

Abstract
Excess dietary salt intake contributes to or exacerbates some forms of hypertension by increasing sympathetic nerve activity (SNA) and arterial blood pressure (ABP) through angiotensin II (Ang II) type 1 receptor activation in the rostral ventrolateral medulla (RVLM). Despite this interaction among dietary salt, Ang II, and the RVLM, no studies have directly examined whether dietary salt by itself alters Ang II–dependent responses and regulation of RVLM neurons, SNA, and ABP. Therefore, the present study directly tested this hypothesis. Male Sprague-Dawley rats were fed normal chow and given access to water or 0.9% NaCl solution for 14 days. Unilateral injection of Ang II (0.6, 6, and 60 pmol) into the RVLM produced a significantly greater increase in renal SNA and mean ABP of rats drinking 0.9% NaCl versus water. However, dietary salt did not alter mRNA levels of RVLM Ang II type 1a receptors or the SNA and ABP responses to stimulation of the dorsolateral funinculus. Additional experiments demonstrate that blockade of RVLM Ang II type 1 receptors significantly reduced renal SNA, splanchnic SNA, and mean ABP of rats drinking 0.9% NaCl but not water. Blockade of iontotropic glutamate receptors had no effect. Altogether, these findings suggest that elevated dietary salt enhances the sympathoexcitatory actions of Ang II in the RVLM via changes in the intrinsic properties of RVLM neurons. Moreover, elevated dietary salt intake differentially affects the tonic activity of the peripheral versus brain RVLM Ang II type 1 receptors to regulate baseline SNA and ABP.
Keywords: sympathetic nervous system, blood pressure, hypertension, brain, sodium
The brain rennin-angiotensin system plays an important role in cardiovascular regulation through its ability to modulate sympathetic nerve activity(SNA) and arterial blood pressure (ABP). One of the major centers postulated to mediate the sympathoexcitatory actions of brain angiotensin II (Ang II) is the rostral ventrolateral medulla (RVLM).1,2 The RVLM contains tonically active, bulbospinal neurons that mediate a number of sympathetically mediated reflexes and contribute to sympathoexcitatory disease states.3 The RVLM contains a high density of Ang II type 1 (AT1) receptors,1 and microinjection of Ang II into the RVLM produces an AT1 receptor–mediated increase in SNA and ABP.2 Overexpression of constitutively active AT1a receptors in the RVLM increases ABP,4 and blockade of RVLM AT1 receptors has been reported to reduce ABP in several forms of experimental hypertension5,6 and other sympathoexcitatory conditions.7,8
A major factor that contributes to the pathogenesis of hypertension and modulates the excitability of RVLM sympathetic-regulatory neurons is dietary salt intake. Evidence from both clinical and experimental models indicate that excess dietary salt intake exacerbates the level of hypertension, including Ang II–dependent models, via increases in sympathetic vasomotor tone.9,10 In several instances, findings from experimental models suggest that the elevated sympathetic tone depends on altered neurotransmission and AT1 receptor activation in the RVLM.5,6 However, few studies have directly examined whether excess dietary salt intake by itself affects the regulation of RVLM sympathetic-regulatory neurons. To date, the limited data indicate that elevated dietary salt intake enhances both sympathoexcitatory and sympathoinhibitory responses evoked by a number of neurotransmitters exogenously applied to the RVLM.11–13 However, these enhanced responses could not be attributed to changes in downstream sympathetic pathways13,14 or vascular reactivity.12,13,15
Despite the ability of dietary salt intake to modulate the excitability of RVLM neurons and the role of RVLM AT1 receptors in salt-dependent hypertension, no studies have determined whether dietary salt intake by itself alters Ang II–dependent responses or regulation of RVLM neurons, SNA, and ABP. The present findings indicate that excess dietary salt intake enhances the sympathoexcitatory responses evoked by exogenously applied Ang II in the RVLM without changes in mRNA levels for RVLM AT1 receptors or downstream sympathoexcitatory pathways. Additional experiments indicate that elevated dietary salt intake tonically activates RVLM AT1 receptors to maintain baseline SNA and ABP.
Materials and Methods
Animals
All of the experimental procedures conform to the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the University of Kentucky Institutional Animal Care and Use Committee. Male Sprague-Dawley rats (200 to 250 g; Charles River Laboratories) were housed in a temperature-controlled room (22±1°C) with a 14:10-hour light/dark cycle. Rats were fed standard rat chow containing 0.23% NaCl (Harlan Teklad Global Diet #2018) and given access to deionized water for ≥7 days before experiments began. Then rats were fed standard chow and given access to deionized water or 0.9% NaCl for 14 days. This protocol produces reliable and physiological increases in daily salt intake of rodents.11
General Procedures and RVLM Microinjections
Rats were anesthetized with a mixture of urethane (750 mg/kg IV) and α-chloralose (75 mg/kg IV) and prepared for recordings of renal or splanchnic SNA and ABP as described previously.11 Animals were artificially ventilated with oxygen-enriched room air and paralyzed with gallamine triethiodide (25 mg/kg per hour, 25 μL/h IV). End-tidal CO2 and body temperature were maintained at 4% to 4.5% and 37±1°C, respectively. An adequate depth of anesthesia was assessed by either the absence of a withdrawal reflex (before neuromuscular blockade) or a pressor response to foot pinch. Supplemental doses of anesthetic (10% initial dose) were given as necessary. RVLM microinjections were performed as described previously in our laboratory.11 Initially, L-glutamate (1 nmol) was injected into the RVLM at 3 different sites separated by 300 μm in the rostral–caudal plane to identify the site that produced the largest increase in ABP; subsequent injections were performed at these coordinates. For all experiments, injections (60 nL) were performed over 5 seconds. Injection sites were marked at the end of experiments with 0.2% rhodamine beads.
