

Abstract
Peroxynitrite activates the cyclooxygenase activities of constitutive and inducible prostaglandin endoperoxide synthases by serving as a substrate for the enzymes’ peroxidase activities. Activation of purified enzyme is induced by direct addition of peroxynitrite or by in situ generation of peroxynitrite from NO coupling to superoxide anion. Cu,Zn-superoxide dismutase completely inhibits cyclooxygenase activation in systems where peroxynitrite is generated in situ from superoxide. In the murine macrophage cell line RAW264.7, the lipophilic superoxide dismutase-mimetic agents, Cu(II) (3,5-diisopropylsalicylic acid)2, and Mn(III) tetrakis(1-methyl-4-pyridyl)porphyrin dose-dependently decrease the synthesis of prostaglandins without affecting the levels of NO synthase or prostaglandin endoperoxide synthase or by inhibiting the release of arachidonic acid. These findings support the hypothesis that peroxynitrite is an important modulator of cyclooxygenase activity in inflammatory cells and establish that superoxide anion serves as a biochemical link between NO and prostaglandin biosynthesis.
Prostaglandins and thromboxanes are important mediators of inflammation, hyperalgesia, cell growth, and hemostasis inter alia. The committed step in prostaglandin biosynthesis is the oxygenation of arachidonic acid (AA) by prostaglandin endoperoxide (PGH) synthase, a bifunctional, membrane-bound hemeprotein (1–4). Inhibition of the cyclooxygenase activity of PGH synthase is the basis for the pharmacological action of nonsteroidal antiinflammatory drugs (5, 6). Two different PGH synthases exist in vertebrates—PGH synthase-1, which is expressed constitutively and occurs in many tissues, and PGH synthase-2, which is inducible and expressed transiently (7–10). PGH synthase-2 is present at high levels in monocytes/macrophages, where it appears to play a major role in the production of inflammatory prostaglandins (7, 11).
Several recent reports demonstrate that NO stimulates prostaglandin biosynthesis in vivo, in perfused organs, and in macrophages (12–17). The stimulatory effect of NO is rapid and appears to be the result of direct activation of cyclooxygenase activity (16). However, conflicting reports exist regarding the ability of NO to stimulate purified PGH synthase, and it is possible that a derivative of NO is responsible for the activation in inflammatory cells (16, 18–20).
The principal mechanism described for direct activation of the cyclooxygenase activity of PGH synthase is reaction of fatty acid hydroperoxides with the heme prosthetic group to generate a protein radical. This protein radical (probably Tyr-385) serves as the catalytic oxidant of AA (21–23). The identity of the hydroperoxides that activate PGH synthase in different cell types is uncertain because fatty acid hydroperoxides are excellent substrates for glutathione peroxidase (GSH-Px)-catalyzed reduction by glutathione (GSH) (24). The potential for control of prostaglandin biosynthesis by GSH-Px/GSH reduction of hydroperoxide activators exhibits substantial tissue-to-tissue variation (25).
NO couples to superoxide anion (O2−) at a
diffusion-controlled rate to produce peroxynitrite (Eq. 1)
(26).
![]()
The occurrence of this reaction in macrophages contributes to their cytotoxic activity toward invading pathogens because peroxynitrite and its conjugate acid, peroxynitrous acid, are potent oxidizing agents (26–32). Because peroxynitrite is an inorganic hydroperoxide, it is a potential substrate for the peroxidase activity of PGH synthase and an activator of the enzyme’s cyclooxygenase activity. Therefore, we investigated the interaction of peroxynitrite with PGH synthase and its effect on cyclooxygenase and peroxidase catalysis. The results indicate that peroxynitrite is an efficient peroxidase substrate and cyclooxygenase activator. Both PGH synthase-1 and PGH synthase-2 are activated by peroxynitrite added directly to the enzymes or generated in situ from NO and O2−. Activation in vitro by the combination of NO and O2− is inhibited by Cu,Zn-superoxide dismutase (SOD). Membrane-permeant SOD-mimetic agents reduce prostaglandin biosynthesis in a cultured macrophage-like cell line by up to 80%. Thus, O2− appears to link NO synthesis to prostaglandin synthesis in macrophages through the intermediacy of peroxynitrite.
