

Abstract
It is a goal of cancer chemotherapy to achieve the selective killing of tumor cells while minimizing toxicity to normal tissues. We describe the design of selective toxins forming DNA adducts that attract the estrogen receptor (ER), a transcription factor that is overexpressed in many human breast and ovarian tumors. The compounds consist of 4-(3-aminopropyl)-N,N-(2-chloroethyl)-aniline linked to 2-(4′-hydroxyphenyl)-3-methyl-5-hydroxy-indole. The former moiety is a DNA damaging nitrogen mustard and the latter is a ligand for the ER. The connection between these groups was refined to permit DNA adducts formed by the mustard portion of the molecule to present the ligand domain so that it was able to interact efficiently with the ER. By using 16-mers containing specific DNA adducts, it was determined that monoadducts and putative intrastrand crosslinks were preferred targets for the ER over interstrand crosslinks. A series of structurally related 2-phenylindole mustards was prepared, some of which were selectively toxic to the ER-positive breast cancer cell line MCF-7, as compared with the ER(−) negative line MDA-MB231. The ability both to bind to DNA and to interact significantly with the ER were essential to achieve selective lethality toward ER(+) cells. Compounds forming DNA adducts without the ability to bind receptor showed similar toxicities in the two cell lines. Several models could explain the selective toxicity of the mustard–phenylindole compounds toward ER(+) cells. The favored model suggests that a mustard–DNA adduct is shielded by the ER from DNA repair enzymes and hence cells possessing an abundance of the ER selectively retain the adduct and are killed.
Keywords: molecular recognition, DNA repair, breast cancer
DNA repair proteins recognize genetic damage by chemicals and radiation (1). The protein–DNA lesion-recognition event is the prelude to enzymatic reaction(s) restoring the structural and informational integrity of the genome. It recently came as a surprise when proteins not involved in DNA repair were discovered that bind to specific DNA lesions formed by some chemical carcinogens (2) and anticancer drugs (3–7). The discovery that most of these lesion-binding proteins are transcription factors suggests some similarity between the structure of DNA in the vicinity of certain types of DNA damage and the architectural features of DNA recognized by the proteins that affect gene expression. The binding of transcription factors to DNA lesions can be tight (Kd values in the picomolar range have been observed), suggesting that these protein–adduct interactions might negatively affect cellular welfare. For example, the binding may be so tight that DNA repair proteins are unable to access the lesion (Fig. 1B), resulting in lesion persistence and accompanying toxicity (5, 8). It is also possible that the adducts may disrupt gene expression by titrating or “hijacking” transcription factors from normal sites of action (9). If the transcription factor is necessary for cellular survival or growth, its diminished concentration could result in death or retarded progression through the cell cycle (Fig. 1C).
Figure 1.

DNA adducts act as decoy binding sites for nuclear proteins: possible biological consequences. (A) A normal cell with a nuclear protein [the estrogen receptor (ER) in this example] interacting with DNA in a sequence specific manner. (B) Shielding model. DNA adducts (lollipop symbol) form (typically 104 per cell) and they attract the nuclear protein. The adduct is shielded from DNA repair enzymes, promoting the longevity of the adduct and sensitizing the cell to the toxin. (C) Hijacking model. The adducts titrate the nuclear protein away from its normal site of binding, resulting in diminished expression of an important gene.
The models described above were developed to explain components of the toxicity profile of known DNA damaging agents (2, 3, 9). These models also present opportunities for the rational design of novel selective toxins. As an exercise in molecular recognition, and to begin testing the models above, we set out to design a genotoxin capable of forming a DNA lesion that would attract a transcription factor. If successful in vitro, it would also be possible to determine if the interaction between the DNA adduct and the transcription factor in vivo would affect the growth or viability of specific cells. The model system described here focused upon ER, a transcription factor modulated in vivo by endogenous estrogens (10). The ER is expressed at high levels in many breast and ovarian tumors (11, 12) and the abundance of this protein is exploited in diagnosis (13) and in hormonal therapies against breast cancer (14). Derivatives of 2-phenylindole (2PI) interact well with the ER in competition assays with the natural ligand, estradiol (15). In this work, derivatives of 2PI were linked to an aniline mustard whose chemistry and ability to alkylate DNA are well understood (16). A means of linking the ER recognition domain to the mustard was discovered, which preserved the ability of the recognition domain to interact with the ER, even after the mustard had formed covalent lesions with DNA.
MATERIALS AND METHODS
Synthesis and Characterization of Phenylindole–Aniline Mustards.
