

Summary
Huntington's disease (HD) is caused by a polyglutamine expansion mutation in huntingtin that makes the protein toxic and aggregate-prone. The subcellular localisation of huntingtin and many of its interactors suggest a role in endocytosis, and recently it has been shown that huntingtin interacts indirectly with the early endosomal protein, Rab5, via HAP40. Here we show that Rab5 inhibition enhanced polyglutamine toxicity, while Rab5 overexpression attenuated toxicity in our HD cell and fly models. We have here tried to identify a mechanism for the Rab5 effects on our HD models. Our data suggest that Rab5 acts at an early stage of autophagosome formation in a macromolecular complex containing Beclin-1 and Vps34. Interestingly chemical or genetic inhibition of endocytosis also impeded macroautophagy and enhanced mutant huntingtin aggregation/toxicity. However, in contrast to Rab5, inhibition of endocytosis by various means suppressed autophagosome-lysosome fusion (final step in macroautophagy pathway) similar to BafilomycinA1. Thus, Rab5, previously thought to be exclusively involved in endocytosis, has a novel role in macroautophagy. We have previously shown that macroautophagy is an important clearance route for several aggregate-prone proteins including mutant huntingtin. Thus, better understanding of Rab5-regulated autophagy may lead to rational therapeutic targets for HD and other protein-conformation diseases.
Keywords: Huntington's disease, Autophagy, Rab5
Introduction
Huntington's disease (HD) is a late onset, inherited neurodegenerative condition caused by an expansion of CAG trinucleotide repeats (>35 repeats) in exon-1 of the IT15 gene. The mutation results in the production of an abnormally long polyglutamine (polyQ) tract in the N-terminus of the huntingtin protein (Rubinsztein, 2002). Full-length mutant huntingtin undergoes several proteolytic cleavages giving rise to N-terminal fragments comprising the first 100-150 residues containing the expanded polyQ tract. These N-terminal fragments are also the toxic species found in aggregates in HD mouse models and human brains (Lunkes et al., 2002). Thus, HD pathogenesis is frequently modelled with exon-1 fragments containing expanded polyQ repeats, which cause aggregate formation and toxicity in cell models and in vivo (Rubinsztein, 2002).
A key pathway regulating the degradation of aggregate-prone cytosolic proteins like mutant huntingtin (both full-length and exon-1 forms) is macroautophagy (henceforth referred to simply as autophagy) (Ravikumar et al., 2002; Ravikumar et al., 2004; Shibata et al., 2006). Wild-type huntingtin (full-length or exon-1) has a very low dependence on autophagy for its clearance. Autophagy, a process conserved from yeast to man, typically clears long-lived proteins and organelles by engulfing cytoplasmic contents in double-membrane structures called autophagosomes, which later fuse with lysosomes, where their contents are degraded (Klionsky and Ohsumi, 1999). It is believed that isolation membranes (phagophores) which later give rise to autophagosomes are formed at the phagophore assembly site (also referred to as pre-autophagosomal structures) (PAS) in yeast (Suzuki et al., 2001). PAS however have not been defined in mammalian autophagy. Several ATG (autophagy) genes that regulate yeast autophagy have been identified, and many of these genes have mammalian orthologues. A protective role of autophagy has been implicated in some neurodegenerative disorders, cancers and infectious diseases (Rubinsztein et al., 2007). Conditional knockouts of the key autophagy genes ATG5 or ATG7 in the brains of mice result in a neurodegenerative phenotype caused by aberrant accumulation of ubiquitinated proteins (Hara et al., 2006; Komatsu et al., 2006). In addition to HD, where enhanced clearance of mutant huntingtin fragments by autophagy induction attenuates toxicity in cell, fly and mouse models (Ravikumar et al., 2002; Ravikumar et al., 2004), this pathway appears to be equally important for the clearance of several other intracellular proteins that cause neurodegeneration, including the A53T α-synuclein mutation that causes autosomal dominant Parkinson's disease (Berger et al., 2006; Ravikumar et al., 2002; Webb et al., 2003).
The subcellular localisation of huntingtin, the nature of many of its normal interactors and its indirect interaction with Rab5 via HAP40 (Pal et al., 2006), suggested that it may have roles in endocytosis. Rab5, a member of a family of small GTPases, is a key regulator of the early endocytic pathway in mammalian cells (Bucci et al., 1992; Stenmark et al., 1994). Accordingly we initially tested the role of Rab5 in the regulation of mutant huntingtin toxicity. We show that Rab5 inhibition enhances polyglutamine toxicity, while Rab5 overexpression attenuates toxicity in HD cell and fly models. Our data suggest that Rab5, in addition to its role in endocytosis, modifies mutant huntingtin aggregation/toxicity by having a novel role in the early stage of autophagosome formation. Interestingly, inhibition of endocytosis by a variety of means also influenced mutant huntingtin toxicity by inhibiting autophagy at a downstream step (autophagosome-lysosome fusion) distinct from Rab5 inhibition. A possible therapeutic approach for HD and other proteinopathies would thus be to enhance Rab5 activity.
Results and Discussion
Rab5 modifies polyglutamine toxicity and aggregation
We initially tested whether Rab5 could modify the toxicity of a mutant huntingtin exon-1 fragment with 74 polyglutamine repeats (Q74) (Narain et al., 1999) in COS-7 cells. Dominant-negative inhibition of Rab5 by overexpression of a GTP-binding defective S34N mutant Rab5 (Stenmark et al., 1994) (DN-Rab5) increased the toxicity of Q74 (Fig. 1a), while overexpression of wild-type (WT) Rab5 or constitutively active (CA) Q79L Rab5 (Stenmark et al., 1994) significantly decreased Q74-induced toxicity (Fig. 1a).
Figure 1.

Rab5 modulates mutant huntingtin toxicity/aggregation. a. Quantification of proportions of GFP-expressing cells with cell death in COS-7 cells transiently transfected with dominant-negative (DN), constitutive active (CA), wild-type (WT) Rab5 or empty vector control and EGFP-tagged huntingtin exon-1 with 74 polyglutamine repeats (Q74) (3:1 ratio) for 48h. *** - P < 0.0001, ** - P < 0.001, * - P < 0.05. b. Quantification of proportions of GFP-expressing cells with aggregates in COS-7 cells that were quantified for toxicity shown above. *** - P < 0.0001. c. Quantification of proportions of GFP-expressing cells with aggregates in HeLa cells transiently transfected with siRNA for Rab5a, Rab5b, Rab5c or all three siRNA simultaneously (Rab5abc) for 72h and further with EGFP-tagged huntingtin exon-1 with 74 polyglutamine repeats for the last 24h of the 72h siRNA transfection. *** - P < 0.0001, * - P < 0.05. d. Rab5 overexpression increases the numbers of rhabdomeres in ommatidia of mutant huntingtin-expressing flies. Frequency distribution of ommatidia with different numbers of rhabdomeres, 3 days after eclosion (hatching), in progeny of flies expressing mutant huntingtin exon-1 (gmrQ120) crossed to either a control stock (w1118) (white minus) (have huntingtin transgene only) or to Rab5-EGFP flies (have huntingtin and Rab5 transgenes). P ≤ 0.001 t-test; P ≤ 0.001 also if tested by Mann-Whitney U test. The rhabdomere frequency of Rab5 flies crossed to a control stock is also shown. e. Proportion of GFP-expressing cells with Q74 aggregates in wild-type (Atg5+/+) vs Atg5 knockout (Atg5−/−) MEFs. *** - P < 0.0001. f. Quantification of proportions of Q74-expressing Atg5−/− (Atg5 knockout) or Atg5+/+ (wild-type) MEF cells with aggregates, after transient transfection with dominant-negative Rab5 (DN-Rab5), constitutive active Rab5 (CA-Rab5) or empty vector control and EGFP-tagged huntingtin exon-1 with 74 polyglutamine repeats (Q74) (3:1 ratio) for 48h. *** - P < 0.0001. Odds ratios are used to provide comparable pooled summary statistics across multiple independent experiments (see methods). Control conditions are fixed at 1 in both cell lines to facilitate comparisons. g. Quantification of proportions of GFP-expressing cells with aggregates in HeLa cells transiently transfected with dominant-negative Rab5 (DN-Rab5) or empty vector (Cont) and huntingtin exon-1 with 74 polyglutamine repeats (Q74) (3:1 ratio) for 48h and either left untreated (-Rap) or treated with 0.2μg/ml rapamycin (Rap) to induce autophagy. ** - P < 0.001. Error bars in all the above graphs represent S.E.M.
We then examined the effect of Rab5 on mutant huntingtin aggregation. Mutant huntingtin aggregation correlates with its expression levels (Narain et al., 1999). Also, the proportion of cells with inclusions formed by mutant huntingtin fragments generally correlates with its toxicity in cell culture models (although inclusions may not themselves be as toxic as diffusely distributed mutant huntingtin) (Arrasate et al., 2004; Ravikumar et al., 2002). DN-Rab5 increased Q74 aggregation, while CA-Rab5 and WT-Rab5 decreased the proportions of Q74-expressing cells with inclusions (Fig. 1b), mirroring the toxicity data (Fig. 1a). We next confirmed that the effects of DN-Rab5 were mirrored by depletion of endogenous Rab5 using RNAi. There are three isoforms of Rab5 namely Rab5A, 5B and 5C. siRNA against individual Rab5 isoforms significantly increased Q74 aggregation while simultaneous knockdown of all three isoforms increased Q74 aggregation even further (Fig. 1c; Supp. Fig. 1). Thus, Rab5 modulation affects aggregation and toxicity of the mutant huntingtin fragments.
We next tested whether Rab5 could modify huntingtin toxicity in vivo using a Drosophila HD model. Fly photoreceptors that express a mutant huntingtin fragment with 120 polyglutamine repeats exhibit degeneration that is not observed in flies that express the wild-type fragment with 23 polyglutamine repeats (Jackson et al., 1998). The Drosophila compound eye consists of many ommatidia, each comprising of eight photoreceptor neurons with light-gathering parts called rhabdomeres, seven of which can be visualised by light microscopy using the pseudopupil technique (Franceschini and Kirschfeld, 1971). Neurodegeneration in the HD flies is progressive and is associated with a decrease in the number of visible rhabdomeres in each ommatidium with time (Jackson et al., 1998). We crossed the HD flies with flies transgenic for a wild-type Rab5-EGFP fusion and looked at the number of visible rhabdomeres in the double transgenic flies. Degeneration of photoreceptors due to Q120 overexpression was greatly rescued by overexpression of Rab5-EGFP (Fig. 1d).