MATERIALS AND METHODS
Materials.
Unlabeled AA was from Nu Chek Prep (Elysian, MN). Hematin, reduced GSH, xanthine, xanthine oxidase grade III, catalase (from bovine liver), SOD (from bovine erythrocytes), NADPH, butylated hydroxyanisole, oxidized glutathione (GSSG) reductase type IVB, S-nitrosoglutathione and S-nitroso-N-acetyl-penicillamine (SNAP) were from Sigma. 3-Morpholino sydnonimine (SIN-1) was from Molecular Probes. Phenol and hydrogen peroxide were from Fisher. Guaiacol and Cu(II) (3,5-diisopropylsalicylate)2 (CuDips) were from Aldrich. Mn(III)tetrakis(1-methyl-4-pyridyl)porphyrin (MnTMPyP) was from Porphyrin Products (Logan, UT). Nitrate reductase from Aspergillus, glutamate dehydrogenase from beef liver and α-ketoglutarate were from Boehringer Mannheim. Bovine erythrocyte GSH-Px and lipopolysaccharide (LPS) serotype 0111:B4 from Escherichia coli were obtained from Calbiochem. S,S′-1,4-phenylene-bis-(1,2-ethanediyl) bis-isothiourea·(dihydrobromide) was obtained from Cayman Chemical (Ann Arbor, MI). DMEM, minimal essential medium, and mouse recombinant interferon γ (IFN-γ) were obtained from GIBCO/BRL. Fetal bovine serum was from HyClone. Antibody to PGH synthase-2 was kindly provided by David Dewitt (Michigan State University). The enhanced chemiluminscence Western detection kit was from Amersham Life Science (Cleveland). [1-14C]AA (55 mCi/mmol; 1 Ci = 37 GBq) was from DuPont/NEN. 15-Hydroperoxyeicosatetraenoic acid (15-HPETE) was synthesized from AA using soybean lipoxygenase as described (33). apoPGH synthase-1 was purified from ram seminal vesicles as described and stored at −80°C (34, 35). Recombinant human PGH synthase-2 was expressed in Sf9 cells and purified as described (36). The specific activities of PGH synthase-1 and -2 were 15–20 μmol AA min−1·mg−1 and 10–12 μmol AA min−1·mg−1, respectively. Oxygen uptake was measured with a Gilson model 5/6 oxygraph equipped with a Clark electrode and a thermostated cuvette.
Synthesis of Peroxynitrite.
Peroxynitrite was synthesized in a quenched-flow apparatus as described with modifications (37). The concentration of H2O2 used in the synthesis was decreased to 0.2 M and the concentration of sodium nitrite was increased to 1 M. The concentration of peroxynitrite was determined by measuring the absorbance at 302 nm in 1.2 M NaOH (ɛ302 = 1670 M−1·cm−1). Before use, peroxynitrite solutions were assayed for residual H2O2 as described (38). The concentration of H2O2 in peroxynitrite stocks was ≈1%.
Guaiacol Peroxidase Activity.
PGH synthase peroxidase assays contained 100 nM apoPGH synthase, reconstituted with 100 nM hematin and 500 μM guaiacol in 0.1 M NaPO4 (pH 8.0). The peroxidase reaction was initiated by the addition of peroxide substrate. Guaiacol oxidation was monitored at 436 nm (ɛ436 = 6390 M−1·cm−1) (39). The peroxidase of PGH synthase undergoes rapid inactivation which makes rate determination difficult (40). V*max represents the maximal number of turnovers catalyzed by the peroxidase at saturating concentrations of peroxide. Previous studies have shown this to be an accurate reflection of Vmax (41). The units of V*max are mol guaiacol oxidized per mol PGH synthase. The rates of guaiacol oxidation were determined over a range of peroxide concentrations, and control assays were performed in the absence of enzyme to correct for nonenzymatic oxidation.
GSH-Px Activity.