The conjugate compounds, with the exception of 2PI(OH)-C6NC2, were composed of 2-(4′-hydroxyphenyl)-3-methyl-5-hydroxy-indole linked to 4-(3-aminopropyl)-N,N-(2-chloroethyl)-aniline. The 2PI domain was prepared as described (15); its hydroxyl groups were protected as methylmethoxy ethers. Reaction of the sodium salt of the protected phenylindole with an excess of the corresponding alkyldibromide provided a bromoalkylphenylindole. The amino group of an O-tert-butyldimethylsilyl-protected amino alcohol was protected as the diphenylphosphinamide and coupled to the bromoalkylphenylindole as described (17). The aniline mustard moiety, 4-(3-aminopropyl)-N,N-(2-chloroethyl)-aniline (18), was then coupled to the 2PI linker by tetra-n-butylammonium fluoride deprotection of the alcohol and reaction with p-nitrobenzyl chloroformate. The final compounds were obtained by removal of the diphenylphosphinic and methylmethoxy ether protecting groups with 2 N HCl in MeOH at ambient temperature. All new compounds were characterized by high-resolution field-desorption mass spectrometry (MS), as well as 300 MHz 1H NMR spectroscopy. Data for one representative compound are given as follows. High-resolution field-desorption MS for 2PI-C6NC2 mustard (C37H48N4O4Cl2); m/z 682.30558. 1H NMR; (CD3OD): δ = 1.04–1.68 (m, 8H, CH2), 1.70 (t, J = 7 Hz, 2H, CH2), 2.07 (s, 3H, CCH3), 2.48 (t, J = 7 Hz, 2H, N-CH2), 2.65 (t, J = 7 Hz, 2H, N-CH2), 2.97–3.11 (m, 4H, N-CH2), 3.41–3.44 (m, 2H, Ar-CH2), 3.54–3.64 (m, 8H, -CH2CH2Cl), 3.94 (t, J = 7 Hz, 2H, CONH-CH2), 4.17 (t, 2H, J = 7 Hz, CO2-CH2), 6.58–7.15 (m, 11H, ArH).
Relative Affinity of Compounds for the ER.
The relative affinities of the 2PI–mustard compounds for the calf uterine ER were measured by a competitive binding assay (19) with 17β-[3H]estradiol (New England Nuclear). The dextran-coated charcoal method was used to determine the ratio of the molar concentrations of 17β-estradiol and the test compound necessary to reduce receptor-bound radioactivity by 50%. The relative binding affinity (RBA) is that molar ratio × 100.
Preparation and Analysis of Mustard–DNA Adducts.
The self-complementary oligonucleotide 5′-d(AATATTGGCCAATATT)-3′ was synthesized on an Applied Biosystems model 391, purified by denaturing PAGE (20%/19:1 acrylamide/bisacrylamide/40% urea), and 5′ end-labeled with [γ-32P]ATP (6000 Ci/μmol) using T4 polynucleotide kinase (10 units) or 3′-labeled with [α-32P]dTTP (6000 Ci/μmol) using Klenow DNA polymerase (20). Duplex DNA (3.25 nmol) in 40 mM sodium cacodylate buffer (pH 8) was allowed to react for 15 h at 37°C with the respective mustards [dissolved in dimethyl sulfoxide (DMSO)] at a concentration of 0.1 mM; final volume was 40 μl containing 10% DMSO. DNA products were recovered by ethanol precipitation. After denaturation in 90% formamide, 1 mM EDTA (pH 8) at ambient temperature, modified DNAs were separated by using a 20% denaturing gel at 30°C. Quantitation of reaction products was obtained from two-dimensional scans by using a PhosphorImager (Molecular Dynamics). After recovery from denaturing gels, DNAs labeled at the 3′ or 5′ ends were treated with piperidine (1 M, 90°C, 1 h). In some experiments, DNAs were heated at 70°C for 1 h at neutral pH prior to piperidine treatment to produce apurinic sites at any N3-modified adenines. Sites of alkylation were determined by comparison of fragment sizes to those produced by the Maxam–Gilbert sequencing reactions for G or G+A bases (21).
Preparative reactions (4 ml) containing 125 A260 units of duplex DNA, 40 mM sodium cacodylate buffer (pH 8), 0.24 mM test compound, and 10% DMSO were incubated at 37°C for 19 h. DNA was recovered by ethanol precipitation, resuspended in formamide at 25°C, and analyzed by 20% denaturing PAGE. Separated DNA bands were visualized by UV shadowing, excised and eluted from the gel in 0.5 M NH4OAc, 10 mM Mg(OAc)2, 1.0 mM EDTA (pH 8) using a crush-and-soak technique at ambient temperature (20). Recovered DNAs were loaded onto a C18 Sep-Pak (Waters) and desalted products were eluted with 25% (aq) CH3CN and dried in vacuo.