In the first set of experiments, Ang II (0.6, 6, and 60 pmol) was injected unilaterally into the RVLM. The Ang II dose was selected randomly, and 2 doses were tested per animal (1 per side). Injections were separated by a minimum of 30 minutes. In a second set of experiments, the AT1 receptor antagonist losartan (1 nmol/side), kynurenic acid (3 nmol/side), or isotonic saline was injected bilaterally into the RVLM. Each drug was tested in a separate group of rats. In kynurenic acid experiments, a blood sample (0.2 mL) was also collected from the arterial line for analysis of plasma electrolytes with an I-STAT1 analyzer and 6+ cartridges (Abbott; East Windsor, NJ). In preliminary experiments (n=4), the dose of losartan completely eliminated the sympathoexcitatory response to 60 pmol Ang II in control rats: renal SNA (control, 50±6% versus losartan, 0±0%; P<0.001) and mean ABP (control, 20±2 versus losartan, 0±0 mm Hg; P< 0.001). The dose of kynurenic acid was based on a previous study.16 To control for any effect of AT1 receptor blockade in the circulation, a third experiment administered the identical dose of losartan intravenously (2 nmol/200 μL).
Spinal Cord Stimulation
Rats were prepared for renal SNA and ABP recording as described above. The spinal cord was stabilized with a spinal clamp, and a pneumothorax was performed to limit respiratory movements. Current trains (5 seconds, 1-ms pulse; 300 μA) of various frequencies (10, 20, and 50 Hz) were delivered through a monopolar stimulating electrode lowered into the T1 dorsolateral funiculus. To quantify changes in renal SNA, spike-triggered averages of renal SNA were constructed from 60 sweeps (0.5 Hz; 1-ms pulse) at different stimulus intensities (50, 100, and 300 μA).
RT-PCR Analysis of Ang II Receptors
Rats were deeply anesthetized with 5% isoflurane and perfused transcardially with cold oxygenated artificial cerebrospinal fluid (124 mmol/L NaCl, 26 mmol/L NaHCO3, 0.6 mmol/L NaH2PO4,3 mmol/L KCl, 1.6 mmol/L MgCl2, 1.5 mmol/L CaCl2, 11 mmol/L glucose, pH 7.4). The brain stem was rapidly removed and sectioned at 300 μm in oxygenated artificial cerebrospinal fluid (4°C) using a vibratome. The RVLM was isolated under a microscope, immediately frozen in liquid nitrogen, and stored at −80°C. Additional samples were collected from the renal cortex.
Total RNA was isolated using TRIzol (Invitrogen; Carlsbad, Calif). All RNA samples were treated with TURBO DNase (Ambion; Austin, Tex) to remove genomic DNA. Non–RT-PCR using 50 ng of total RNA and Taqman Gene Expression Assay for β-actin confirmed DNase treatment successfully removed genomic DNA because no signal was detected after 40 cycles. First-strand cDNA synthesis from total RNA was performed using SuperScript III First-Strand Synthesis SuperMixRT (Invitrogen). Taqman Gene Expression Assays (Applied Biosystems; Foster City, Calif) were used to determine the receptor expression of AT1a (Agtr1a, Rn01435427_s1), AT1b (Agtr1b, Rn02132799_m1), and AT2 (Agtr2, Rn00560677_s1). Gene expression was normalized to 3 endogenous control genes β-actin (Actb, Rn00667869_m1), β-2 microglobulin (B2m, Rn00560865_m1), and hydroxymethylbilane synthase (Hmbs, Rn01421880_g1) and then quantified using the comparative CT method (ΔΔCT).
Data Analysis
All data are expressed as mean±SE. Changes in integrated SNA were calculated by subtracting background noise after hexamethonium (30 mg/kg IV). Successful recordings of renal or splanchnic SNA were not obtained in every experiment for technical reasons. For Ang II injections, the 1-second peak SNA and ABP response was compared with a 30-second baseline segment immediately before the injection. SNA responses to Ang II are only reported for those injections performed ipsilateral to the nerve isolation. Therefore, n values for ABP are much greater than SNA and are reported in the figure legends. Data from losartan and kynurenic acid experiments were averaged in 1-minute bins and compared with baseline values. For spinal cord stimulation studies, peak mean ABP (1 second) or renal SNA (10 ms) was compared with baseline values (30 seconds and 250 ms, respectively). All data were analyzed by a 1- or 2-way ANOVA with repeated measures when appropriate (time factor). All post hoc tests were performed with independent or paired t tests with a layered Bonferonni correction. A P<0.05 was statistically significant.
