErbB inhibitor ZD1839 promote rapid PP1 phosphatase-dependent inactivation of AKT in ErbB2 overexpressing breast cancer cells. Cancer Res. 2003;63:7777–7784. [PubMed] [Google Scholar]
- Yang H, Chen D, Cui QC, Yuan X, Dou QP. Celastrol, a triterpene extracted from the Chinese “Thunder of God Vine,” is a potent proteasome inhibitor and suppresses human prostate cancer growth in nude mice. Cancer Res. 2006;66:4758–4765. doi: 10.1158/0008-5472.CAN-05-4529. [DOI] [PubMed] [Google Scholar]
- Yun BG, Matts RL. Differential effects of Hsp90 inhibition on protein kinases regulating signal transduction pathways required for myoblast differentiation. Exp. Cell Res. 2005;307:212–223. doi: 10.1016/j.yexcr.2005.03.003. [DOI] [PubMed] [Google Scholar]
- Zhang T, Hamza A, Cao X, Wang B, Yu S, Zhan CG, et al. A novel Hsp90 inhibitor to disrupt Hsp90/Cdc37 complex against pancreatic cancer cells. Mol. Cancer Ther. 2008;7:162–170. doi: 10.1158/1535-7163.MCT-07-0484. [DOI] [PubMed] [Google Scholar]
- Zhang W, Hirshberg M, McLaughlin SH, Lazar GA, Grossmann JG, Nielsen PR, et al. Biochemical and structural studies of the interaction of Cdc37 with Hsp90. J. Mol. Biol. 2004;340:891–907. doi: 10.1016/j.jmb.2004.05.007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.