The identity of the major adduct formed by the reaction of 2PI-C6NC2 with DNA in vitro was investigated after hydrolysis of modified calf thymus DNA with 0.1 N HCl at 70°C for 1 h. Hydrolysis products, isolated by C18 Sep-Pak followed by reversed-phase HPLC, were analyzed by electrospray MS using a Hewlett–Packard 5989B in the positive ion mode. The sample was introduced by direct infusion (20 μl/min) in a mixture of methanol and water (90:10).
Toxicity of Phenylindole–Mustard Compounds.
Toxicity experiments were performed on breast cancer cell lines MCF-7 and MDA-MB231 obtained from the American Type Culture Collection. Both cell lines were grown in MEM containing phenol red/10% fetal calf serum supplemented with 2 mM glutamine and 2 mM pyruvate in a 5% CO2/95% air atmosphere. Cells were free of mycoplasma. Cells were seeded at 2 × 105 in 6-well plates and, 24 h later, exposed to test compounds in growth medium for 2 h. After treatment, cells were washed once and incubated in growth medium for 24 h after which they were trypsinized and replated at 103 cells per 6-cm dish. Seven to 10 days later colonies were fixed with acetic acid/ethanol and stained with crystal violet.
RESULTS
Synthesis of 2PI Mustards.
A series of bifunctional alkylating agents was prepared consisting of a DNA damaging agent covalently connected to a molecular recognition domain for a transcription factor. The linkage between these moieties was designed to permit the aromatic nitrogen mustard to create potentially lethal damage in DNA while preserving the ability of the 2PI group to interact noncovalently with the ER (15). Control molecules of similar structure were also prepared in which the DNA damaging portion of the molecule was intact, but the ER recognition domain was unable to bind effectively to the ER. The compounds prepared and evaluated for biological activity are shown in Fig. 2, along with a depiction of one, 2PI-C6NC2, attached to a potential binding site in DNA. Three are composed of 2PI linked to 4-(3-aminopropyl)-N,N-(2-chloroethyl)-aniline via an alkylaminocarbamate chain. A fourth compound incorporates 2-(4′-hydroxyphenyl)-3-methyl-indole; this compound serves as a control in that monohydroxylated phenylindoles interact poorly with the ER. A carbamate was chosen to join the two halves of the molecules together because it provides a relatively rigid connection between the two functional portions of the molecule and it is resistant to hydrolytic enzymes (22). The secondary amino group was introduced to increase water solubility. The anticipated positive charge of this group at physiological pH could also promote association with DNA.
Figure 2.

(A) Structures of aromatic mustard–2PI conjugates and their RBA for the ER. (B) Molecular model illustrating the dimensions of the 2PI-C6NC2 monoadduct in the context of an alkylation site in duplex DNA. 2PI-C6NC2 was attached through one arm of the nitrogen mustard to the N7 of the first guanine in the duplex 5′-TTGGCCAA-3′. The second arm contains a 2-hydroxyl group. Molecular model created on a Silicon Graphics Indigo xs24 workstation running insight ii 95.0 software (Biosym Technologies, San Diego). 2PI-C6NC2 was energy minimized prior to attachment to DNA (final complex not minimized) with the discover 3.0.0 program using the consistent valence force field.
Affinity of Phenylindole–Aniline Mustards for the ER.
The molecular composition and length of the linker between the aromatic mustard and 2PI groups was varied to modulate the affinity of the compounds for the ER and their ability to react with DNA. The length of the hydrocarbon chains flanking the secondary amino group in the linker (Fig. 2) markedly influenced receptor affinity. 2PI-C3NC3 mustard had little, if any, ability to compete with [3H]estradiol for the ER; RBA = 0 (RBA = 100 for estradiol). The addition of two methylene groups in 2PI-C5NC3 mustard produced a compound with an RBA of 0.6 compared with estradiol. The isomer 2PI-C6NC2 mustard, in which the secondary amine was displaced from the ER recognition domain by one additional methylene residue, showed a more than a 10-fold increase in affinity for the receptor (RBA = 7.1). The 2PI(OH)-C6NC2 mustard, which lacks the 5-hydroxy group on the ER recognition domain, only weakly interacted with the ER (RBA = 0.1).
Alkylation of DNA and Affinity of Damaged Sites in DNA for the ER.