Results
The RVLM was initially identified by the site that produced the largest increase in ABP in response to L-glutamate. As reported previously,11,13 injection of glutamate into the RVLM of rats drinking 0.9% NaCl versus water evoked a significantly larger increase in renal SNA (253±4 versus 201±6%, respectively; P<0.01) and ABP (48±2 versus 31±3 mm Hg, respectively; P<0.01). There were no differences in baseline mean ABP, heart rate, and renal or splanchnic SNA between groups despite a significant elevation in 24-hour fluid ingestion and daily sodium intake (Table). The elevation in sodium intake did not alter hematocrit or plasma Na+, K+, and Cl− concentrations.
Table.
Characteristics of Rats Drinking Water or 0.9% NaCl for 14 Days
| Characteristic | Water | 0.9% NaCl |
|---|---|---|
| Body weight, g | 375±6 (50) | 383±4 (50) |
| Daily food intake, g | 28±1 (50) | 28±1 (50) |
| Daily ingested fluid, mL | 36±1 (50) | 57±2* (50) |
| Daily Na+ intake, mEq/day | 2.6±0.1 (50) | 11.6±0.3* (50) |
| Baseline mean ABP, mm Hg | 118±2 (45) | 116±2 (45) |
| Baseline heart rate, bpm | 397±5 (45) | 391±5 (45) |
| Baseline renal SNA, mV | 0.115±0.009 (42) | 0.123±0.009 (43) |
| Baseline splanchnic SNA, mV | 0.058±0.009 (15) | 0.046±0.004 (16) |
| Plasma Na+, mEq/L | 140±1 (5) | 139±1 (5) |
| Plasma K+, mEq/L | 4.7±0.2 (5) | 5.0±1.0 (5) |
| Plasma Cl−, mEq/L | 109±2 (5) | 107±1 (5) |
| Hematocrit, % | 43±1 (5) | 44±1 (5) |
Values (mean±SE) were pooled from all experiments including the online supplement. Value in parentheses indicates number of animals. Note that n values for renal and splanchnic SNA are smaller than those for mean ABP and heart rate because SNA recordings were not obtained in all experiments for technical reasons. Plasma electrolytes were analyzed in animals receiving injection of kynurenic acid.
Indicates significant difference from rats drinking water (P<0.001).
Elevated Dietary Salt Enhances SNA and ABP Response to Microinjection of Ang II Into the RVLM
A major goal of the present study was to determine whether elevated dietary salt intake enhanced the SNA and ABP responses evoked by Ang II in the RVLM. Injection of Ang II into the RVLM produced dose-dependent increases in renal SNA and ABP of both groups (Figure 1). However, rats drinking 0.9% NaCl versus water displayed a significantly greater increase in renal SNA and ABP after injection of 0.6 and 6 pmol Ang II (Figure 1). In fact, injection of 0.6 pmol Ang II in rats drinking 0.9% NaCl produced a similar increase in renal SNA and ABP of rats drinking water and receiving 6 pmol Ang II. There were no significant differences in renal SNA or mean ABP between groups at 60 pmol Ang II.
Figure 1.

A, Peak increase in mean ABP (n=8 to 13 per group per dose) and renal SNA (n=5 to 6 per dose per group) after injection of Ang II (0.6 and 6.0 pmol) into the RVLM was significantly greater in rats drinking 0.9% NaCl (^) vs water (○). SNA values are reported only for injections performed ipsilateral to the nerve recording. B, Individual example of ABP, mean ABP, and renal SNA during unilateral injection of Ang II (6 pmol) into the RVLM (Figure S1). * indicates significant difference between rats drinking water and 0.9% NaCl (P<0.01);▾, microinjection of Ang II.
To determine whether the enhanced SNA and ABP responses to RVLM injection of Ang II could be attributed to differences downstream of RVLM cell bodies, descending sympathoexcitatory spinal pathways were activated by electric stimulation of the dorsolateral funiculus. Rats drinking water or 0.9% NaCl displayed frequency-dependent increases in mean ABP (Figure 2); however, there were no significant differences in the magnitude of these responses between groups (P>0.7 from overall ANOVA). Spike-triggered averaging of renal SNA indicated that both groups had stimulus-dependent increases in renal SNA (Figure 2), but the magnitude of these increases were not different between rats drinking water versus 0.9% NaCl (P>0.6 from overall ANOVA).
Figure 2.

Top, Peak increase in mean ABP of rats drinking water (open; n=5) or 0.9% NaCl (filled; n=4) during electric stimulation (5-second train, 1-ms pulse) of the dorsolateral funiculus. Bottom, Peak increase in renal SNA of rats drinking water or 0.9% NaCl during electric stimulation of the dorsolateral funiculus (1 Hz; 1-ms pulse). There were no significant differences between groups.
Elevated Dietary Salt Does not Change AT1 mRNA Expression in the RVLM
In a separate set of experiments, we examined whether elevated dietary salt altered expression of AT1a, AT1b, or AT2 receptor levels in the RVLM. AT1a and AT2 receptor mRNA were expressed in the RVLM but not AT1b mRNA (CT values >35). However, there were no significant differences in mRNA levels of RVLM AT1a and AT2 receptors between rats drinking water versus 0.9% NaCl (Figure 3), despite a significant reduction of AT1a mRNA in the renal cortex of rats drinking 0.9% NaCl. These observations were consistent regardless of the housekeeping gene used for normalization and calculation of the 2−(ΔΔCt) values (data not shown for β2 microglobulin or hydroxymethylbilane synthase). Dietary salt did not alter the expression of any housekeeping gene (data not shown).