Since our previous studies suggest that mutant huntingtin aggregation can be regulated by autophagy (Ravikumar et al., 2002), we next tested whether the effects of Rab5 on Q74 aggregation were autophagy-dependent using either wild-type (Atg5+/+) or autophagy-deficient Atg5-knockout (Atg5−/−) mouse embryonic fibroblasts (MEFs) (Mizushima et al., 2001). As previously observed with autophagy inhibitors (Ravikumar et al., 2002), Q74 aggregation was higher in autophagy incompetent Atg5−/− compared to Atg5+/+ MEFs (Fig. 1e). (These MEFs show minimal Q74-induced toxicity with the transfection conditions used in these experiments.) As expected, DN-Rab5 caused an increase and CA-Rab5 resulted in a decrease in Q74 aggregation in wild-type MEFs (Fig. 1f, left), similar to the results seen in COS-7 cells (Fig. 1b). However, overexpression of DN-Rab5 or CA-Rab5 had no effects on Q74 aggregation in autophagy-incompetent Atg5−/− MEFs (Mizushima et al., 2001) (Fig. 1f, right). Furthermore, Rab5 inhibition abrogated the ability of the autophagy enhancers, rapamycin or L-690,330 (an inositol monophosphatase inhibitor) (which target mTOR-dependent and -independent pathways, respectively), to clear Q74 aggregates (Fig. 1g; Supp. Fig. 2) (Ravikumar et al., 2002; Sarkar et al., 2005). This suggests that the effect of Rab5 on Q74 aggregation and toxicity were due to alterations in the autophagic pathway rather than its role in endocytosis.
Rab5 regulates autophagosome formation in mammalian cells
Since the above data suggest a possible role of Rab5 on autophagy, we tested whether Rab5 influenced the formation of autophagosomes. Two ubiquitin-like modifications are involved in the elongation and completion of autophagosome formation. The first involves conjugation of Atg12 to Atg5 in a reaction requiring Atg7 (E1-like) and Atg10 (E2-like). Atg5-Atg12 conjugates are localised onto the PAS and dissociate upon completion of autophagosome formation. The second modification involves conjugation of microtubule-associated protein 1 light chain 3 (MAP-LC3/Atg8/LC3) to phosphatidylethanolamine (PE). LC3 (cytosolic) is cleaved at its C-terminus by Atg4 to form LC3-I. LC3-I is covalently conjugated to PE to form LC3-II, a process requiring the activities of Atg7 and Atg3. LC3-II (membrane associated) is specifically targeted to Atg5-Atg12-associated, expanded phagophores and remains associated with autophagosomes even after fusion with lysosomes, after which LC3-II can be delipidated and recycled. LC3 is the only known protein that specifically associates with autophagosomes and not with other vesicular structures (Kabeya et al., 2000). Thus, LC3-II levels correlate with autophagic vacuole number, which can also be assessed by scoring LC3-positive vesicle numbers (Kabeya et al., 2000). We first tested the effect of Rab5 on LC3. Inhibition of Rab5 decreased the proportion of COS-7 cells with approximately >20 LC3-labelled autophagic vesicles (Fig. 2a). On the other hand overexpression of CA-Rab5 or WT-Rab5 significantly increased the proportion of cells with >20 LC3-positive autophagic vesicles (Fig. 2a). We however did not observe any change in the size or morphology of the LC3-vesicles under any of the above conditions (data not shown). The above results also correlated with decrease (upon DN-Rab5 overexpression) or increase (upon CA-rab5 or WT-Rab5 overexpression respectively) with autophagosome-associated LC3-II levels on western blots when we blocked LC3-II clearance by inhibiting autophagosome-lysosome fusion with bafilomycin A1 (BafA1) (Fig. 2b). As LC3-II accumulation can occur not only due to increased autophagosome formation, but also due to impaired autophagosome-lysosome fusion, we have assayed LC3-II in the presence of BafA1 thus enabling one to assess autophagosome formation. BafA1 increases LC3-II and the concentration used is saturating for LC3-II levels in this assay (Sarkar et al., 2006). Fig. 2b suggests that Rab5 increased autophagosome synthesis, rather than decreasing autophagosome-lysosome fusion/autophagosome degradation. Further blockage of autophagosome-lysosome fusion via a BafA1-independent mechanism, using the dynein inhibitor erythro-9-[3-(2-hydroxynonyl)] adenine (EHNA), along with this dose of BafA1, results in no increase in LC3-II compared to BafA1 alone (Sarkar et al., 2006). Thus, Rab5 appears to regulate early stages of the autophagic pathway probably at the initiation of autophagic vacuole formation.
Figure 2.
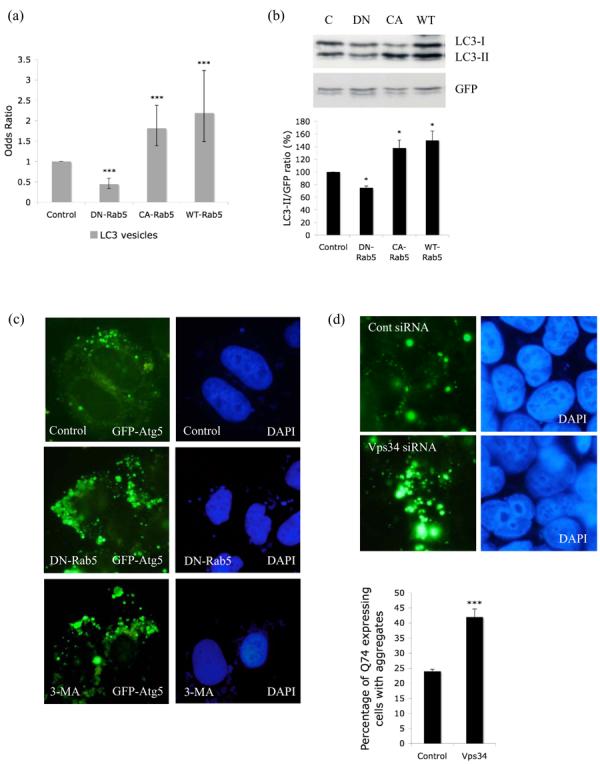
a. COS-7 cells were transiently transfected with empty vector (Control), DN-, CA- or WT-Rab5 and GFP-LC3/mRFP-LC3 (3:1 ratio) for 24h. GFP-/mRFP-positive cells with increased LC3-positive vesicles (> 20 vesicles per cell) were quantified. The proportion of control cells with >20 vesicles per cell is 29%. *** - P < 0.0001. b. Western blot analysis of COS-7 cells transfected with empty vector (C), DN-, CA- or WT-Rab5 and myc-LC3 for 24h in the presence of 200nM Bafilomycin A1 (treated for last 15h), with anti-myc antibody. GFP was used as a transfection control. Quantitation of the band intensities from 3 independent experiments is shown * - P < 0.05. c. Analysis of GFP-Atg5 structures (green) in HeLa cells transfected with control vector (either untreated or treated for 24h with 10mM 3-methyladenine (3-MA)) or dominant-negative Rab5 (DN-Rab5) and GFP-Atg5, after saponin extraction. Nuclei are shown in blue. d. HeLa cells were transfected with siRNA against Vps34 or a control siRNA for 48h after which GFP-Atg5 together with siRNA was transfected for further 24h. The cells were fixed following saponin extraction to visualise GFP-Atg5 (green) structures. Quantification of proportions of GFP-expressing cells with Q74 aggregates in HeLa cells transiently transfected with siRNA for control or Vps34 for 48h and further with HA-tagged huntingtin exon-1 with 74 polyQ repeats for the next 24h. *** - P < 0.0001. e. Co-localisation of GFP-Atg5 structures (green) with myc-tagged FYVE (red) in Hela cells transfected with DN-Rab5 together with GFP-Atg5 and myc-FYVE for 24h. f. Co-localisation of GFP-Atg5 structures (green) with Beclin-1 (red) in Hela cells transfected with DN-Rab5 together with GFP-Atg5 and Flag-tagged wild-type (WT) Beclin-1. g. Co-localisation of endogenous Atg5 (red) and endogenous Rab5 (green) in HeLa cells treated with 3MA for 15h. In panels e-g we observed more than 30% co-localisation between GFP-Atg5 structures and saponin extracted, membrane associated, FYVE, Beclin-1 or Rab5 in cells expressing both the relevant proteins. h. Co-localisation of GFP-Atg5 structures (green) with Atg12 (red) in HeLa cells transfected with DN-Rab5 together with GFP-Atg5 and HA-tagged Atg12 for 24h. Nuclei labelled with DAPI are in blue.
Rab5 and its effector Vps34 regulate early steps of autophagosome formation
Autophagosome formation begins with a nucleation step, where membranes of unknown origin forms the phagophores, which then expands and fuses to form completed autophagosomes. The formation of autophagosome precursors is regulated by a macromolecular complex containing the class-III PI-3-kinase, Vps34, Beclin-1 (the ortholog of yeast Atg6p), Atg14 and Vps15 (Kihara et al., 2001b). Vps34, which generates phosphatidyl-inositol-3-phosphate (PI-3-P), directly interacts with Beclin-1 (Kihara et al., 2001a). 3-methyl adenine (3MA) (Kovacs et al., 1998) or wortmannin (Blommaart et al., 1997) inhibit PI-3-kinases, including Vps34, and block autophagy by preventing the formation of autophagosomes. Conjugation of Atg12 with Atg5 initiates the elongation process. This conjugation however is not required for membrane targeting of Atg5, but is necessary for membrane elongation. This is followed by conjugation of PE to LC3-I to form LC3-II. Atg5-Atg12 conjugates co-localise with LC3-II on the PAS. Mutation of K130R in Atg5 inhibits its conjugation with Atg12; as a result, membranes with unconjugated Atg5K130R that do not co-localise with LC3-II accumulate upon autophagy induction (Mizushima et al., 2001). Atg5-Atg12 conjugate only localises to phagophores and dissociates just before or after completion of autophagic vacuole formation (Mizushima et al., 2001). So Atg5 and Atg12 are not associated with completed autophagosomes.