GSH-Px activity was measured in a coupled enzyme assay containing 0.25 unit GSH-Px, 1 mM GSH, 1 unit GSSG reductase, 250 μM NADPH, and 150 μM peroxide substrate in 0.1 M NaPO4 (pH 8.0). The oxidation of NADPH was monitored at 340 nm for 60 sec. Parallel experiments were conducted in the absence of GSH-Px, and the extent of NADPH oxidation was subtracted from the value determined in the presence of GSH-Px.
Activation of Cyclooxygenase Activity by Peroxynitrite.
Purified PGH synthase-1 (22 nM) was incubated at 37°C with 8 units GSH-Px, varying concentrations of GSH, and 10 μg catalase in 0.1 M NaPO4 (pH 8.0) containing 500 μM phenol (200 μl reaction volume). Following addition of 50 μM [1-14C]AA, the activator (peroxynitrite or SIN-1) was added. Reactions were terminated at varying time points by addition of an equal volume of ethyl ether/methanol/1 M citric acid (pH 4.0) (30:4:1) containing 10 μg butylated hydroxyanisole and 10 μg unlabeled AA. The organic layers were spotted onto silica gel plates and the plates were developed with ethyl acetate/methylene chloride/glacial acetic acid (75:25:1) at 4°C. Radiolabeled prostaglandin products were quantitated using a radioactivity scanner (Bioscan, Washington, DC).
Alternatively, PGH synthase-1 (75 nM) was incubated at 37°C in 1.3 ml 0.1 M NaPO4 (pH 8.0) containing 500 μM phenol, varying concentrations of GSH, 10 μg catalase, and 120 units GSH-Px in an oxygraph cell equipped with an oxygen electrode. Following addition of 50 μM [1-14C]AA, 300 μM SNAP, and/or 100 μM xanthine plus 0.2 unit xanthine oxidase were added. Aliquots (200 μl) of the reaction mixture were removed at varying time points, terminated, and analyzed as described above.
Inhibition of AA Metabolism in RAW264.7 Cells.
Stocks of RAW264.7 cells were maintained at low passage number in DMEM plus 10% fetal bovine serum. Cells (3.5 × 106 cells/T25 flask) were activated with 500 ng/ml LPS and 10 units/ml IFN-γ in serum-free supplemented minimal essential medium (SMEM) for 7 hr. The composition of SMEM was 1 liter of minimal essential medium containing 3.5 g glucose, 110 mg pyruvate, 584 mg l-glutamine, and 3.57 g Hepes (pH 7.4); this medium did not contain phenol red. The cell monolayers were washed with PBS at t = 7 hr, then incubated in fresh PBS containing 0–10 μM CuDips for 60 min or 0–10 μM Mn(III)TMPyP for 30 min. The dimethyl sulfoxide vehicle was kept constant at 1% in all flasks. At t = 8 hr of activation, the PBS was removed and analyzed for prostaglandin content by gas chromatography/negative ion chemical ionization mass spectrometry (42, 43). For determination of effects on exogenous AA metabolism, [1-14C]AA (20 μM) was incubated for 15 min at 25°C with LPS/IFN-γ-activated RAW264.7 cells that were preincubated for 30–60 min in 1 ml PBS with or without CuDips or Mn(III)TMPyP. An aliquot of the PBS was removed and mixed with an equal volume of ethyl ether/methanol/1 M citric acid (pH 4.0) (30:4:1) containing 10 μg butylated hydroxyanisole and 10 μg unlabeled AA. The organic layer was analyzed for radiolabeled metabolites as described above.
Nitrite/Nitrate Measurement.
Total nitrite/nitrates in the culture medium were measured using the Griess reagent as described (44). Medium samples were treated with 0.1 unit nitrate reductase and 100 μM NADPH for 10 min to reduce nitrate to nitrite. NH4Cl (500 mM), 4 mM α-ketoglutarate, and 20 μg glutamate dehydrogenase were then added to oxidize any remaining NADPH that can interfere with the Griess reaction. After 3 min, 500 μl of Griess reagent was added (total volume = 1 ml), and the absorbance at 540 nm was measured.