Figure 3.
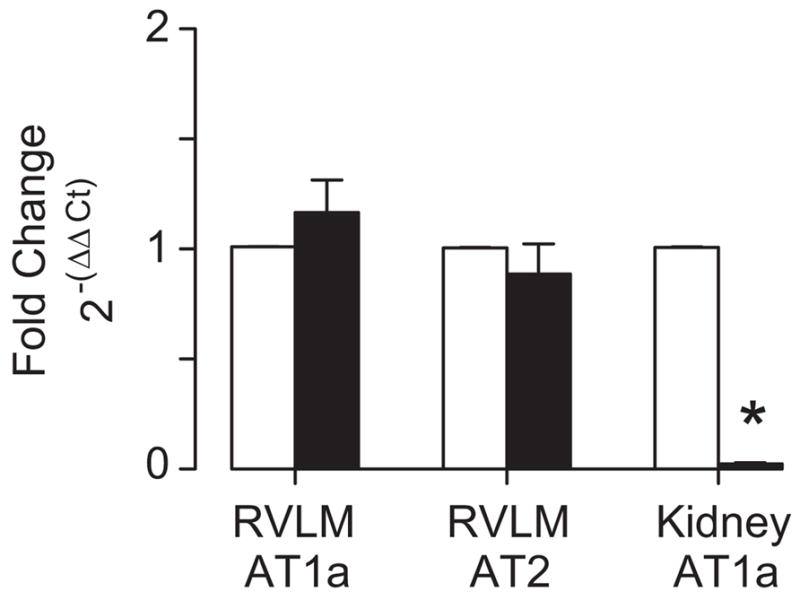
Quantitative RT-PCR of AT1a and AT2 receptor expression in the RVLM and AT1a receptor expression in the renal cortex of rats drinking water (open; n=5) or 0.9% NaCl (filled; n=5) for 14 days. Values were calculated using the 2−(ΔΔCt) method and normalized to β-actin. * indicates significant difference from rats drinking water (P<0.01).
Bilateral Injection of Losartan, but not Kynurenic Acid, Lowers SNA and ABP in Rats Drinking 0.9% NaCl
A final set of experiments examined whether elevated dietary salt intake altered the contribution of AT1 receptor activation to the maintenance of baseline SNA and ABP. Bilateral injection of the AT1 receptor antagonist losartan into the RVLM of rats drinking water did not significantly alter mean ABP and renal SNA from baseline values (P<0.05 for time factor) but did increase splanchnic SNA (P<0.05; Figure 4A) at 2 to 30 minutes. In marked contrast, blockade of RVLM AT1 receptors in rats drinking 0.9% NaCl significantly decreased mean ABP, renal SNA, and splanchnic SNA below baseline values and gradually returned after 30 minutes. In fact, mean ABP, renal SNA, and splanchnic SNA was significantly lower in rats drinking 0.9% NaCl versus water after injection of losartan. Although losartan tended to transiently increase renal and splanchnic SNA in rats drinking 0.9% NaCl, this increase was only significant at 2 minutes for splanchnic SNA. In contrast to the effects of losartan, bilateral injection of the ionotropic glutamate antagonist kynurenic acid did not alter any of these variables in either group (P>0.6 from overall ANOVA for all factors; Figure 4B). Injection of losartan or kynurenic acid into the RVLM did not alter heart rate in either group (P>0.8 from overall ANOVA).
Figure 4.

Mean ABP, renal SNA, and splanchnic SNA response to bilateral injection of losartan (n=11 per group; A) or kynurenic acid (n=5 per group; B) into the RVLM of rats drinking water (○) or 0.9% NaCl (^) for 14 days. Injections were performed at 0 minutes and separated by 2 to 3 minutes. SNA values are based on a smaller number of animals (losartan, n≥7 per group; kynurenic acid, n≥4). * indicates significant difference between groups (P<0.05).
Injection of isotonic saline into the RVLM or the identical dose of losartan intravenously did not affect any variable in either group (Figure S2, available online at http://www.hypertensionaha.org).
Histology
All injection sites were centered in the RVLM defined as the triangular region located 0 to 600 μm caudal to the caudal pole of the facial nucleus and bordered dorsally by nucleus ambiguus, medially by the inferior olive or pyramidal tracts, and laterally by the spinal trigeminal nucleus (Figure S3).