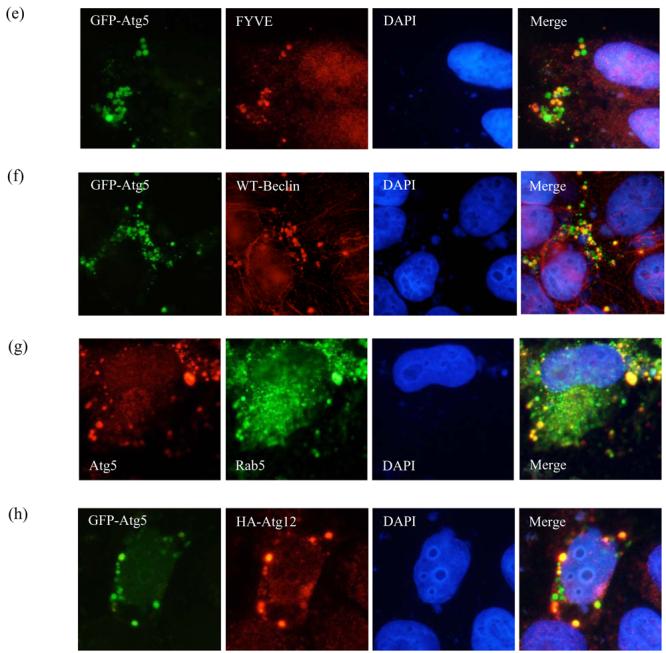
In order to investigate how Rab5 influenced early steps in autophagosome formation, we investigated the distribution of GFP-Atg5, a marker for autophagosomal precursor structures (George et al., 2000; Mizushima et al., 2001). The cells were permeabilised with saponin to remove soluble cytosolic contents and reveal membrane-associated GFP-Atg5. After Rab5 inhibition, we noted an increased abundance of large punctate Atg5 structures (Fig. 2c). These contrast with the smaller and less abundant Atg5 puncta seen in the great majority of untreated cells (Fig. 2c). We found similar results when we performed immunostaining for endogenous Atg5 (Supp. Fig. 3). In control cells however, these structures were rare (Fig. 2c; Supp. Fig. 3). While saponin extraction precludes accurate quantitation (as it does not allow visualisation of transfected versus untransfected cells), we saw these Atg-5 structures in 3% of all control cells and about 12% of Rab5-inhibited cells (approximately 4-fold increase, p<0.001 from triplicate slides).
Previous studies have shown that Rab5 interacts with and activates the PI3-kinase Vps34 (Christoforidis et al., 1999). Thus, we tested whether Vps34 inhibition had similar effects to Rab5 inhibition. As with Rab5 inhibition, inhibition of Vps34 with the PI-3-kinase inhibitors, 3MA (Fig. 2c; Supp. Fig. 3) or wortmannin (data not shown) (which are both established blockers of autophagosome formation) or siRNA knockdown of Vps34 (Fig. 2d; Supp. Fig. 4) resulted in increases of Atg5 structures. Similar to Rab5 inhibition, inhibition of Vps34 also increased the proportion of cells with Q74 aggregates, consistent with autophagy inhibition (Fig. 2d) (Ravikumar et al., 2002). To further characterise the Atg5 structures we tested whether they co-localised with other markers or proteins associated with phagophores. A subset of the punctate Atg5 structures co-localised with a PI-3-P marker, myc-FYVE (Fig. 2e) (Gaullier et al., 1998) and they also overlapped with Beclin-1 (Fig. 2f) but did not co-localise with LC3 (Supp. Fig. 5) suggesting that these were indeed early autophagic structures. We also did not observe any co-localisation of the Atg5 structures with the golgi markers p230 (Supp. Fig. 6) (Derby et al., 2007) or golgin-84 (Satoh et al., 2003) and the Atg5 in these structures was not associated with the ER proteins BiP (Haas, 1994) or Grp94 (Argon and Simen, 1999) (data not shown). However, Rab5 associated with these Atg5-positive puncta compatible with a role for Rab5 in early mammalian autophagy (Fig. 2g). We only observed very rare co-localisation of Atg5 and Atg12 in such puncta (Fig. 2h). It should be noted that these experiments were performed under constitutive autophagy conditions (full medium) in cells with Rab5 inhibition. Rab5 inhibition or 3MA treatment was required in order to allow visualisation of sufficient numbers of these structures for characterisation. The accumulation of Atg5 structures by Vps34 or Rab5 inhibition might reflect a block in the progression of Atg5-positive membranes to the formation of autophagic vacuoles suggesting a possible role of Rab5 in very early steps of autophagy. The observation that the Atg5 in these puncta was only rarely associated with Atg12 led to the hypothesis that defective Atg5-Atg12 conjugation may be causally related to puncta formation.
Both Rab5 and Vps34 regulate Atg5-Atg12 conjugation
The possibility that defective Atg5-Atg12 conjugation may lead to defective autophagosome formation associated with an accumulation of phagophores enriched in conjugated Atg5 was compatible with the similarity of the above results and the phenotypes seen in apg7Δ or apg12Δ yeast strains, which typically show one to five large Atg5-positive punctate structures per cell, (a phenomenon that is very rare in wild-type cells) (George et al., 2000). Thus when we knocked down Atg7 (E1-like enzyme crucial for Atg5-Atg12 conjugation) (Supp. Fig. 7) in mammalian cells, we observed increased GFP-Atg5 structures, consistent with what had been noted in yeast (Fig. 3a). These Atg5-positive structures also co-localised with Beclin-1 (Supp. Fig. 8). Accordingly, we tested whether the increase in Atg5 structures seen with Rab5/Vps34 inhibition was associated with aberrant Atg5-Atg12 conjugation. Consistent with this hypothesis and with the only very rare co-localisation of Atg12 with Atg5 in the puncta that accumulated with Rab5 inhibition (Fig. 2h), we found that the ratio of Atg5-Atg12 complex formed compared to the free Atg12 pool was significantly lower in cells expressing DN-Rab5 (Fig. 3b). 3MA (a PI-3-kinase inhibitor that inhibits Vps34) treatment and siRNA knockdown of Vps34 also had similar effects on Atg5-Atg12 conjugation to those seen with loss-of-function of Rab5, a Vps34 activator (Fig 3c, left and right respectively). We also found decreased Atg5-Atg12 complex compared to free Atg12 when we knocked down all Rab5 isoforms simultaneously (Fig. 3d, left). This effect was similar to the conjugation defect we observed with Atg7 knockdown (Fig. 3d, right). Thus, Rab5 inhibition and loss of activity of Vps34 (which is activated by Rab5) have a range of similar phenotypes: suppression of autophagy, increased numbers of Atg5-positive structures and decreased Atg5-Atg12 conjugation.
Figure 3.
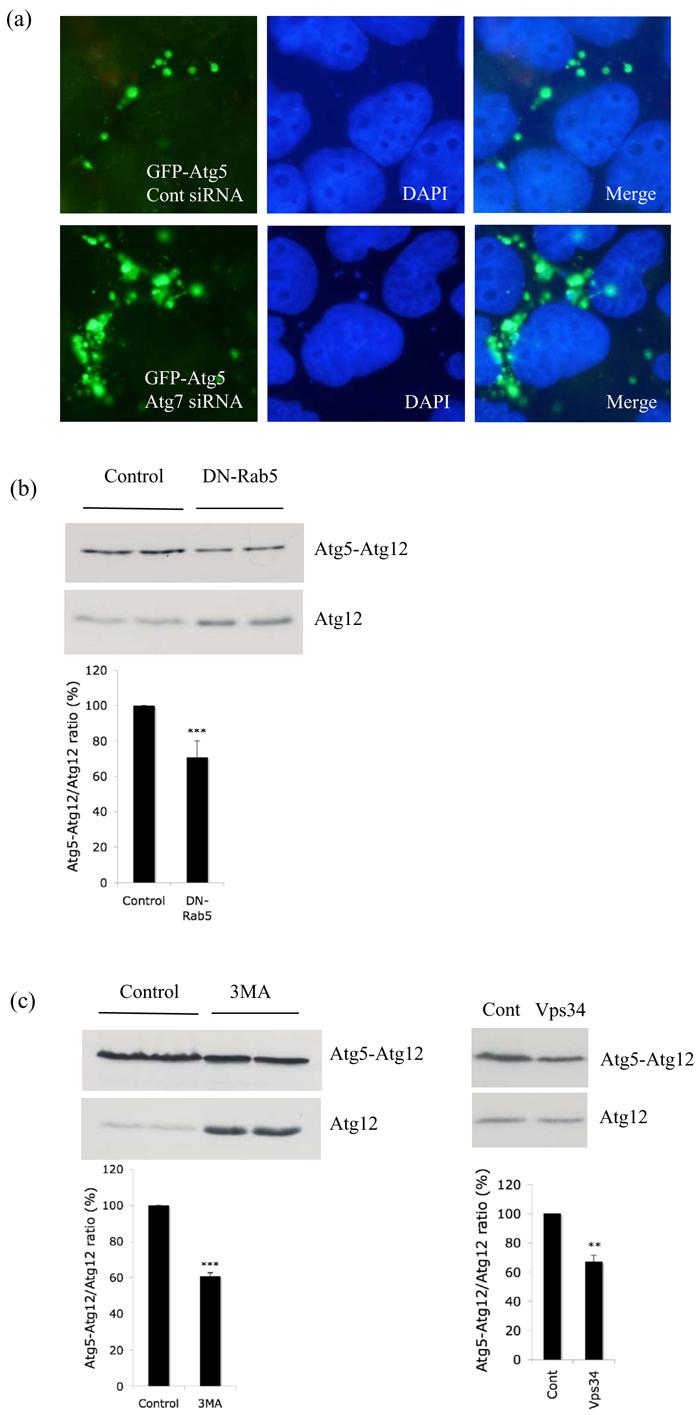
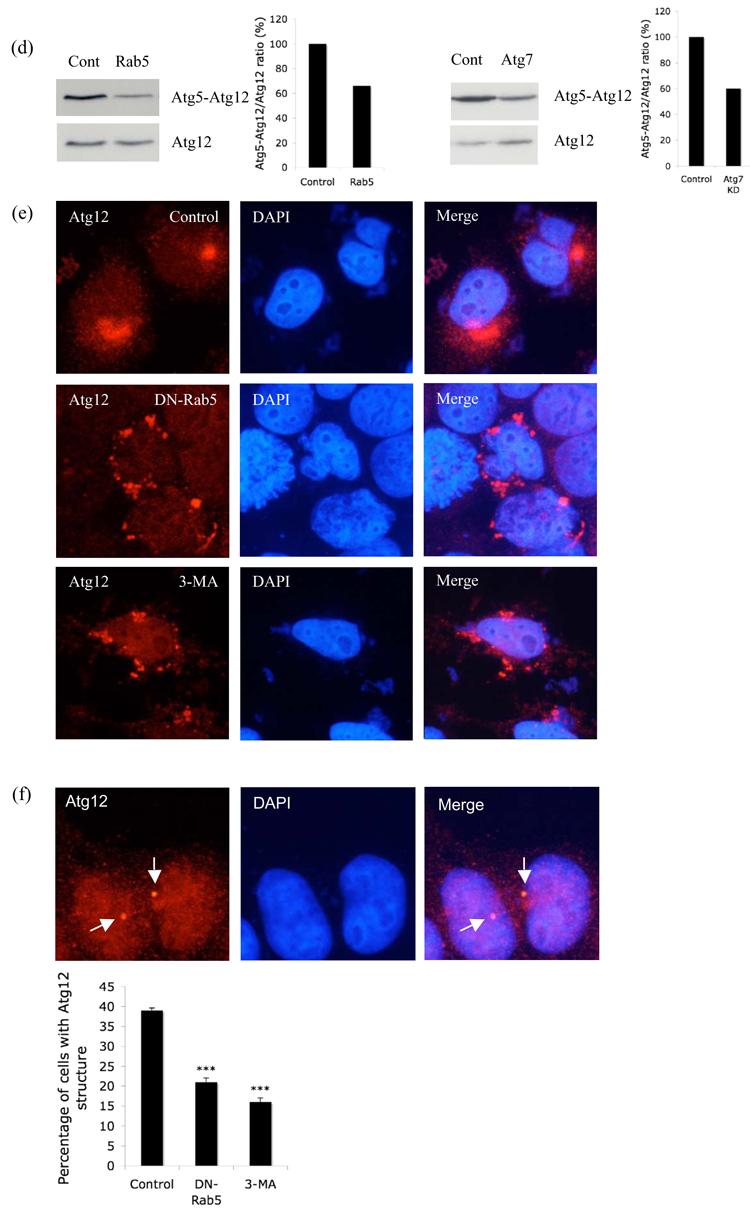