The compounds shown in Fig. 2 were allowed to react with the duplex oligonucleotide shown in Fig. 3. The self-complementary 16-mer contained the trinucleotide GNC, which is the preferred site for the formation of interstrand crosslinks by aliphatic nitrogen mustards (23). Overall, the order of reactivity toward DNA was 2PI-C3NC3 > 2PI-C5NC3 > 2PI-C6NC2 > chlorambucil > 2PI(OH)-C6NC2 as determined by PhosphorImager analysis of alkylated products. Chemical fragmentation of the alkylated DNAs indicated guanine as the primary base alkylated by these compounds. Based upon previous studies (16, 24), it is likely that the adducts were mainly at the N7 atoms of the guanyl residues. Analysis of covalently modified DNAs revealed both monoalkylated and bis-alkylated products including interstrand crosslinks of duplex DNA (Fig. 3). Some modification was also detected at adenine residues (data not shown), likely at N3, consistent with previous reports on adducts of bifunctional aniline mustards (25).
Figure 3.

Phosphorimage of 32P-labeled 16-mer separated by denaturing PAGE after reaction of duplex DNA with 2PI-C6NC2, 2PI-C3NC3, 2PI-C5NC3, 2PI(OH)-C6NC2, or chlorambucil. Numbered bands identify modified DNA species that were isolated and used in the competition assay of Fig. 4. The data show the formation of interstrand crosslinks (band 1), alkylated single strands (bands 2 and 3), and unmodified DNA.
The identity of the major DNA adduct of 2PI-C6NC2 in vitro was investigated after acidic hydrolysis of modified calf thymus DNA. Electrospray MS of the major liberated adduct revealed m/z values of 781 (M+H)+ and 391 (M+2H)+2, which are consistent with a monoadduct in which the 2PI-C6NC2 core (Fig. 2A) was attached through one ethylene arm to guanine, whereas the other 2-chloroethyl group had undergone hydrolysis to the alcohol. This structural analysis demonstrated the physical linkage between the 2PI group and a guanine base in DNA, thus providing evidence for the attachment of the ER recognition domain to the oligonucleotide. To show the scale of the adduct in perspective to the DNA major groove binding site, Fig. 2B presents a molecular model of the octamer core of the synthetic sequence used in binding studies (5′-TTGGCCAA-3′) containing the 2PI-C6NC2 monoadduct at the 5′ guanine.
We next examined the ability of the ER to bind to the various types of DNA damage produced by several of the 2PI-linked mustards. After electrophoretic purification of alkylated single strands and interstrand crosslinked (bifunctional) products produced by the 2PI-C3NC3 and 2PI-C6NC2 mustards (Fig. 3), we assessed the ability of the adduct-containing self-complementary oligonucleotide to compete with [3H]-estradiol for binding to the ER. Fig. 4 shows that oligonucleotides isolated as alkylated single strands (bands 2 and 3 of Fig. 3), as well as the interstrand crosslinked DNA (band 1) formed by 2PI-C6NC2 mustard, competed with estradiol for binding to the ER. Oligonucleotides isolated as alkylated single strands (monofunctional adducts and possibly intrastrand crosslinks) competed best (RBA = 0.4–0.6), whereas interstrand crosslinks showed a significantly lower affinity for the ER (RBA = 0.2). Unmodified DNA did not affect the interaction of estradiol with the ER. Furthermore, none of the alkylated DNA products formed by 2PI-C3NC3 mustard showed affinity for the ER (data not shown). These data established that the 2PI moiety of the 2PI-C6NC2 adducts was presented in a manner that enabled it to act as a decoy binding site for the ER.
Figure 4.

Specific DNA adducts of 2PI-C6NC2 mustard compete with estradiol for the ER. Bands 1, 2, and 3 from Fig. 3 were excised and the DNA extracted. The modified or control 16-mers were added to a calf uterine extract along with [3H]estradiol, and the ability of each of the modified DNAs to compete with estradiol for the ER was determined. For reference, the mid-point of the binding isotherm of 2PI-C6NC2, before reaction with DNA, is indicated by the open arrow.
Toxicity of Phenylindole–Aniline Mustards to Breast Cancer Cells.
The selective toxicities of the phenylindole–mustard compounds and chlorambucil were evaluated by comparing their relative abilities to kill cell lines that differ in the expression of the ER. Preliminary experiments (not shown) revealed that if the mustard moiety in 2PI-C6NC2 mustard were inactivated by hydrolysis, some toxicity to MCF-7 cells was still evident, but only if the compound were present continuously in the growth medium. This result suggested that the free compound could act as an ER antagonist. Consequently, in the experiments shown, cells were exposed to the test compounds for a brief period (2 h) to minimize receptor antagonism by the unreacted or hydrolyzed compound. Under these conditions the hydrolyzed 2PI-C6NC2 mustard had a minimal effect on survival (≈10% reduction in survival), ruling out the possibility that ER antagonism contributed in a major way to the results observed.