Discussion
Excess dietary salt intake contributes to or exacerbates many forms of hypertension, including Ang II–dependent models, by increasing SNA and vasomotor tone.9,10 Evidence in anesthetized animals indicates that the elevated SNA and ABP depend, in part, on AT1 receptor activation in the RVLM.5,6 Despite the interaction among dietary salt, Ang II, and the RVLM in the pathogenesis of hypertension, no studies have directly examined whether dietary salt by itself alters Ang II–dependent regulation of RVLM neurons, SNA, and ABP. The present study provides several novel observations: (1) elevated dietary salt intake enhances the SNA and ABP responses to exogenously applied Ang II in the RVLM; (2) these exaggerated sympathoexcitatory responses were not associated with changes in RVLM AT1a mRNA levels or the excitability of downstream sympathoexcitatory pathways; and (3) blockade of RVLM AT1, but not iontotropic glutamate, receptors significantly lowered SNA and ABP in rats drinking 0.9% NaCl. Altogether, these findings suggest that elevated dietary salt enhances the sympathoexcitatory actions of Ang II in the RVLM via changes in the intrinsic properties of RVLM neurons. Furthermore, excess dietary salt intake activates an angiotensinergic pathway to the RVLM to regulate baseline SNA and ABP.
Injection of Ang II into the RVLM produces an AT1 receptor–mediated increase in SNA and ABP.1,2 These sympathoexcitatory effects are likely attributable to direct postsynaptic actions because Ang II–induced depolarization of bulbospinal RVLM neurons persists after synaptic blockade.17,18 Although Ang II has been reported to presynaptically modulate glutamate release in other brain regions,2 the available data for the RVLM,5,19 including this study, indicate that AT1 and glutamatergic receptors independently regulate the activity of RVLM neurons. Interestingly, the present findings provide strong evidence that excess dietary salt intake enhances the sympathoexcitatory actions of Ang II (and L-glutamate). In fact, the plot of the SNA and ABP responses to Ang II (Figure 1) suggest the sensitivity, not the maximal response, has changed. These exaggerated responses cannot be attributed to an alteration in downstream spinal sympathetic pathways or neuroeffector junctions because electric stimulation of the dorsolateral funiculus produced similar increases in both renal SNA and ABP of rats drinking water versus 0.9% NaCl. Moreover, several investigators have demonstrated that dietary salt does not alter vascular reactivity.12,13,15
The mechanism(s) underlying the greater responsiveness of RVLM neurons during elevated dietary salt intake is not known. Although changes in dietary salt intake have been reported previously to alter brain AT1 mRNA levels,20–22 analysis of RVLM AT1a (and AT2) mRNA levels in the present study revealed no difference between rats drinking water versus 0.9% NaCl. Thus far, we have been unsuccessful extending this observation to the protein level because Western blot analyses reveal dense bands at the predicted molecular weight of ≈45 kDa in the RVLM and renal cortex, but these bands were still present in the renal cortex of AT1a−/− mice and the brain region dorsolateral to the RVLM in rats, where no Ang II binding has been reported23 (Figure S4). A second noteworthy observation is that excess dietary salt intake enhances the sympathoexcitatory and sympathoinhibitory responses to a number of neurotransmitters (>5) in the RVLM.11–13 Therefore, elevated dietary salt either alters the expression of every receptor or, more likely, alters the intrinsic excitability of RVLM sympathetic-regulatory neurons. Such possibilities may include a general change in membrane conductance (ie, potassium) because this would permit enhanced responses to both excitatory and inhibitory synaptic inputs.
Blockade of AT1 receptors in the RVLM decreases ABP in models of experimental hypertension including Dahl salt–sensitive hypertensive rats.5,6 The present findings indicate that excess dietary salt by itself increases the contribution of RVLM AT1 receptor activation to the maintenance of baseline SNA and ABP. This seems at odds with a previous report that systemic administration of losartan decreases ABP in animals maintained on a low-salt but not a high-salt diet.24 However, the dose of systemic losartan needed to block RVLM AT1 receptors has not been defined, and the hypotensive actions of systemic losartan in animals on a low-salt diet may be attributed to tissues other than the RVLM. Despite a greater activation of AT1 receptors in the RVLM of rats drinking 0.9% NaCl, baseline SNA and ABP were not different between groups. The greater AT1 receptor activation may be offset by a withdrawal of other excitatory inputs or an increase in inhibitory input. The transient increase in SNA and ABP observed in rats drinking water or 0.9% NaCl after losartan injection is likely attributed to the potassium salt. However, SNA tended to remain above baseline levels in control rats. The explanation for this observation is not clear, but previous studies have indicated that bilateral injection of losartan into the RVLM did not change8,19 or increased25 SNA or ABP in normal animals.
One intriguing question that arises from the current findings is how does excess dietary salt increase AT1 receptor activation in the RVLM? The ability of AT1 receptor blockade in the RVLM to lower SNA and ABP is reminiscent to those findings reported in water-deprived rats with elevated plasma sodium concentration.7 Although we did not observe differences in plasma sodium concentration between groups, excess dietary salt intake has been reported to increase plasma sodium concentration in rodents at night26 and humans.27 Interestingly, preliminary data from our laboratory indicate that lesion of osmosensitive sites in ventral forebrain lamina terminalis prevents the salt-induced changes in the responsiveness of RVLM neurons.28 Yet, how does activation of osmotically sensitive sites in the forebrain increase AT1 receptor activation in the RVLM? First, forebrain osmosensitive regions densely innervate neurons of the hypothalamic paraventricular nucleus (PVH),29 and acute hyperosmolality increases the discharge of RVLM-projecting PVH neurons.30 Second, previous studies have reported that PVH neurons are Ang II immunoreactive,31 and the sympathoexcitatory response produced by disinhibition of the PVH is significantly attenuated by blockade of AT1 receptors in the RVLM.19 Thus, excess dietary salt may activate osmosensitive neurons in the forebrain lamina terminalis to drive an angiotensinergic pathway from the PVH to the RVLM. At this time, we cannot exclude the possibility that dietary salt activates other afferent pathways or sources of Ang II input to the RVLM.