Aberrant conjugation of Atg5 with Atg12 after Rab5 inhibition. a. HeLa cells transfected with control or Atg7 siRNA for 48h were subjected to second round of transfection with GFP-Atg5 together with the siRNA for another 24h. Then the cells were fixed following saponin extraction to visualise the GFP-Atg5 structures (green). b. HeLa cells transfected with control vector or DN-Rab5 together with HA-Atg12 and Atg5 were subjected to western blot analysis with anti-HA antibody to detect free Atg12 and the Atg5-Atg12 complex. The experiment assays the proportion of transfected Atg12 that gets conjugated to Atg5 by assessing the ratio of Atg5-Atg12 complex to free Atg12 as shown in the graph. Atg5-Atg12/Atg12 ratio from 4 independent experiments is shown *** - P < 0.0001. c. Similar experiments to Fig. 3b were performed in the presence or absence (control) of 3-methyladenine (3-MA; left) or with control or Vps34 siRNA (right). Atg5-Atg12/Atg12 ratio from 3 (for Vps34 siRNA) and 4 (for 3MA) independent experiments is shown. *** - P < 0.0001, ** - P < 0.001. d. HeLa cells transfected simultaneously with Rab5a, Rab5b and Rab5c siRNA (Rab5; left), Atg7 (right) or control siRNA for 48h were subjected to second round of transfection with Atg5 and HA-Atg12 together with the siRNA for another 24h. Western blot analysis was performed with anti-HA antibody to detect free Atg12 and the Atg5-Atg12 complex. Representative images from two independent, reproducible experiments are shown. e. Immunostaining of HeLa cells transfected with control (with or without 3-MA) or DN-Rab5 and HA-Atg12 for 24h after saponin extraction with anti-HA antibody (red). DAPI stained nuclei are in blue. f. Immunostaining of HeLa cells transfected with control (with or without 3-MA) or DN-Rab5 for 24h after saponin extraction with anti-Atg12 antibody (red). DAPI stained nuclei are in blue. Punctate Atg12 structures in control cells are shown in arrow. Quantitation of the percentage of cells with the punctate Atg12 structures in control, 3MA or DN-Rab5 is shown in graph. *** - P < 0.0001.
As both Rab5 and Vps34 are likely to regulate Atg5-Atg12 conjugation, which, in turn, regulates formation of autophagic vacuoles from Atg5-rich phagophores, we also looked at the distribution of Atg12 in saponin-extracted cells with Rab5 inhibition. In control cells, overexpressed Atg12 was located in a juxtanuclear single large domain (Fig. 3e), which did not change with the presence or absence of Atg5 overexpression (data not shown). However with Rab5 inhibition, Atg12 was re-distributed to several peripheral small punctate structures (Fig. 3e), a phenomenon also seen with 3MA treatment (Fig. 3e). Similarly, when we looked at the distribution of endogenous Atg12, we saw it distributed in a single juxtanuclear domain (reminiscent of the yeast PAS) in wild-type cells (Fig. 3f). However, either dominant-negative inhibition of Rab5 or 3MA treatment significantly decreased the proportion of cells with these punctate endogenous Atg12 structures (Fig. 3f). The significance of the Atg12 re-distribution is still a matter of ongoing research, but leads to the hypothesis that Rab5 inhibition or 3MA treatment might affect the accessibility of Atg12 for the conjugation process, probably by re-distributing it away from its normal cellular localisation. Alternatively, the decrease in Atg5-Atg12 conjugation that we observed with Rab5 inhibition or 3MA treatment might result in aberrant accumulation of unconjugated Atg12.
Rab5 is found in a macromolecular complex containing Vps34 and Beclin-1
Since a macromolecular complex containing Vps34 and Beclin-1 regulates the early nucleation step in autophagy, we next tested whether Rab5 was found in such a complex. We tested whether Rab5 interacted with Beclin-1, since both proteins interact with Vps34 (Christoforidis et al., 1999; Kihara et al., 2001a), Beclin-1 is associated with phagophores, and our data suggests that Rab5 acts at the autophagosome precursor stage and is also associated with such structures (Fig. 2g). In 3MA-treated cells, we observed co-localisation of endogenous Rab5 and Beclin-1 (Fig 4a), particularly in structures like those that accumulated when Rab5 or Vps34 were inhibited (e.g. Fig 2). These likely autophagosome-precursors are precisely where we expected such co-localisation to occur. We also found strong co-localisation of Beclin-1 with an activated Rab5 mutant (Fig. 4b). We used activated Rab5 as this forms large clear vesicles that allow unambiguous co-localisation, a strategy frequently used for Rab5-interactor immunocytochemistry studies (Shin et al., 2005). We next looked for wildtype Rab5 interaction with Beclin-1 by immunoprecipitation in cells with no exogenous Rab5. Rab5 interacted with Beclin-1 (immunoprecipitated for Beclin-1 and detected for Rab5) only in the presence of Vps34 suggesting that Rab5 is part of the macromolecular complex containing Beclin-1 and Vps34 (Fig. 4c). Indeed, Beclin-1 knockdown (Supp. Fig. 9) enhances mutant huntingtin (Q74) aggregation (Fig. 4d) (Shibata et al., 2006) compatible with the knowledge that this will impair autophagosome formation. Critically, Beclin-1 knockdown led to decreased Atg5-12 conjugation (Fig. 4e), compatible with the concept that both Beclin-1 and Rab5 activity are required for Vps34 function in autophagosome formation. Thus, Rab5 activation of Vps34 probably regulates not only endosome maturation and multivesicular body formation but also, by recruitment of Beclin-1, initiation of autophagic vacuole formation.
Figure 4.
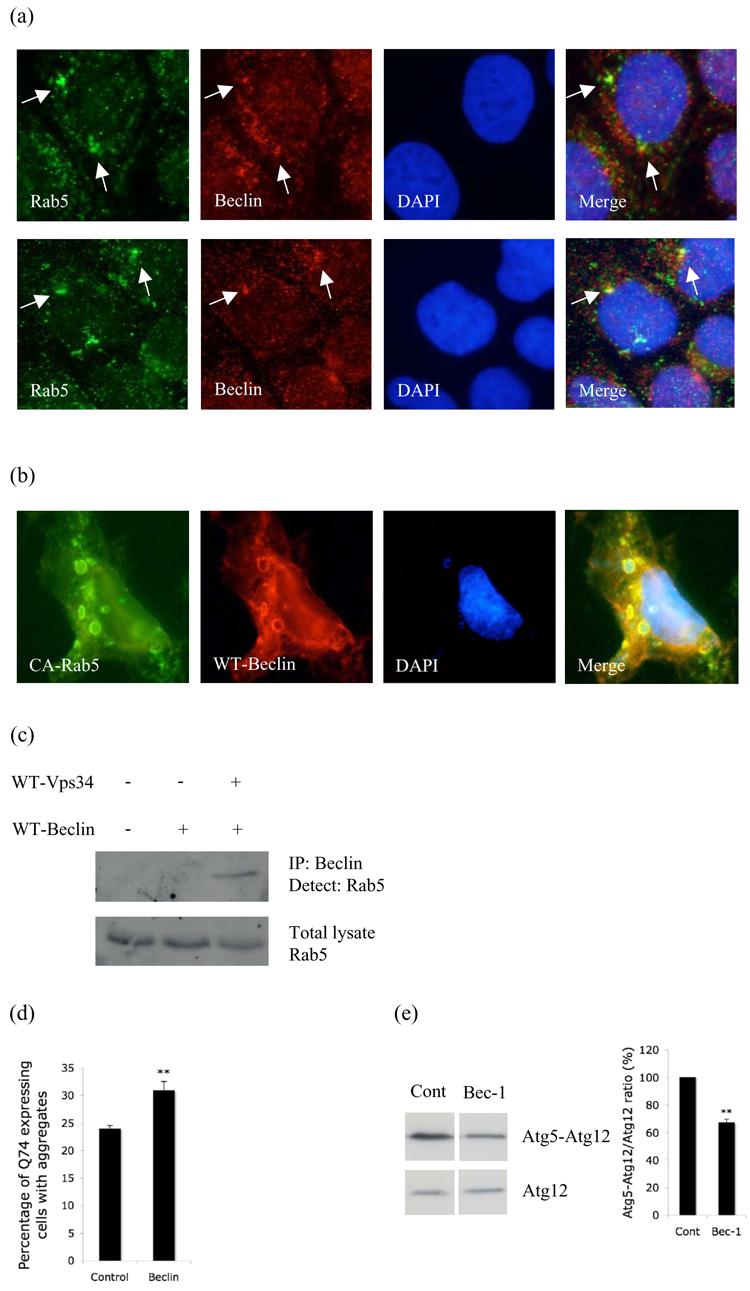
Rab5 is part of a macromolecular complex containing Vps34 and Beclin-1. a. Co-localisation of endogenous Rab5 (green) and endogenous Beclin-1 (red) in 3MA treated HeLa cells after saponin extraction. DAPI-stained nuclei are in blue. b. Co-localisation of constitutive active Rab5 (CA-Rab5; green) with wild-type (WT) Beclin-1 (red). c. COS-7 cells transfected with control vector alone (lane 1) or Flag-tagged WT-Beclin-1 alone (lane 2) or WT-Beclin-1 with WT-Vps34 (lane 3) were immunoprecipitated with anti-Flag antibody (to immunoprecipitate Beclin-1) and blotted for Rab5 using an anti-Rab5 antibody. d. Q74-HA aggregation in HeLa cells transfected with control or Beclin-1 siRNA as in Fig. 2d. p < 0.005. e. Atg5-Atg12 conjugate to Atg12 ratio (from 3 independent experiments) in HeLa cells transfected with control or Beclin-1 siRNA for 72h. Atg5 and HA-Atg12 were transfected for the last 24h. p = 0.0003.