Fig. 5 shows survival curves for ER(+) and ER(−) cells treated with mustards shown in the earlier experiments to have varying affinities for the receptor. The 2PI-C6NC2 and 2PI-C5NC3 mustards, which interacted best with the ER in competitive binding assays, were more toxic to the ER-expressing MCF-7 cells than to MDA-MB231 cells, which do not express receptor (Fig. 5 C and D). Compounds with minimal or no ability to interact with the ER, such as chlorambucil, 2PI(OH)-C6NC2 mustard, and 2PI-C3NC3 mustard, showed similar toxicities in the two cell lines (Fig. 5 A and B). The results with these control compounds made it unlikely that the differences in sensitivity of the ER(+) and ER(−) cells to 2PI-C6NC2 and 2PI-C5NC3 were due to cell line specific variations in mustard detoxification or in DNA repair capability. Taken together, these results provided support for the role of the ER in the observed differential toxicity.
Figure 5.
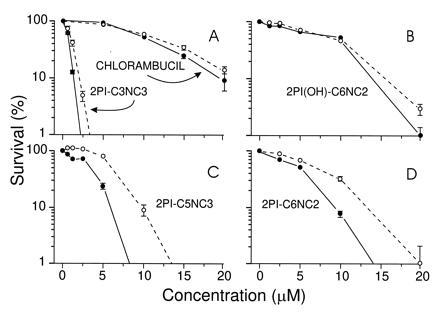
Survival of MCF-7 [ER(+); •] and MDA-MB231 [ER(−); ○] cells following exposure to 2PI–mustard compounds or chlorambucil for 2 h, as determined by colony formation. (A and B) The toxic effects of chlorambucil, 2PI(OH)-C6NC2, and 2PI-C3NC3, which do not interact or interact poorly with the ER. (C and D) The toxicities of 2PI-C5NC3 and 2PI-C6NC2, which do interact with the ER. Error bars indicate ± SD; error bars are not shown if they were eclipsed by the data point.
The absolute toxicities of all of the compounds tested in either cell line were consistent with their abilities to alkylate DNA in vitro, as shown in Fig. 3. 2PI-C3NC3 mustard and 2PI-C5NC3 mustard, both of which have three methylene groups separating the secondary amine and carbamate functions, were significantly more toxic to cells in culture and more reactive with DNA in vitro than was the 2PI-C6NC2 mustard, which has two methylene groups at this position in the linker. We have also prepared 2PI-C6NC3 mustard and likewise found it to have greater toxicity than 2PI-C6NC2 mustard (data not shown). It is clear that slight changes in the linker very significantly alter reactivity toward DNA and toxicity to cells in culture.
DISCUSSION
Synthetic toxins forming DNA adducts that attract the ER have been designed. By comparison of a series of molecules of similar structure, we were able to probe the features of molecular architecture that maintain both the ability to form DNA adducts and to interact specifically with the ER. For one of the genotoxins studied in detail, the 2PI-C6NC2 mustard, the binding of the ER to oligonucleotides isolated as alkylated single strands was more favorable than binding to interstrand crosslinks. In view of the significant affinity of the DNA adducts for the ER, we postulated that the binding might be tight enough in ER(+) cells to disrupt cellular homeostasis in vivo. The biological relevance of these associations can be gauged by an estimate of the fraction of the adducts formed in vivo expected to exist in a complex with the ER, in the presence of the natural ligand, estradiol. This estimate was obtained by application of Eq. 1 (26, 27), which describes the competitive binding of two ligands to a peptide,
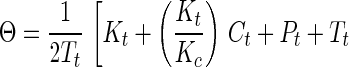 |
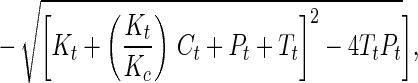 |
1 |
where Θ = fractional saturation of DNA adducts by the ER, Pt = concentration of ER protein, Tt = concentration of DNA adducts, Ct = concentration of estradiol, Kt = Kd of DNA adduct for the ER, and Kc = Kd of estradiol for ER. Our calculation required estimates of the concentrations of estradiol and 2PI-C6NC2 mustard adducts in damaged cells as well as their respective Kd values for the ER. In cells of humans treated with therapeutic alkylating drugs, a DNA adduct concentration of ≈1 μM is observed (28). It is assumed that the intracellular concentration of estradiol is close to its Kd for the ER (0.35 nM) (29). It is difficult to obtain a precise Kd of the ER for 2PI-C6NC2 mustard DNA adducts because purified ER was not available and multiple adducts were present in the damaged DNA. From our data, the apparent dissociation constant of the monoadducted single strands for the ER (Fig. 4) was estimated to be ≈0.1% of that of estradiol (Kd = 0.35 nM), implying a Kd of ≈350 nM for the most abundant 2PI-C6NC2 mustard adducts. Finally, the concentration of the ER in the nucleus was estimated to be 0.5 μM based on its reported abundance in exponentially growing MCF-7 cells (30). Using these estimated parameters one can calculate from the equation above that ≈20-25% of the 2PI-C6NC2 mustard adducts would be associated with the ER in vivo.