Previous studies have reported that sympathetic outflow is inversely related to the level of dietary salt intake,32–35 but the present study did not observe any difference in baseline renal and splanchnic SNA or ABP between groups. The reason for the differences between our findings and previous studies is unclear but may be related to the experimental conditions or species studied. However, our findings are consistent with a recent report in which chronic recordings of renal SNA and ABP in awake rabbits did not reveal any effect of dietary salt on baseline sympathetic vasomotor activity or ABP.36 Regardless of the effect of dietary salt intake on baseline SNA, the present findings together with previous reports11–13 suggest that dietary salt intake adjusts the gain or responsiveness of RVLM neurons. Therefore, a small increase in excitatory drive to the RVLM now produces an exaggerated increase in SNA and ABP. In fact, activation of somatic afferents produces a significantly greater increase in ABP of animals maintained on a high-salt diet;12 this response depends on glutamatergic neurotransmission in the RVLM.16 Similarly, dietary salt has been reported to potentiate the sympathoexcitatory response to hyperinsulinemia.37 These observations indicate that these functional changes in the responsiveness of RVLM sympathoexcitatory neurons have physiological significance in the regulation of SNA and ABP. Future studies will need to extend these observations and identify the level of dietary salt intake needed for such an effect; however, we suspect that the responsiveness of RVLM neurons is proportional to the level of dietary salt rather than a threshold because both dietary salt restriction and loading decreases or enhances, respectively, the pressor responses evoked from the RVLM.12
Perspectives
The present findings identify a new factor that influences the potency of the brain renin-angiotensin system in sympathetic-regulatory networks. This is particularly intriguing because excess dietary salt inhibits peripheral renin secretion, thereby suggesting that the excess dietary salt differentially modulates the activity of the peripheral versus brain renin-angiotensin system. Based on these findings, administration of AT1 receptor blockers at doses that functionally block RVLM AT1 receptors should reduce SNA and ABP in salt-sensitive hypertensive subjects and experimental models of hypertension. In fact, blockade of AT1 receptors in the RVLM of the Dahl salt–sensitive hypertensive rats lowered ABP.5 Subsequent identification of the factors that underlie the ability of dietary salt to alter the intrinsic excitability of RVLM sympathetic neurons during changes in dietary salt intake may represent a novel therapeutic target for the treatment of salt-sensitive hypertension.
Supplementary Material
Acknowledgments
We thank Dr Christopher Madden for constructive comments on this manuscript.
Sources of Funding
This research was supported by an American Heart Association scientist development grant (S.D.S) and postdoctoral fellowship (J.M.A.), National Institutes of Health Heart Lung and Blood Institute grant HL090826 (S.D.S.), and National Institute of Arthritis and Musculoskeletal and Skin Diseases grant AR053641 (J.J.M.).
Footnotes
Disclosures
None.
Publisher's Disclaimer: This is an un-copyedited author manuscript that was accepted for publication in Hypertension, copyright The American Heart Association. This may not be duplicated or reproduced, other than for personal use or within the “Fair Use of Copyrighted Materials” (section 107, title 17, U.S. Code) without prior permission of the copyright owner, The American Heart Association. The final copyedited article, which is the version of record, can be found at Hypertension. The American Heart Association disclaims any responsibility or liability for errors or omissions in this version of the manuscript or in any version derived from it by the National Institutes of Health or other parties.