Rab5 effect on autophagy is not due to a general inhibition of the endocytic pathway
Since Rab5 is an important regulator of the endocytic pathway, we further tested whether the effects we saw with Rab5 inhibition were indirect consequences of endocytosis inhibition. We first perturbed the function of dynamin, a large GTPase essential for clathrin-mediated endocytosis, using dominant-negative dynamin (DN-Dyn). DN-Dyn increased the proportion of COS-7 cells with aggregates and cell death similar to DN-Rab5 (Fig. 5a). However, unlike Rab5, overexpression of DN-Dyn increased the size and also the number of LC3-positive autophagic vacuoles (Fig. 5b, c, d) similar to what we and others observe after treatment with BafA1, an inhibitor of autophagosome-lysosome fusion (Bampton et al., 2005). This suggests that DN-Dyn inhibits autophagy at the level of autophagosome-lysosome fusion similar to BafA1. Accordingly we found decreased co-localisation of LC3-labelled vesicles with the lysosomal marker, lgp120, similar to BafA1 (Fig. 5e). Previous studies have shown that autophagosomes may fuse with endosomes to form intermediary compartments called amphisomes, which subsequently fuse with lysosomes. Thus, it is possible that inhibition of the endocytic flux may prevent the formation of amphisomes, thereby also inhibiting a subsequent step in the autophagic pathway, amphisome-lysosome fusion. Again, unlike Rab5, we did not see any increase in the Atg5 structures (Supp. Fig. 10) nor could we observe any change in the Atg5-Atg12 complex to free Atg12 ratio with inhibition of dynamin (Fig. 5f).
Figure 5.
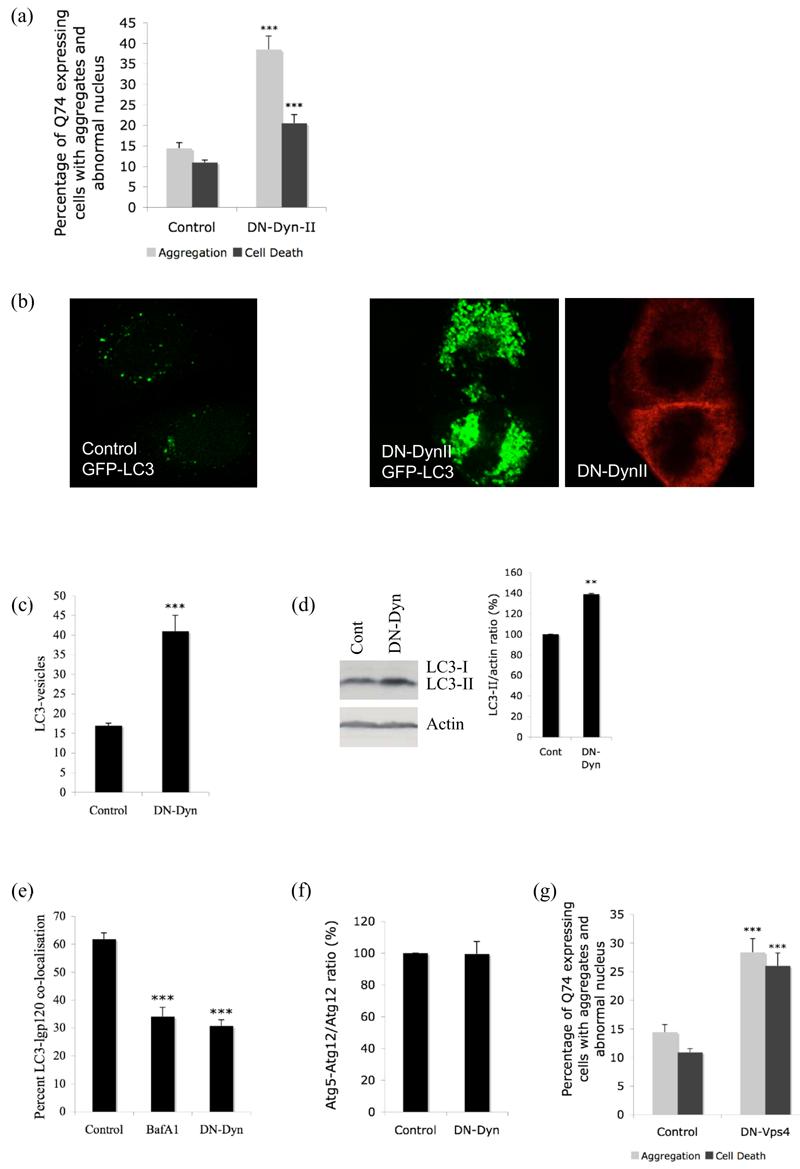
a. Quantification of proportions of GFP-expressing cells with aggregates and cell death in COS-7 cells transiently transfected with dominant-negative dynamin II (DN-Dyn-II) or empty vector control and EGFP-tagged huntingtin exon-1 with 74 polyglutamine repeats (3:1 ratio) for 48h. *** - P < 0.0001. b. Distribution of GFP-LC3 vesicles in control (left) or DN-dynamin (red) transfected cells (right). c. COS-7 cells were transiently transfected with empty vector (Control), DN-dynamin (DN-Dyn) and GFP-LC3 (3:1 ratio) for 24h. GFP-positive cells with increased LC3-positive vesicles (> 20 vesicles per cell) were quantified. *** - P < 0.0001. d. Lysates from HeLa cells expressing empty vector control (Cont) or DN-dynamin (DN-Dyn) were blotted for endogenous LC3 and actin as control. Quantification of band intensities from four independent experiments is shown ** - P < 0.001. Under exposure conditions allowing endogenous LC3-II quantitation in these cells, the LC3-I signal is frequently too low to detect. e. NRK cells was transiently transfected for 15h with either empty vector (control) (with or without bafilomycin A1 (BafA1)) or DN-dynamin (DN-Dyn), and mRFP-LC3 and GFP-lgp120. The proportion of mRFP-LC3 and GFP-lgp120 double stained vesicles in individual cells were scored (Jahreiss et al., 2008). *** - P < 0.0001. f. HeLa cells transfected with control vector or DN-dynamin (DN-Dyn) together with HA-Atg12 and Atg5 were subjected to western blot analysis with anti-HA antibody to detect free Atg12 and the Atg5-Atg12 complex. Atg5-Atg12/Atg12 ratio from 3 independent experiments is shown. g. Quantification of proportions of GFP-expressing cells with aggregates and cell death in COS-7 cells transiently transfected with dominant-negative (DN) Vps4 or empty vector control and EGFP-tagged huntingtin exon-1 with 74 polyglutamine repeats (3:1 ratio) for 48h. *** - P < 0.0001.
These results together suggest that DN-Dyn inhibits autophagy at the level of autophagosome-lysosome fusion similar to what has been previously reported with Vps4, an AAA ATPase involved in trafficking through the endocytic pathway (Nara et al., 2002). Overexpression of dominant-negative Vps4 showed defect in autophagy-dependent bulk protein degradation due to an impairment of autolysosome formation (Nara et al., 2002). Similar to DN-Dyn we did not see an increase in Atg5 structures (data not shown) or decrease in Atg5-Atg12 complex with DN-Vps4 (Supp. Fig. 11) but similar to DN-Dyn (Fig. 5a) or BafA1 (Ravikumar et al., 2002) we observed an increase in Q74-aggregation and associated cell death with DN-Vps4 (Fig. 5g).
We obtained identical results as seen with DN-Dyn when we used siRNA against clathrin heavy chain (Supp. Fig. 12), which is involved in formation of clathrin-coated vesicles. siRNA against clathrin heavy chain increased the proportion of HeLa cells with aggregates (Fig. 6a), increased the number and size of LC3-labelled autophagic vacuoles (Fig. 6b, c), did not increase Atg5 structures (data not shown) and had no effect on Atg5-Atg12 conjugation (Fig. 6d). Finally, we also looked at the effect of methyl-β-cyclodextrin (β–CD, an inhibitor of endocytosis; Supp. Fig. 13) on LC3-II levels both in the presence or absence of BafA1. β–CD increased the levels of LC3-II in the absence of BafA1, but had no effect in the presence of BafA1 (Fig. 6e) again suggesting that endocytosis inhibition blocks autophagosome-lysosome fusion. DN-Dyn and siRNA against clathrin had similar effect on LC3-II levels in the presence of BafA1 as β–CD (Supp. Fig. 14, 15). Thus, Rab5 inhibition, which impairs Atg5-Atg12 conjugation, autophagosome (and LC3-II) formation and is associated with an accumulation of Atg5-positive structures, has different effects to a range of endocytosis inhibitors (β–CD, DN-Dyn, DN-Vps4 and siRNA for clathrin), which do not affect Atg5-Atg12 conjugation, but block autophagosome-lysosome fusion and increase LC3-II levels. (The specificity of the effect of Rab5 inhibition versus endocytosis inhibition on LC3-II levels is clearly demonstrated in our experiments where we have treated cells with BafA1). This argues that the effect of loss of Rab5 function on autophagosome formation is not due to effects on endocytosis.