RESULTS
Purified ram seminal vesicle PGH synthase-1 and human recombinant PGH synthase-2 were incubated with H2O2-free peroxynitrite to assay for guaiacol peroxidase activity. Parallel incubations were performed with H2O2 and 15-HPETE (Table 1). Peroxynitrite was an excellent substrate for the peroxidase of both PGH synthase-1 and PGH synthase-2 and exhibited the highest Vmax* of the three peroxide substrates. 15-HPETE exhibited the lowest Km and the highest Vmax*/Km.
Table 1.
Substrate specificity of PGH synthase peroxidases and GSH-Px
| Substrate | PGH
synthase-1*
|
PGH synthase-2
|
GSH-Px† NADPH oxidation, mol × 10−7 | ||||
|---|---|---|---|---|---|---|---|
| Km, μM | V*max | V*max/Km | Km, μM | V*max | V*max/Km | ||
| Peroxynitrite | 140 | 759 | 5.4 | 100 | 287 | 2.9 | 0.08 |
| 15-HPETE | 24 | 237 | 9.7 | 9 | 59 | 6.3 | 1.54 |
| H2O2 | 287 | 692 | 2.4 | 109 | 167 | 1.5 | 1.69 |
PGH synthase peroxidase assays were performed as described. The units of V*max are mol guaiacol oxidized per mol PGH synthase. The rates of guaiacol oxidation were determined over a range of peroxide concentrations, and control assays were performed in the absence of enzyme to correct for nonenzymatic oxidation.
GSH-Px assays were conducted as described.
The ability of peroxynitrite to serve as a substrate for GSH-Px was tested in a coupled enzyme assay in which the oxidation of NADPH was monitored spectrophotometrically at 340 nm in the presence of peroxide substrate, GSH, GSH-Px, and GSSG reductase (45). Control assays were performed in the absence of GSH-Px to assess nonenzymatic GSSG formation. Although peroxynitrite induced significant nonenzymatic GSSG formation, the data in Table 1 illustrate that peroxynitrite was not an efficient substrate for GSH-Px. Pretreatment of GSH-Px with peroxynitrite in the presence of GSH did not affect the ability of GSH-Px to react with H2O2, demonstrating that peroxynitrite did not inactivate GSH-Px under these conditions.
Because peroxynitrite was an excellent substrate for the PGH synthase peroxidase activities, we evaluated its ability to activate the enzymes’ cyclooxygenase activities. PGH synthase-1 or PGH synthase-2 was incubated with GSH, catalase, and [1-14C]AA in the absence or presence of GSH-Px. In the presence of GSH-Px and GSH, AA is not converted into prostaglandins by the cyclooxygenase (25). Following addition of varying amounts of peroxynitrite, the incubation mixtures were extracted with acidic methanol/ether and the radiolabeled products were separated by thin-layer chromatography and quantitated with a radioactivity scanner. Fig. 1 plots the conversion of AA to total products as a function of peroxynitrite concentration at two different concentrations of GSH. The reaction of peroxynitrite with GSH competes with the reaction of peroxynitrite with the PGH synthase peroxidase; thus, maximal cyclooxygenase activity is achieved following GSH depletion (peroxynitrite/GSH ≈ 2:1) (46). However, stimulation of cyclooxygenase activity also is observed at concentrations of peroxynitrite that are not sufficient to deplete GSH completely. The ratio of individual AA metabolites did not change with increasing concentrations of peroxynitrite. However, PGH synthase activity was inhibited by higher concentrations of peroxynitrite presumably through oxidative inactivation (40).
Figure 1.