The possibility of a significant association of the ER with adducts in vivo prompted us to determine if ER(+) cells would be selectively affected by the phenylindole mustards in vivo. Both 2PI-C6NC2 mustard and its isomer 2PI-C5NC3 mustard, compounds that alone and in DNA attracted the ER, were selectively toxic to ER(+) cells (Fig. 5 C and D). Compounds with little or no affinity for the receptor failed to show selective toxicity. The generality of these findings has been extended in studies of two additional ER(+) and one ER(−) negative human breast cancer lines, where it was found that the mustards that attracted the ER best were selectively toxic to the ER(+) lines (data not shown). Importantly, under the conditions of dosing used, an anti-estrogenic mechanism explaining the selective toxicity of the compounds was ruled out (vide supra).
Several models could explain the selective toxicity of the phenylindole mustards for ER(+) breast cancer cells. The first model derives from studies in which alkylating agents and platinum derivatives have been tethered to ligands for steroid receptors with the aim of using the affinity of the compounds for the receptor to facilitate delivery of the molecules to receptor-positive cells (31–33). Most such agents have an intentionally hydrolyzable internal linkage. It was anticipated that the molecules, once delivered to ER(+) cells, would jettison the bulky ER ligand, liberating the alkylating group to damage DNA and kill the cell. This strategy for the selective delivery of alkylating drugs differs from the present system in which the objective was the selective retention of the lesion within DNA of target cells. Selective delivery is unlikely to be the mechanism by which the present molecules preferentially kill ER(+) cells on the basis of preliminary data showing that the levels of interstrand crosslinks of 2PI-C6NC2 mustard in ER(+) (MCF-7) and ER(−) (MDA-MB231) cells are identical shortly after dosing (R.G.C. and J.M.E., unpublished results).
A second and more plausible model to explain the selective toxicity toward ER(+) cells is hindered repair of mustard adducts in vivo brought on by the bulky ER when it is in a complex with the phenylindole portion of the DNA adducts (Fig. 1B). Support for this model comes from Eq. 1, which suggests that up to one-quarter of the adducts of 2PI-C6NC2 would be expected to be associated with the ER in vivo. There are several examples of proteins that shield structurally flawed sites in DNA from repair, and evidence exists that such shielding can increase cytotoxicity. Increased toxicity of cisplatin was observed in yeast cells that express Ixr1, a high mobility group protein that binds to intrastrand cisplatin 1,2-d(GpG) adducts (5). The implications of that result are further suggested by the demonstration that repair of cisplatin adducts was inhibited in vitro by the HMG1 protein (34). Similarly, the association of DNA photolyase with damaged sites produced by several alkylating drugs has been implicated in their toxic effects (7). In vitro, proteins that bind uracil in DNA can inhibit uracil removal by uracil glycosylase (35). It has also been suggested that, in the absence of poly-(ADP)ribosylation, the nuclear enzyme poly(ADP)ribose polymerase binds tightly to DNA strand breaks blocking access by repair enzymes (36). The repair shielding model would predict that the adducts of phenylindole mustards that interacted best with the ER would be selectively retained in ER(+) cells. This result has been observed in studies using alkaline elution to monitor the kinetics of removal of interstrand crosslinks over a 48-h time period; the results showed an almost 2-fold slower rate of removal of 2PI-C6NC2 mustard adducts in MCF-7 cells as compared with MDA-MB231 cells (R.G.C. and J.M.E., unpublished results).