Figure 6.
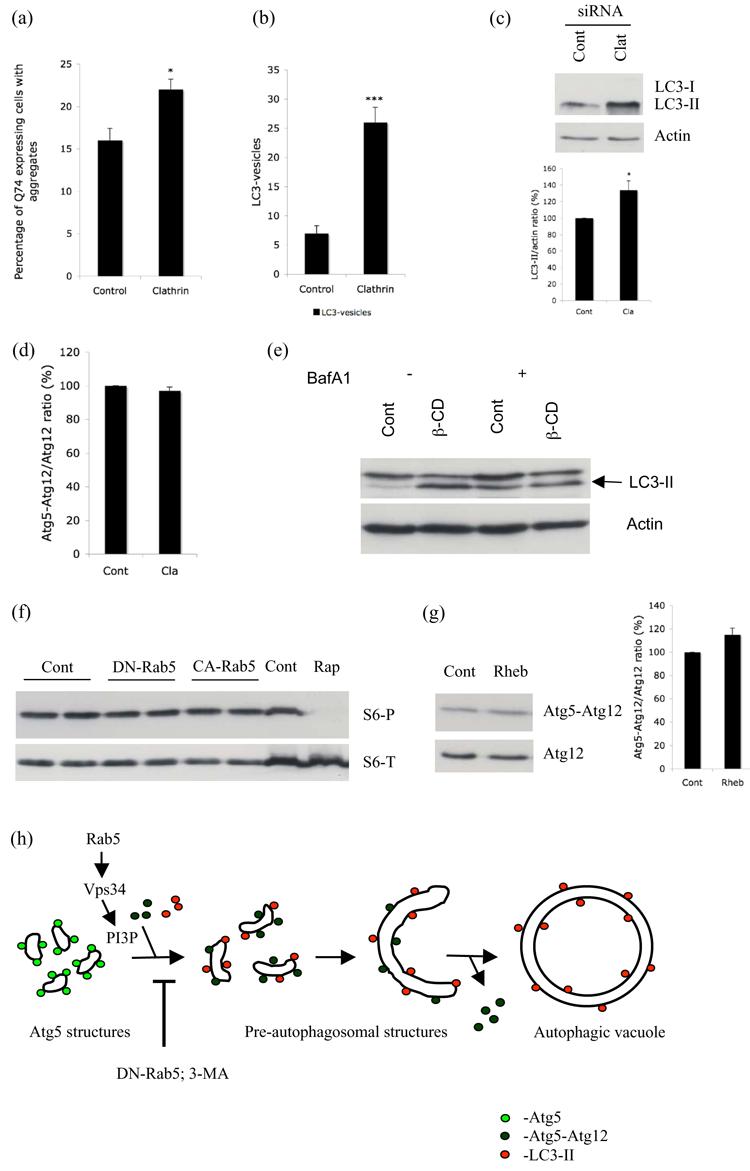
a. HeLa cells transfected with control or clathrin heavy chain siRNA for 48h were subsequently transfected with EGFP-tagged huntingtin exon-1 with 74 polyglutamine repeats (Q74) for 24h. Quantification of proportion of Q74 expressing cells with aggregates is shown. * - P < 0.01. b. GFP-LC3 stable Hela cells transfected with control or clathrin siRNA for 72h were scored for increased LC3-positive vesicles (> 20 vesicles per cell). *** - P < 0.0001. c. HeLa cells transfected with control or clathrin heavy chain siRNA for 72h were subjected to western blot analysis with anti-LC3 and anti-actin antibodies. Under exposure conditions allowing endogenous LC3-II quantitation in these cells, the LC3-I signal is frequently too low to detect. Quantification of band intensities from three independent experiments is shown. * - P < 0.05. d. HeLa cells transfected with control (Cont) or clathrin (Cla) heavy chain siRNA for 48h were subsequently transfected with HA-Atg12 and Atg5 for a further 24h. Western blot analysis was performed with anti-HA antibody to detect free Atg12 and the Atg5-Atg12 complex. Atg5-Atg12/Atg12 ratio from 3 independent experiments is shown. e. GFP-LC3 stable HeLa cells left untreated (Cont) or treated with 5mM methyl-β-cyclodextrin (β-CD), in the presence (+) or absence (−) of Bafilomycin A1 (BafA1) for 6h were subjected to western blot analysis with anti-GFP (to detect LC3) and anti-actin antibodies. f. Lysates from HeLa cells transfected with empty vector (Cont), DN- or CA-Rab5 were blotted for phospho- (S6-P) and total- (S6-T) ribosomal protein S6. Lysate from cells treated with Rapamycin (Rap) was used as a positive control. g. HeLa cells transfected with control vector (Cont) or wild-type Rheb together with HA-Atg12 and Atg5 were subjected to western blot analysis with anti-HA antibody to detect free Atg12 and the Atg5-Atg12 complex. Atg5-Atg12/Atg12 ratio from 3 independent experiments is shown. h. Schematic hypothetical representation of regulation of autophagy by Rab5. Our data suggests that the Atg5 structures we observed are probably precursors to the pre-autophagosomal structures. The accumulation of Atg5 structures that we observed with Rab5 or Vps34 inhibition might be due to a block in the progression from early Atg5-positive autophagosomal structures to formation of autophagic vacuoles.
Rab5 effect on autophagy is not due to mTOR signalling
Previous studies in Drosophila have shown that disruption of endocytosis can lead to changes in target of rapamycin (TOR) signalling, a key process regulating autophagy (Hennig et al., 2006). Since Rab5 regulates endocytosis, we tested if the conditions of Rab5 inhibition or activation we used altered the mTOR-signalling pathway. mTOR directly phosphorylates at least two effectors, S6 kinase-1 and 4EBP1. S6 kinase-1 phosphorylates the ribosomal protein S6. The levels of phosphorylation of any of these downstream targets are recognized indicators of mTOR activity in the cell (Jacinto and Hall, 2003). Accordingly, we tested if DN-Rab5 or CA-Rab5 altered the mTOR pathway by looking at the phosphorylation of S6. We did not observe any change in the phosphorylation of S6 (Fig. 6f). We also did not see any change in the phosphorylation of S6 Kinase or 4EBP1 with DN-Rab5 or CA-Rab5 (data not shown). Thus, the effects we observed with Rab5 were not due to alterations in the mTOR signalling. Furthermore, activation of mTOR signalling by overexpression of Rheb (Supp. Fig. 16) did not show the effects that we observed with DN-Rab5, as we observed no increase in Atg5 structures (data not shown) and did not see any decrease in the Atg5-Atg12 conjugate compared to free Atg12 pool (Fig. 6g). Taken together, our results suggest that the role of Rab5 on autophagy was independent of its effects on endocytosis and is not due to perturbations in mTOR signalling.
Conclusions
In conclusion, we show that Rab5 is a modifier of polyglutamine toxicity in cell and fly models. These modifying effects are due to Rab5 regulating autophagy-dependent clearance of the mutant toxic protein. Our data suggest that Rab5, previously considered as a “specific” endosome marker, also influences mammalian autophagy. Inhibition of Rab5 resulted in a decrease in LC3-positive vesicles suggesting a defect in the formation of autophagic vacuoles. Rab5 is an activator of Vps34, a PI-3-kinase essential for autophagy initiation, and we have shown that Rab5 is a new member of a complex containing Vps34 and Beclin-1 that is associated with autophagosome precursors. Thus, the most parsimonious explanation for our data is that Rab5 acts as an activator for Vps34 in autophagy as it is known to do in endocytosis. This hypothesis is consistent with the various similarities we observed in cells with loss-of-function of Rab5 or Vps34. Loss of either of these activities decreased Atg5-Atg12 conjugation, a critical step in early phagophore elongation. We speculate that the accumulation of Atg5 structures with Rab5 or Vps34 inhibition might be due to a block in the progression from early Atg5-positive autophagosomal structures to the formation of autophagosomes (Fig. 6h). This block might be due to a defect in Atg12 recruitment. Indeed, decreasing Atg5 conjugation (which is known to block autophagosome formation) also resulted in increased numbers of Atg5-positive autophagosome precursors.
Our data do not exclude the possibility that membranes for autophagosome biogenesis may be derived from endosomes. However, the effect of Rab5 inactivation on Atg5-Atg12 conjugation and autophagosome synthesis are not seen with a wide range of other endocytosis inhibitors (β–CD, DN-Dyn, DN-Vps4 and siRNA for clathrin), which instead impede autophagic flux by inhibiting autophagosome-lysosome fusion directly or by inhibiting the autophagosome-amphisome fusion step. This does suggest that endocytosis inhibition through different mechanisms will also enhance polyglutamine aggregation/toxicity by blocking autophagy. It is possible that loss of Rab5 activity has effects on autophagy by perturbing other unrelated/unknown membrane trafficking pathways (distinct from endocytosis). However, it is important to point out that overexpression of CA-Rab5 or WT-Rab5 enhanced autophagosome synthesis and suppressed huntingtin aggregation/toxicity in cells and in vivo.
We believe our data suggest a hypothetical sequential model in mammalian cells where PI-3-P generated by Vps34 in a complex comprising at least Beclin-1 and active Rab5, is a key regulator of Atg12 conjugation to Atg5, a rate-limiting step in the conversion of Atg5-positive autophagosome precursors to Atg5-negative autophagosomes. On the one hand, inhibition of this putative cascade at a number of points will lead to impaired autophagy and enhance polyglutamine toxicity. On the other hand, better understanding of the initial rate-limiting steps of autophagy may provide opportunities for rational therapeutic design of more specific and safer autophagy-inducing drugs than rapamycin (which affects many pathways). This may have relevance to HD and also to a range of related neurodegenerative diseases caused by intracytosolic aggregate-prone proteins.