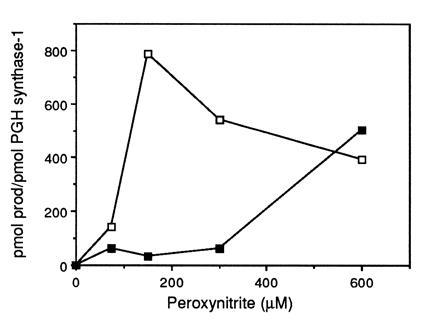
Peroxynitrite stimulation of AA metabolism by PGH synthase-1. Purified ram seminal vesicle PGH synthase-1 (22 nM) was incubated at 37°C with 8 units bovine erythrocyte GSH-Px, GSH, and 10 μg catalase in 0.1 M NaPO4 (pH 8.0) containing 500 μM phenol (200 μl reaction volume). Following addition of 50 μM [1-14C]AA, peroxynitrite was added and reactions were terminated with ethyl ether/methanol/1 M citric acid (pH 4.0) (30:4:1) after 2 min. Radiolabeled arachidonate metabolites were separated by thin-layer chromatography and quantitated using a radioactivity scanner. Control PGH synthase-1 activity in the absence of GSH-Px was 870 and 559 pmol product/pmol PGH synthase-1 at 0.2 and 1 mM GSH, respectively. □, 0.2 mM GSH; ▪, 1 mM GSH.
Peroxynitrite decomposes rapidly in physiological buffers with a half-life of ≈1 sec; thus the ability of peroxynitrite added as a bolus to serve as a peroxidase substrate or to activate cyclooxygenase is limited (29). SIN-1, which decomposes to generate both NO and O2−, was used to provide a continuous source of peroxynitrite (47). Incubation of GSH-Px/GSH-inhibited PGH synthase-1 with [1-14C]AA and 750 μM SIN-1 resulted in substantial prostaglandin formation (Fig. 2). SIN-1 activation of GSH-Px/GSH inhibited PGH synthase-1 was observed even at 2.5 mM GSH although prostaglandin formation was greatest at 0.25 mM GSH. SIN-1 activation of prostaglandin formation was completely inhibited by 1 μM SOD, indicating that NO alone was unable to activate GSH-Px/GSH-inhibited PGH synthase.
Figure 2.
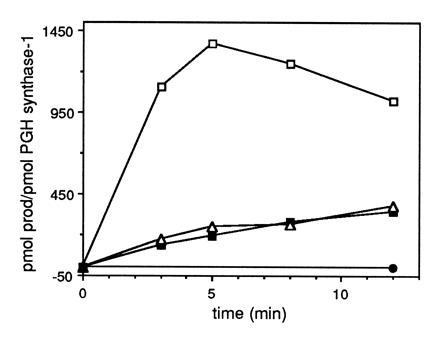
SIN-1 activation of GSH-Px/GSH inhibited PGH synthase-1. The assay conditions were identical to those described in the legend to Fig. 1 except that SIN-1 (750 μM) was added instead of peroxynitrite. Control PGH synthase-1 activity in the absence of GSH-Px was 1155 and 813 pmol product/pmol PGH synthase-1 at 0.25 and 2.5 mM GSH, respectively. □, 0.25 mM GSH; ▪, 1.0 mM GSH; ▵, 2.5 mM GSH; •, 0.25 mM GSH plus 1 μM SOD. The rate of formation of peroxynitrite from 750 μM SIN-1 in 0.1 M NaPO4 (pH 8.0) containing 500 μM phenol was estimated to be 30 μM/min following a 60-sec lag by monitoring the rate of oxygen uptake.
PGH synthase also was activated by in situ generation of peroxynitrite by the combination of the NO donor, SNAP, with the superoxide-generating system, xanthine/xanthine oxidase. Addition of either SNAP or xanthine/xanthine oxidase alone did not activate PGH synthase (Fig. 3). Thus, three independent methods for generating and administering peroxynitrite—direct addition, gradual production from SIN-1, or in situ generation from SNAP plus xanthine/xanthine oxidase—stimulated the cyclooxygenase activity of PGH synthase whereas the addition of NO or O2− alone did not. The results summarized in Figs. 1, 2, 3 were obtained with PGH synthase-1 purified from ram seminal vesicles, but identical results were obtained with recombinant human PGH synthase-2. No enhancement of the cyclooxygenase activity of either enzyme was observed if the peroxynitrite decomposition products, nitrite or nitrate, were added instead of peroxynitrite. Also, S-nitrosoglutathione, a putative product of the reaction of GSH with peroxynitrite, did not stimulate product formation (48).
Figure 3.