References
- 1.Allen AM, Moeller I, Jenkins TA, Zhuo J, Aldred GP, Chai SY, Mendelsohn FA. Angiotensin receptors in the nervous system. Brain Res Bull. 1998;47:17–28. doi: 10.1016/s0361-9230(98)00039-2. [DOI] [PubMed] [Google Scholar]
- 2.Dampney RA, Tan PS, Sheriff MJ, Fontes MA, Horiuchi J. Cardiovascular effects of angiotensin II in the rostral ventrolateral medulla: the push-pull hypothesis. Curr Hypertens Rep. 2007;9:222–227. doi: 10.1007/s11906-007-0040-4. [DOI] [PubMed] [Google Scholar]
- 3.Guyenet PG. The sympathetic control of blood pressure. Nat Rev Neurosci. 2006;7:335–346. doi: 10.1038/nrn1902. [DOI] [PubMed] [Google Scholar]
- 4.Allen AM, Dosanjh JK, Erac M, Dassanayake S, Hannan RD, Thomas WG. Expression of constitutively active angiotensin receptors in the rostral ventrolateral medulla increases blood pressure. Hypertension. 2006;47:1054–1061. doi: 10.1161/01.HYP.0000218576.36574.54. [DOI] [PubMed] [Google Scholar]
- 5.Ito S, Hiratsuka M, Komatsu K, Tsukamoto K, Kanmatsuse K, Sved AF. Ventrolateral medulla AT1 receptors support arterial pressure in Dahl salt-sensitive rats. Hypertension. 2003;41:744–750. doi: 10.1161/01.HYP.0000052944.54349.7B. [DOI] [PubMed] [Google Scholar]
- 6.Ito S, Komatsu K, Tsukamoto K, Kanmatsuse K, Sved AF. Ventrolateral medulla AT1 receptors support blood pressure in hypertensive rats. Hypertension. 2002;40:552–559. doi: 10.1161/01.hyp.0000033812.99089.92. [DOI] [PubMed] [Google Scholar]
- 7.Freeman KL, Brooks VL. AT(1) and glutamatergic receptors in paraventricular nucleus support blood pressure during water deprivation. Am J Physiol Regul Integr Comp Physiol. 2007;292:R1675–R1682. doi: 10.1152/ajpregu.00623.2006. [DOI] [PubMed] [Google Scholar]
- 8.Mayorov DN, Head GA. AT1 receptors in the RVLM mediate pressor responses to emotional stress in rabbits. Hypertension. 2003;41:1168–1173. doi: 10.1161/01.HYP.0000064574.29317.45. [DOI] [PubMed] [Google Scholar]
- 9.Brooks VL, Haywood JR, Johnson AK. Translation of salt retention to central activation of the sympathetic nervous system in hypertension. Clin Exp Pharmacol Physiol. 2005;32:426–432. doi: 10.1111/j.1440-1681.2005.04206.x. [DOI] [PubMed] [Google Scholar]
- 10.Osborn JW, Fink GD, Sved AF, Toney GM, Raizada MK. Circulating angiotensin II and dietary salt: converging signals for neurogenic hypertension. Curr Hypertens Rep. 2007;9:228–235. doi: 10.1007/s11906-007-0041-3. [DOI] [PubMed] [Google Scholar]
- 11.Adams JM, Madden CJ, Sved AF, Stocker SD. Increased dietary salt enhances sympathoexcitatory and sympathoinhibitory responses from the rostral ventrolateral medulla. Hypertension. 2007;50:354–359. doi: 10.1161/HYPERTENSIONAHA.107.091843. [DOI] [PubMed] [Google Scholar]
- 12.Ito S, Gordon FJ, Sved AF. Dietary salt intake alters cardiovascular responses evoked from the rostral ventrolateral medulla. Am J Physiol Regul Integr Comp Physiol. 1999;276:R1600–R1607. doi: 10.1152/ajpregu.1999.276.6.R1600. [DOI] [PubMed] [Google Scholar]
- 13.Pawloski-Dahm CM, Gordon FJ. Increased dietary salt sensitizes vasomotor neurons of the rostral ventrolateral medulla. Hypertension. 1993;22:929–933. doi: 10.1161/01.hyp.22.6.929. [DOI] [PubMed] [Google Scholar]
- 14.Bush EN, Vollmer RR. Reduced sympathetic responsiveness resulting from a high dietary sodium intake in the rat. Clin Exp Hypertens A. 1983;5:759–774. doi: 10.3109/10641968309081806. [DOI] [PubMed] [Google Scholar]
- 15.Kaufman LJ, Vollmer RR. Low sodium diet augments plasma and tissue cate-cholamine levels in pithed rats. Clin Exp Hypertens A. 1984;6:1543–1558. doi: 10.3109/10641968409044068. [DOI] [PubMed] [Google Scholar]
- 16.Kiely JM, Gordon FJ. Role of rostral ventrolateral medulla in centrally mediated pressor responses. Am J Physiol Heart Circ Physiol. 1994;267:H1549–H1556. doi: 10.1152/ajpheart.1994.267.4.H1549. [DOI] [PubMed] [Google Scholar]
- 17.Li YW, Guyenet PG. Neuronal excitation by angiotensin II in the rostral ventrolateral medulla of the rat in vitro. Am J Physiol Regul Integr Comp Physiol. 1995;268:R272–R277. doi: 10.1152/ajpregu.1995.268.1.R272. [DOI] [PubMed] [Google Scholar]
- 18.Li YW, Guyenet PG. Angiotensin II decreases a resting K+ conductance in rat bulbospinal neurons of the C1 area. Circ Res. 1996;78:274–282. doi: 10.1161/01.res.78.2.274. [DOI] [PubMed] [Google Scholar]
- 19.Tagawa T, Dampney RA. AT(1) receptors mediate excitatory inputs to rostral ventrolateral medulla pressor neurons from hypothalamus. Hypertension. 1999;34:1301–1307. doi: 10.1161/01.hyp.34.6.1301. [DOI] [PubMed] [Google Scholar]