A final proposed model is based on the observation that the DNA adducts of an anticancer drug and a chemical carcinogen can hijack transcription factors in vitro (Fig. 1C). hUBF, which helps regulate rRNA expression, binds to the G–G intrastrand crosslink of the anti-cancer drug cisplatin (9). The binding event is very favorable, suggesting that transcriptional regulation of rRNA synthesis may be disrupted if the adducts hijack hUBF in vivo. Moreover, the adducts of an electrophilic form of benzo(a)pyrene similarly attract a transcription factor, Sp1 (2). The participation of the hijacking mechanism in the toxic effects of our mustards cannot be ruled out. If adduct concentrations approach 1 μM in treated cells (vide supra), Eq. 1 predicts that a 50% reduction in the level of free ER would occur. The present data do not allow us to predict the functional significance of such a reduction.
The selective toxicities observed so far toward ER(+) cells for compounds such as the 2PI-C6NC2 mustard could be improved by systematic refinements of molecular architecture. Ligands showing much higher binding affinities are known (37). Moreover, one could change the DNA damaging domain in order, for example, to reposition the adduct from the major groove to the minor groove of DNA. Finally, we note that molecules of the generic design shown in Fig. 1 can be re-programmed to address targets other than the ER. Many proteins are known to be selectively expressed in diseased cells and for many of these proteins ligands are known, or could be discovered in combinatorial or other libraries. Those ligands appropriately attached to a chemical alkylating group could enable one to produce toxins that would selectively kill diseased cells while sparing the host.
Note Added in Proof.
We have recently shown that the toxicity of 2PI-C6NC2-mustard to MCF-7 cells was markedly reduced upon addition of estradiol (1 μM) to the growth medium following exposure. This result provides additional evidence for the direct role of the estrogen receptor in the sensitivity of MCF-7 cells to this mustard.
Acknowledgments
We thank D. Treiber for helpful discussions and J. Wishnok for performing the electrospray MS analyses, which were supported by National Institutes of Health Grants CA26731 and ES05622. This work was supported by National Institutes of Health Grants CA52127 and ES07020.
Footnotes
Abbreviations: ER, estrogen receptor; ER(+) and ER(−), estrogen receptor positive and negative, respectively; 2PI, 2-phenylindole; RBA, relative binding affinity; MS, mass spectrometry.
References
- 1.Friedberg E C, Walker G C, Siede W. DNA Repair and Mutagenesis. Washington, DC: Am. Soc. Microbiol.; 1995. [Google Scholar]
- 2.MacLeod M C, Powell K L, Tran N. Carcinogenesis. 1995;16:975–983. doi: 10.1093/carcin/16.5.975. [DOI] [PubMed] [Google Scholar]
- 3.Donahue B A, Augot M, Bellon S F, Treiber D K, Toney J H, Lippard S J, Essigmann J M. Biochemistry. 1990;29:5872–5880. doi: 10.1021/bi00476a032. [DOI] [PubMed] [Google Scholar]
- 4.Pil P M, Lippard S J. Science. 1992;256:234–237. doi: 10.1126/science.1566071. [DOI] [PubMed] [Google Scholar]
- 5.Brown S J, Kellett P J, Lippard S J. Science. 1993;261:603–605. doi: 10.1126/science.8342024. [DOI] [PubMed] [Google Scholar]
- 6.Bruhn S L, Pil P M, Essigmann J M, Housman D E, Lippard S J. Proc Natl Acad Sci USA. 1992;89:2307–2311. doi: 10.1073/pnas.89.6.2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fox M E, Feldman B J, Chu G. Mol Cell Biol. 1994;14:8071–8077. doi: 10.1128/mcb.14.12.8071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McA’Nulty M M, Lippard S J. Mutat Res. 1996;362:75–86. doi: 10.1016/0921-8777(95)00037-2. [DOI] [PubMed] [Google Scholar]
- 9.Treiber D K, Zhai X, Jantzen H-M, Essigmann J M. Proc Natl Acad Sci USA. 1994;91:5672–5676. doi: 10.1073/pnas.91.12.5672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Evans R M. Science. 1988;240:889–895. doi: 10.1126/science.3283939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ferno M, Johansson B U, Olsson N H, Ryden S, Sellberg G. Acta Oncol. 1990;29:129–135. doi: 10.3109/02841869009126532. [DOI] [PubMed] [Google Scholar]
- 12.Slotman B J, Rao B R. Anticancer Res. 1988;8:417–434. [PubMed] [Google Scholar]
- 13.Katzenellenbogen, J. A. (1995) J. Nucl. Med. 36, Suppl. 6, 8s–13s. [PubMed]
- 14.Katzenellenbogen B S, Montano M M, Le Goff P, Schodin D J, Kraus W L, Bhardwaj B, Fujimoto N. J Steroid Biochem Mol Biol. 1995;53:387–393. doi: 10.1016/0960-0760(95)00084-d. [DOI] [PubMed] [Google Scholar]
- 15.von Angerer E, Prekajac J, Strohmeier J. J Med Chem. 1984;27:1439–1447. doi: 10.1021/jm00377a011. [DOI] [PubMed] [Google Scholar]
- 16.Lawley P D. Prog Nucleic Acid Res Mol Biol. 1966;5:89–131. doi: 10.1016/s0079-6603(08)60232-9. [DOI] [PubMed] [Google Scholar]
- 17.Zwierzak, A. (1982) Synthesis, 920–922.