Materials and methods
Mammalian cell culture and transfection
COS-7 and HeLa cells were grown in Dulbecco's Modified Eagle Medium (DMEM) (Sigma) supplemented with 10% Fetal Bovine Serum (FBS) (Sigma), 100 U/ml Penicillin/Streptomycin (Sigma), 2mM L-Glutamine (Sigma) at 37°C, 5% CO2. DNA transfections were performed using lipofectAMINE reagent (Invitrogen). 20nmol of control (Cat. no: 4611), Atg7 (ID 135756), Rab5a (ID 120372), Rab5b (ID 120273), Rab5c (ID 120809), Clathrin heavy chain (ID 107565), Vps34 (ID 143802) or Beclin (ID 137200) siRNA (all from Ambion) was transfected using lipofectAMINE 2000 according to manufacturer's instructions.
Western Blot Analysis
Western blot analysis was done using standard techniques with ECL detection kit (Amersham). The primary antibodies used include anti-GFP (Clontech); anti-HA (Covance); anti-myc and anti-actin (Sigma); anti-Atg7 (Rockland Inc.); anti-Atg5 and anti-Rab5 (Abcam); anti-Vps34 (Zymed); anti-Beclin (Cell Signaling technology). Densitometry analysis was performed using Image J 1.36b or Scion Image Beta 4.02 softwares. For immunoprecipitation, cells were suspended in Lysis buffer (50mM Tris HCl, pH 7.4, 150mM NaCl, 1mM EDTA and 1% Triton-X100) for 30 minutes on ice and supernatant was removed by centrifugation at 13,000 rpm for 7 minutes at 4°C. 30μl of anti-M2 affinity gel (Sigma) was added to the sample and incubated with gentle rocking at 4°C for 2h. After incubation, the tubes were spun for 30 seconds at 4°C at less than 4000×g. The pellet was washed four times with 1ml of chilled buffer A (20mM Tris HCl, pH 7.2; 2mM MgCl2; 150mM NaCl, 0.5% Nonidet P-40). The bound protein was eluted with 100μl of 150ng/μl 3×FLAG peptide in TBS.
Immunocytochemistry
Immunocytochemistry in GFP-Atg5 transfected cells was performed in 0.02% saponin extracted and 4% paraformaldehyde fixed HeLa cells. Saponin was prepared in PHEM buffer (60mM NaPIPES, 25mM NaHEPES, 10mM EGTA 2mM MgCl2, pH 6.9) with 0.19M NaCl. Primary antibodies used: anti-myc, anti-Flag (Sigma), anti-Rab5 and anti-HA. Relevant negative controls without the primary antibodies were performed alongside all the experiments. Nuclei were stained with 4′, 6-diamidino-2-phenylindole (DAPI, 3 mg/ml) (Sigma). Images were acquired with a Nikon Digital Camera DXM1200 using Nikon Eclipse E600 fluorescence microscope. We used Nikon ACT-1 version 2.12 acquisition software. Adobe Photoshop 6.0 (Adobe Systems, Inc.) was used for subsequent image processing.
Quantification of aggregate formation, abnormal nuclear morphologies, LC3-positive vesicles
Aggregate formation and nuclear morphology were assessed using a fluorescence microscope. An example of cells expressing Q74 with aggregates (arrows) and an aggregate-containing cell with abnormal nuclear morphology (arrow head) is shown in Supp. Fig. 17. Two hundred Q74-transfected COS-7 or MEFs cells were selected and the proportion of cells with aggregates are counted. The observer was blinded to the identity of the slides and the experiments were performed in triplicate and repeated twice. Cells are considered dead if the DAPI-stained nuclei showed apoptotic morphology (fragmentation or pyknosis). Pyknotic nuclei are typically <50% the diameter of normal nuclei and show obvious increased DAPI intensity. An example of cells with abnormal nuclear morphologies (arrows) is shown in Supp. Fig. 18. For LC3 quantification, one hundred EGFP/mRFP-positive COS-7 cells were selected and the proportion of cells with approximately more than 20 LC3-labelled vesicles were scored, with the observer blinded to identity of slides. Examples of cells with <20 vesicles (a) and >20 vesicles (b) are shown in Supp. Fig. 19.
Endocytosis assay
HeLa cells were pre-treated with or without 5 mM β-cyclodextrin for 24 h. Cells were washed with serum-free medium containing 0.5% BSA and then incubated at 4°C with 10 μg/ml Alex Fluor 594-labeled human transferrin (Molecular Probes) for 30 min. Excess label was washed off, after which the cells were incubated at 37°C for 10 min to allow uptake of the ligand in serum-free medium containing 0.5% BSA with or without 5 mM β-cyclodextrin. The cells were then fixed with 4% paraformaldehyde in 0.1 M PBS pH 7.6 and images were acquired on a Zeiss LSM510 META confocal microscope (63x 1.4NA plan-apochromat oil immersion lens) at room temperature using Zeiss LSM510 v3.2 software (Carl Zeiss, Inc.).
Drosophila
Recombinant males of genotype w; CyO/If; UAS-Rab5-EGFP elavGal4/MKRS, carrying Rab5-EGFP (Wucherpfennig et al., 2003) and elav-Gal4 3A4(Luo et al., 1994) on the third chromosome (gift of Dr V Korolchuk, University of Cambridge, UK) were crossed with y w; gmr-Htt(exon1)Q120 (Jackson et al., 1998) virgins. As a control both y w; gmr-Htt(exon1)Q120 and w; CyO/If; UAS-Rab5-EGFP-elavGal4/MKRS virgins were crossed with an isogenic w1118 line (Ryder et al., 2004). Rhabdomeres were scored using the pseudopupil technique as previously described (Franceschini and Kirschfeld, 1971). Means, standard errors and statistical comparisons using a t-test are calculated from 6 independent experiments. In each experiment we evaluated 10 eyes for each genotype, and 15 ommatidia for each eye.
Statistics
Pooled estimates for the changes in aggregate formation or cell death, resulting from perturbations assessed in multiple experiments, were calculated as odds ratios with 95% confidence intervals. We have used this method frequently in the past to allow analysis of data from multiple independent experiments (Wyttenbach et al., 2001). Odds ratios and P values were determined by unconditional logistical regression analysis, using the general log-linear analysis option of SPSS 9 software. 95% confidence limits for the odds ratio were obtained by using the robust variance approach. Significance levels for comparisons between groups were determined with t tests, where appropriate, for parametric data and with Mann-Whitney U tests for non-parametric data, using the STATVIEW software, version 4.53 (Abacus Concepts, Berkeley, CA).
Supplementary Material
Acknowledgements
We are grateful to G Jackson (UCLA, USA) for HD flies; M González-Gáitan (Max Planck Institute of Molecular Cell Biology and Genetics, Germany) for UAS-EGFP-Rab5 flies; V Korolchuk (CIMR, UK) for the Rab5 recombinants that we have used in this paper; T Yoshimori (National Institute of Genetics, Japan) for GFP-LC3 and myc-LC3; A Tolkovsky (Cambridge University, UK) for GFP-LC3 stable HeLa cells; K L Guan (University of Michigan, USA) for Rheb; B Beaumelle (University of Montpellier II, France) for WT-, S34N-Rab5 and K44A Dynamin; J P Luzio (CIMR, UK) for Vps34; M Robinson (CIMR, UK) for Alex Fluor 594-labeled human transferring; B Levine (UT Southwestern, USA) for Beclin-1, H Stenmark (The Norwegian Radium Hospital, Norway) for myc-FYVE; N Mizushima (The Tokyo Metropolitan Institute of Medical Science, Japan) for HA-Atg12 and Atg5; Atg5−/− and WT-MEF cells; A Sorkin (University of Colorado Health Sciences Center, USA) for N34S and Q79L Rab5; J Stankova (University of Sherbrooke, Canada) for K44A Dynamin; X Wang (The University of Utah, USA) for DN-Vps4B; Eléonore Mayola for technical support. We are grateful for MRC (DCR & CO'K); EU Framework VI (EUROSCA) (DCR) funding; DCR is a Wellcome Trust Senior Clinical Fellow.