- 20.Chen Y, Liu-Stratton Y, Hassanain H, Cool DR, Morris M. Dietary sodium regulates angiotensin AT1a and AT1b mRNA expression in mouse brain. Exp Neurol. 2004;188:238–245. doi: 10.1016/j.expneurol.2004.04.005. [DOI] [PubMed] [Google Scholar]
- 21.Sandberg K, Ji H, Catt KJ. Regulation of angiotensin II receptors in rat brain during dietary sodium changes. Hypertension. 1994;23:I137–I141. doi: 10.1161/01.hyp.23.1_suppl.i137. [DOI] [PubMed] [Google Scholar]
- 22.Wang JM, Veerasingham SJ, Tan J, Leenen FH. Effects of high salt intake on brain AT1 receptor densities in Dahl rats. Am J Physiol Heart Circ Physiol. 2003;285:H1949–H1955. doi: 10.1152/ajpheart.00744.2002. [DOI] [PubMed] [Google Scholar]
- 23.Song K, Allen AM, Paxinos G, Mendelsohn FA. Mapping of angiotensin II receptor subtype heterogeneity in rat brain. J Comp Neurol. 1992;316:467–484. doi: 10.1002/cne.903160407. [DOI] [PubMed] [Google Scholar]
- 24.Collister JP, Hornfeldt BJ, Osborn JW. Hypotensive response to losartan in normal rats. Role of Ang II and the area postrema. Hypertension. 1996;27:598–606. doi: 10.1161/01.hyp.27.3.598. [DOI] [PubMed] [Google Scholar]
- 25.Fontes MA, Martins Pinge MC, Naves V, Campagnole-Santos MJ, Lopes OU, Khosla MC, Santos RA. Cardiovascular effects produced by micro-injection of angiotensins and angiotensin antagonists into the ventrolateral medulla of freely moving rats. Brain Res. 1997;750:305–310. doi: 10.1016/s0006-8993(96)01476-x. [DOI] [PubMed] [Google Scholar]
- 26.Habecker BA, Grygielko ET, Huhtala TA, Foote B, Brooks VL. Ganglionic tyrosine hydroxylase and norepinephrine transporter are decreased by increased sodium chloride in vivo and in vitro. Auton Neurosci. 2003;107:85–98. doi: 10.1016/S1566-0702(03)00133-4. [DOI] [PubMed] [Google Scholar]
- 27.He FJ, Markandu ND, Sagnella GA, de Wardener HE, MacGregor GA. Plasma sodium: ignored and underestimated. Hypertension. 2005;45:98–102. doi: 10.1161/01.HYP.0000149431.79450.a2. [DOI] [PubMed] [Google Scholar]
- 28.Stocker SD, Adams JM. Forebrain lamina terminalis mediates the enhanced responsiveness of neurons in the rostral ventrolateral medulla during increased dietary salt. Hypertension. 2007;50:e83. [Google Scholar]
- 29.Stocker SD, Osborn JL, Carmichael SP. Forebrain osmotic regulation of the sympathetic nervous system. Clin Exp Pharmacol Physiol. 2008;35:695–700. doi: 10.1111/j.1440-1681.2007.04835.x. [DOI] [PubMed] [Google Scholar]
- 30.Toney GM, Chen QH, Cato MJ, Stocker SD. Central osmotic regulation of sympathetic nerve activity. Acta Physiol Scand. 2003;177:43–55. doi: 10.1046/j.1365-201X.2003.01046.x. [DOI] [PubMed] [Google Scholar]
- 31.Lind RW, Swanson LW, Ganten D. Organization of angiotensin II immunoreactive cells and fibers in the rat central nervous system. An immunohistochemical study. Neuroendocrinology. 1985;40:2–24. doi: 10.1159/000124046. [DOI] [PubMed] [Google Scholar]
- 32.Anderson EA, Sinkey CA, Lawton WJ, Mark AL. Elevated sympathetic nerve activity in borderline hypertensive humans. Evidence from direct intraneural recordings. Hypertension. 1989;14:177–183. doi: 10.1161/01.hyp.14.2.177. [DOI] [PubMed] [Google Scholar]
- 33.DiBona GF, Jones SY, Sawin LL. Effect of endogenous angiotensin II on renal nerve activity and its arterial baroreflex regulation. Am J Physiol Regul Integr Comp Physiol. 1996;271:R361–R367. doi: 10.1152/ajpregu.1996.271.2.R361. [DOI] [PubMed] [Google Scholar]
- 34.Friberg P, Meredith I, Jennings G, Lambert G, Fazio V, Esler M. Evidence for increased renal norepinephrine overflow during sodium restriction in humans. Hypertension. 1990;16:121–130. doi: 10.1161/01.hyp.16.2.121. [DOI] [PubMed] [Google Scholar]
- 35.Grassi G, Dell’Oro R, Seravalle G, Foglia G, Trevano FQ, Mancia G. Short- and long-term neuroadrenergic effects of moderate dietary sodium restriction in essential hypertension. Circulation. 2002;106:1957–1961. doi: 10.1161/01.cir.0000033519.45615.c7. [DOI] [PubMed] [Google Scholar]
- 36.McBryde FD, Barrett C, Guild SJ, Osborn JW, Van Vliet BN, Malpas SC. 2008 Experimental Biology Meeting Abstracts [on CD-ROM] 2008. Increasing dietary salt alone does not affect chronic levels of renal sympathetic nerve activity or the response to stressful stimuli. Abstract #738.8. [Google Scholar]
- 37.Muntzel MS, Crespo R, Joseph T, Onwumere O. Dietary salt loading exacerbates the increase in sympathetic nerve activity caused by IV insulin infusion in rats. Metabolism. 2007;56:373–379. doi: 10.1016/j.metabol.2006.10.020. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.