- 18.Valu K K, Gourdie T A, Boritzki T J, Gravatt G L, Baguley B C, Wilson W R, Wakelin L P G, Woodgate P D, Denny W A. J Med Chem. 1990;33:3014–3019. doi: 10.1021/jm00173a016. [DOI] [PubMed] [Google Scholar]
- 19.Korenman S G. Endocrinology. 1970;87:1119–1123. [Google Scholar]
- 20.Maniatis T, Fritsch E, Sambrook J. Molecular Cloning: A Laboratory Manual. Plainview, NY: Cold Spring Harbor Lab. Press; 1989. [Google Scholar]
- 21.Maxim A M, Gilbert W. Methods Enzymol. 1980;65:499–560. doi: 10.1016/s0076-6879(80)65059-9. [DOI] [PubMed] [Google Scholar]
- 22.Cho C Y, Moran E J, Cherry S R, Stephans J C, Fodor S P A, Adams C L, Sundaram A, Jacobs J W, Schultz P G. Science. 1993;261:1303–1305. doi: 10.1126/science.7689747. [DOI] [PubMed] [Google Scholar]
- 23.Rink S M, Solomon M S, Taylor M J, Rajur S B, McLaughlin L W, Hopkins P B. J Am Chem Soc. 1993;115:2551–2557. [Google Scholar]
- 24.Singer B. Prog Nucleic Acid Res Mol Biol. 1975;15:219–284. [PubMed] [Google Scholar]
- 25.Pieper R O, Futscher B W, Erickson L C. Carcinogenesis. 1989;10:1307–1314. doi: 10.1093/carcin/10.7.1307. [DOI] [PubMed] [Google Scholar]
- 26.Lin S-Y, Riggs A D. J Mol Biol. 1972;72:671–690. doi: 10.1016/0022-2836(72)90184-2. [DOI] [PubMed] [Google Scholar]
- 27.Long K S, Crothers D M. Biochemistry. 1995;34:8885–8895. doi: 10.1021/bi00027a041. [DOI] [PubMed] [Google Scholar]
- 28.Reed E, Parker R J, Gill I, Bicher A, Dabholkar M, Vionnet J A, Bostick-Bruton F, Tarone R, Muggia F M. Cancer Res. 1993;53:3694–3699. [PubMed] [Google Scholar]
- 29.Tora L, Mullick A, Metzger D, Ponglikitmongkol M, Park I, Chambon P. EMBO J. 1989;8:1981–1986. doi: 10.1002/j.1460-2075.1989.tb03604.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jakesz R, Smith C A, Aitken S, Huff K, Schuette W, Shackney S, Lippman M. Cancer Res. 1984;44:619–625. [PubMed] [Google Scholar]
- 31.Roth T, Tang W, Eisenbrand G. Anticancer Drug Des. 1995;10:655–666. [PubMed] [Google Scholar]
- 32.Leclercq G, Devleeschouwer N, Heuson J C. J Steroid Biochem Mol Biol. 1983;19:75–85. [PubMed] [Google Scholar]
- 33.Lam H-Y P, Ng P K T, Goldenberg G J, Wong C-M. Cancer Treat Rep. 1987;71:901–906. [PubMed] [Google Scholar]
- 34.Huang J-C, Zamble D B, Reardon J T, Lippard S J, Sancar A. Proc Natl Acad Sci USA. 1994;91:10394–10398. doi: 10.1073/pnas.91.22.10394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Devchand P R, McGhee J D, van de Sande J H. Nucleic Acids Res. 1993;21:3437–3443. doi: 10.1093/nar/21.15.3437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Satoh M S, Lindahl T. Nature (London) 1992;356:356–358. doi: 10.1038/356356a0. [DOI] [PubMed] [Google Scholar]
- 37.Jordan V C, Mittal S, Gosden B, Koch R, Lieberman M E. Environ Health Perspect. 1985;61:97–110. doi: 10.1289/ehp.856197. [DOI] [PMC free article] [PubMed] [Google Scholar]