References
- Argon Y, Simen BB. GRP94, an ER chaperone with protein and peptide binding properties. Semin Cell Dev Biol. 1999;10:495–505. doi: 10.1006/scdb.1999.0320. [DOI] [PubMed] [Google Scholar]
- Arrasate M, Mitra S, Schweitzer ES, Segal MR, Finkbeiner S. Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature. 2004;431:805–10. doi: 10.1038/nature02998. [DOI] [PubMed] [Google Scholar]
- Bampton ET, Goemans CG, Niranjan D, Mizushima N, Tolkovsky AM. The dynamics of autophagy visualized in live cells: from autophagosome formation to fusion with endo/lysosomes. Autophagy. 2005;1:23–36. doi: 10.4161/auto.1.1.1495. [DOI] [PubMed] [Google Scholar]
- Berger Z, Ravikumar B, Menzies FM, Oroz LG, Underwood BR, Pangalos MN, Schmitt I, Wullner U, Evert BO, O'Kane CJ, et al. Rapamycin alleviates toxicity of different aggregate-prone proteins. Hum Mol Genet. 2006;15:433–42. doi: 10.1093/hmg/ddi458. [DOI] [PubMed] [Google Scholar]
- Blommaart EF, Krause U, Schellens JP, Vreeling-Sindelarova H, Meijer AJ. The phosphatidylinositol 3-kinase inhibitors wortmannin and LY294002 inhibit autophagy in isolated rat hepatocytes. Eur J Biochem. 1997;243:240–6. doi: 10.1111/j.1432-1033.1997.0240a.x. [DOI] [PubMed] [Google Scholar]
- Bucci C, Parton RG, Mather IH, Stunnenberg H, Simons K, Hoflack B, Zerial M. The small GTPase rab5 functions as a regulatory factor in the early endocytic pathway. Cell. 1992;70:715–28. doi: 10.1016/0092-8674(92)90306-w. [DOI] [PubMed] [Google Scholar]
- Christoforidis S, Miaczynska M, Ashman K, Wilm M, Zhao L, Yip SC, Waterfield MD, Backer JM, Zerial M. Phosphatidylinositol-3-OH kinases are Rab5 effectors. Nat Cell Biol. 1999;1:249–52. doi: 10.1038/12075. [DOI] [PubMed] [Google Scholar]
- Derby MC, Lieu ZZ, Brown D, Stow JL, Goud B, Gleeson PA. The trans-Golgi network golgin, GCC185, is required for endosome-to-Golgi transport and maintenance of Golgi structure. Traffic. 2007;8:758–73. doi: 10.1111/j.1600-0854.2007.00563.x. [DOI] [PubMed] [Google Scholar]
- Franceschini N, Kirschfeld K. [Pseudopupil phenomena in the compound eye of drosophila] Kybernetik. 1971;9:159–82. doi: 10.1007/BF02215177. [DOI] [PubMed] [Google Scholar]
- Gaullier JM, Simonsen A, D'Arrigo A, Bremnes B, Stenmark H, Aasland R. FYVE fingers bind PtdIns(3)P. Nature. 1998;394:432–3. doi: 10.1038/28767. [DOI] [PubMed] [Google Scholar]
- George MD, Baba M, Scott SV, Mizushima N, Garrison BS, Ohsumi Y, Klionsky DJ. Apg5p functions in the sequestration step in the cytoplasm-to-vacuole targeting and macroautophagy pathways. Mol Biol Cell. 2000;11:969–82. doi: 10.1091/mbc.11.3.969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas IG. BiP (GRP78), an essential hsp70 resident protein in the endoplasmic reticulum. Experientia. 1994;50:1012–20. doi: 10.1007/BF01923455. [DOI] [PubMed] [Google Scholar]
- Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Saito I, Okano H, et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441:885–9. doi: 10.1038/nature04724. [DOI] [PubMed] [Google Scholar]
- Hennig KM, Colombani J, Neufeld TP. TOR coordinates bulk and targeted endocytosis in the Drosophila melanogaster fat body to regulate cell growth. J Cell Biol. 2006;173:963–74. doi: 10.1083/jcb.200511140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacinto E, Hall MN. Tor signalling in bugs, brain and brawn. Nat Rev Mol Cell Biol. 2003;4:117–26. doi: 10.1038/nrm1018. [DOI] [PubMed] [Google Scholar]
- Jackson GR, Salecker I, Dong X, Yao X, Arnheim N, Faber PW, MacDonald ME, Zipursky SL. Polyglutamine-expanded human huntingtin transgenes induce degeneration of Drosophila photoreceptor neurons. Neuron. 1998;21:633–42. doi: 10.1016/s0896-6273(00)80573-5. [DOI] [PubMed] [Google Scholar]
- Jahreiss L, Menzies FM, Rubinsztein DC. The itinerary of autophagosomes: From peripheral formation to kiss-and-run fusion with lysosomes. Traffic. 2008 doi: 10.1111/j.1600-0854.2008.00701.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. Embo J. 2000;19:5720–8. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kihara A, Kabeya Y, Ohsumi Y, Yoshimori T. Beclin-phosphatidylinositol 3-kinase complex functions at the trans-Golgi network. EMBO Rep. 2001a;2:330–5. doi: 10.1093/embo-reports/kve061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kihara A, Noda T, Ishihara N, Ohsumi Y. Two distinct Vps34 phosphatidylinositol 3-kinase complexes function in autophagy and carboxypeptidase Y sorting in Saccharomyces cerevisiae. J Cell Biol. 2001b;152:519–30. doi: 10.1083/jcb.152.3.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klionsky DJ, Ohsumi Y. Vacuolar import of proteins and organelles from the cytoplasm. Annu Rev Cell Dev Biol. 1999;15:1–32. doi: 10.1146/annurev.cellbio.15.1.1. [DOI] [PubMed] [Google Scholar]
- Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, Ueno T, Koike M, Uchiyama Y, Kominami E, et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441:880–4. doi: 10.1038/nature04723. [DOI] [PubMed] [Google Scholar]
- Kovacs AL, Gordon PB, Grotterod EM, Seglen PO. Inhibition of hepatocytic autophagy by adenosine, adenosine analogs and AMP. Biol Chem. 1998;379:1341–7. doi: 10.1515/bchm.1998.379.11.1341. [DOI] [PubMed] [Google Scholar]
- Lunkes A, Lindenberg KS, Ben-Haiem L, Weber C, Devys D, Landwehrmeyer GB, Mandel JL, Trottier Y. Proteases acting on mutant huntingtin generate cleaved products that differentially build up cytoplasmic and nuclear inclusions. Mol Cell. 2002;10:259–69. doi: 10.1016/s1097-2765(02)00602-0. [DOI] [PubMed] [Google Scholar]
- Luo L, Liao YJ, Jan LY, Jan YN. Distinct morphogenetic functions of similar small GTPases: Drosophila Drac1 is involved in axonal outgrowth and myoblast fusion. Genes Dev. 1994;8:1787–802. doi: 10.1101/gad.8.15.1787. [DOI] [PubMed] [Google Scholar]
- Mizushima N, Yamamoto A, Hatano M, Kobayashi Y, Kabeya Y, Suzuki K, Tokuhisa T, Ohsumi Y, Yoshimori T. Dissection of autophagosome formation using Apg5-deficient mouse embryonic stem cells. J Cell Biol. 2001;152:657–68. doi: 10.1083/jcb.152.4.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nara A, Mizushima N, Yamamoto A, Kabeya Y, Ohsumi Y, Yoshimori T. SKD1 AAA ATPase-dependent endosomal transport is involved in autolysosome formation. Cell Struct Funct. 2002;27:29–37. doi: 10.1247/csf.27.29. [DOI] [PubMed] [Google Scholar]
- Narain Y, Wyttenbach A, Rankin J, Furlong RA, Rubinsztein DC. A molecular investigation of true dominance in Huntington's disease. J Med Genet. 1999;36:739–46. doi: 10.1136/jmg.36.10.739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal A, Severin F, Lommer B, Shevchenko A, Zerial M. Huntingtin-HAP40 complex is a novel Rab5 effector that regulates early endosome motility and is up-regulated in Huntington's disease. J Cell Biol. 2006;172:605–18. doi: 10.1083/jcb.200509091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravikumar B, Duden R, Rubinsztein DC. Aggregate-prone proteins with polyglutamine and polyalanine expansions are degraded by autophagy. Hum Mol Genet. 2002;11:1107–17. doi: 10.1093/hmg/11.9.1107. [DOI] [PubMed] [Google Scholar]
- Ravikumar B, Vacher C, Berger Z, Davies JE, Luo S, Oroz LG, Scaravilli F, Easton DF, Duden R, O'Kane CJ, et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet. 2004;36:585–95. doi: 10.1038/ng1362. [DOI] [PubMed] [Google Scholar]
- Rubinsztein DC. Lessons from animal models of Huntington's disease. Trends Genet. 2002;18:202–9. doi: 10.1016/s0168-9525(01)02625-7. [DOI] [PubMed] [Google Scholar]
- Rubinsztein DC, Gestwicki JE, Murphy LO, Klionsky DJ. Potential therapeutic applications of autophagy. Nat Rev Drug Discov. 2007;6:304–12. doi: 10.1038/nrd2272. [DOI] [PubMed] [Google Scholar]
- Ryder E, Blows F, Ashburner M, Bautista-Llacer R, Coulson D, Drummond J, Webster J, Gubb D, Gunton N, Johnson G, et al. The DrosDel collection: a set of P-element insertions for generating custom chromosomal aberrations in Drosophila melanogaster. Genetics. 2004;167:797–813. doi: 10.1534/genetics.104.026658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar S, Davies JE, Huang Z, Tunnacliffe A, Rubinsztein DC. Trehalose, a novel mTOR-independent autophagy enhancer,accelerates the clearance of mutant huntingtin and alpha-synuclein. J Biol Chem. 2006 doi: 10.1074/jbc.M609532200. [DOI] [PubMed] [Google Scholar]
- Sarkar S, Floto RA, Berger Z, Imarisio S, Cordenier A, Pasco M, Cook LJ, Rubinsztein DC. Lithium induces autophagy by inhibiting inositol monophosphatase. J Cell Biol. 2005;170:1101–11. doi: 10.1083/jcb.200504035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh A, Wang Y, Malsam J, Beard MB, Warren G. Golgin-84 is a rab1 binding partner involved in Golgi structure. Traffic. 2003;4:153–61. doi: 10.1034/j.1600-0854.2003.00103.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibata M, Lu T, Furuya T, Degterev A, Mizushima N, Yoshimori T, MacDonald M, Yankner B, Yuan J. Regulation of intracellular accumulation of mutant Huntingtin by Beclin 1. J Biol Chem. 2006;281:14474–85. doi: 10.1074/jbc.M600364200. [DOI] [PubMed] [Google Scholar]
- Shin HW, Hayashi M, Christoforidis S, Lacas-Gervais S, Hoepfner S, Wenk MR, Modregger J, Uttenweiler-Joseph S, Wilm M, Nystuen A, et al. An enzymatic cascade of Rab5 effectors regulates phosphoinositide turnover in the endocytic pathway. J Cell Biol. 2005;170:607–18. doi: 10.1083/jcb.200505128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenmark H, Parton RG, Steele-Mortimer O, Lutcke A, Gruenberg J, Zerial M. Inhibition of rab5 GTPase activity stimulates membrane fusion in endocytosis. Embo J. 1994;13:1287–96. doi: 10.1002/j.1460-2075.1994.tb06381.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki K, Kirisako T, Kamada Y, Mizushima N, Noda T, Ohsumi Y. The pre-autophagosomal structure organized by concerted functions of APG genes is essential for autophagosome formation. Embo J. 2001;20:5971–81. doi: 10.1093/emboj/20.21.5971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb JL, Ravikumar B, Atkins J, Skepper JN, Rubinsztein DC. Alpha-Synuclein is degraded by both autophagy and the proteasome. J Biol Chem. 2003;278:25009–13. doi: 10.1074/jbc.M300227200. [DOI] [PubMed] [Google Scholar]
- Wucherpfennig T, Wilsch-Brauninger M, Gonzalez-Gaitan M. Role of Drosophila Rab5 during endosomal trafficking at the synapse and evoked neurotransmitter release. J Cell Biol. 2003;161:609–24. doi: 10.1083/jcb.200211087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyttenbach A, Swartz J, Kita H, Thykjaer T, Carmichael J, Bradley J, Brown R, Maxwell M, Schapira A, Orntoft TF, et al. Polyglutamine expansions cause decreased CRE-mediated transcription and early gene expression changes prior to cell death in an inducible cell model of Huntington's disease. Hum Mol Genet. 2001;10:1829–45. doi: 10.1093/hmg/10.17.1829. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.