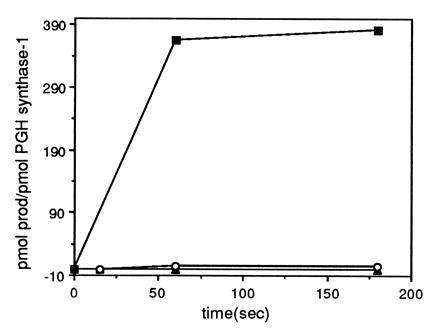
SNAP plus xanthine/xanthine oxidase activation of GSH-Px/GSH inhibited PGH synthase-1. PGH synthase-1 (75 nM) was incubated at 37°C in 1.3 ml 0.1 M NaPO4 (pH 8.0) containing 500 μM phenol, 0.25 mM GSH, and 120 units GSH-Px. Following addition of 50 μM [1-14C]AA, 300 μM SNAP, and 100 μM xanthine plus 0.2 unit xanthine oxidase or both were added. Aliquots of the reaction mixture were removed at the indicated time points and terminated with ethyl ether/methanol/1 M citric acid (pH 4.0) (30:4:1). Radiolabeled prostaglandin products were quantitated as described in Fig. 1. Control PGH synthase-1 activity in the absence of GSH-Px was 395 pmol product/pmol PGH synthase-1. ▴, 300 μM SNAP; ○, 100 μM xanthine plus 0.2 unit xanthine oxidase; •, SNAP plus xanthine/xanthine oxidase.
Relatively high concentrations of PGH synthase were used in the in vitro studies to facilitate cyclooxygenase and peroxidase assays. This necessitated the use of elevated concentrations of peroxynitrite, which makes extrapolation to cellular conditions difficult. To assess the potential cellular role of peroxynitrite as a mediator of prostaglandin biosynthesis, we employed the murine macrophage cell line, RAW264.7. This cell line was used for the experiments that first demonstrated the stimulatory effect of NO on prostaglandin biosynthesis (16). Activation of RAW264.7 cells with LPS (500 ng/ml) and IFN-γ (10 units/ml) for 8 hr resulted in a 10- to 12-fold stimulation of total nitrite and nitrate levels and a 25- to 80-fold increase in PGE2 and PGD2 formation, respectively, from endogenous AA. PGD2 was the major product, which is consistent with other reports demonstrating the production of PGD2 in certain inflammatory cell populations (49, 50). Previous studies have established that PGE2 biosynthesis in LPS-activated RAW264.7 cells is reduced by NO synthase inhibitors (16). Indeed, treatment of LPS/IFN-γ-activated cells with increasing concentrations of S,S′-1,4-phenylene-bis-(1, 2-ethanediyl) bis-isothiourea·(dihydrobromide), an inhibitor of inducible NO synthase (51), led to a progressive decline in the production of PGE2 (not shown).
To test for the involvement of O2− in the control of prostaglandin biosynthesis, activated RAW264.7 cells were incubated with increasing concentrations of CuDips, a lipophilic superoxide scavenger (52, 53). CuDips inhibited the formation of PGE2 and PGD2 in a dose-dependent fashion (Fig. 4). The parallel decrease in PGE2 and PGD2 levels suggested the effect of CuDips was on endoperoxide biosynthesis rather than on endoperoxide metabolism by PGE isomerase or PGD isomerase. Neither CuDips nor MnTMPyP, a structurally distinct SOD-mimetic agent (55), altered the expression of PGH synthase-2 as measured by Western blot analysis (Fig. 5). CuDips did not inhibit inducible NO synthase activity in RAW264.7 cells as determined by measuring nitrite and nitrate concentrations in the cell culture medium nor did it inhibit the oxygenation of AA by purified PGH synthase-1 or PGH synthase-2.
Figure 4.
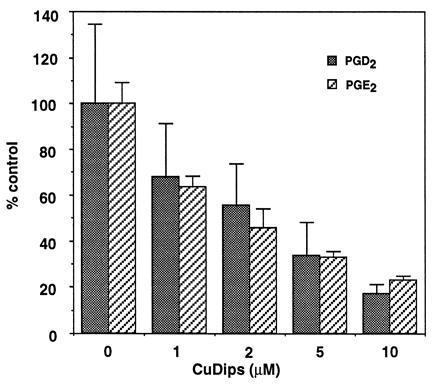
Inhibition of prostaglandin formation in LPS/IFN-γ-treated RAW264.7 cells by CuDips. RAW264.7 cells (3.5 × 106 cells/T25 flask) were activated with 500 ng/ml LPS and 10 units/ml IFN-γ in serum-free SMEM for 7 hr. The cell monolayers were washed with PBS at t = 7 hr and then incubated in fresh PBS containing 0–10 μM CuDips for 1 hr. The dimethyl sulfoxide vehicle was kept constant at 1% in all flasks. At t = 8 hr of activation, the PBS was removed and analyzed for prostaglandin content by gas chromatography/negative ion chemical ionization mass spectrometry (54). The activated cells secreted 138 ng PGD2 and 1.4 ng PGE2 per 106 cells. These values were taken as 100% conversion, respectively. At all concentrations of CuDips, there was a statistically significant difference in eicosanoid production.
Figure 5.
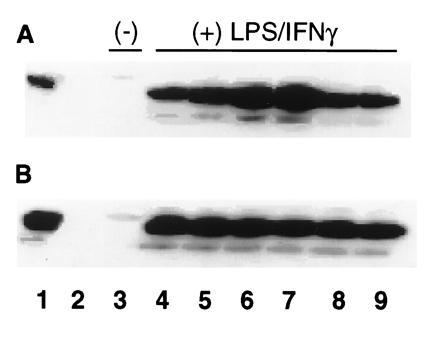
Effect of CuDips or MnTMPyP on PGH synthase-2 protein levels in activated RAW264.7 cells. RAW264.7 cells (4.4 × 106 cells/T25 flask) were activated with 500 ng/ml LPS and 10 units/ml IFN-γ in serum-free SMEM for 7 hr. The activation medium was replaced with 1 ml PBS ± 10 μM CuDips or 20 μM MnTMPyP for 15, 30, 45, or 60 min at 37°C, such that all incubations ended after a total of 1 hr in PBS. The 60-min washout controls were washed twice with PBS, followed by a 15-min incubation in PBS without superoxide scavenger. Cells were scraped off the flasks into microfuge tubes, and the pellets were frozen at −80°C. The pellets were resuspended in 180 μl 100 mM Tris·HCl (pH 8) and 0.25 M sucrose for 5 min, and heated 10 min at 95°C with 100 μl SDS/DTT sample buffer. Samples (25 μl) were run on 10% resolving PAGE and transferred to polyvinylidene difluoride membranes (Millipore). Blots were incubated with PGH synthase-2 antibody 569 (1:3000) as the primary antibody, followed by the Amersham ECL Western detection system and a 7-sec exposure. Lanes: 1, 62 ng recombinant human recombinant PGH synthase-2; 2, blank; 3, nonactivated cells; 4, 8-hr activated cells; 5, activated cells treated 15 min with scavenger; 6, 30-min treatment; 7, 45-min treatment; 8, 60-min treatment; 9, 60-min treatment with washout. (A) CuDips (10 μM). (B) MnTMPyP (20 μM).
CuDips or MnTMPyP also inhibited prostaglandin biosynthesis from exogenous AA. [1-14C]AA (20 μM) was incubated with LPS/IFN-γ-activated RAW264.7 cells for 15 min at 25°C. CuDips (10–50 μM) or MnTMPyP (1–25 μM) effected dose-dependent inhibition of the formation of radiolabeled prostaglandins (mainly PGD2). CuDips did not inhibit mobilization of AA as measured by the release of total radioactivity in prelabeled macrophages. Cells were labeled for 15 hr with 10 μM [1-14C]AA, washed, activated with LPS and IFN-γ for 7 hr, washed, and then stimulated with 100 ng/ml 12-O-tetradecanoylphorbol-13-acetate in the presence or absence of 10 μM CuDips for 1 hr. Although CuDips inhibited the conversion of AA to PGD2, it did not affect the release of total radioactivity from prelabeled phospholipids. The ability of CuDips to inhibit prostaglandin biosynthesis from endogenous or exogenous AA and its inability to inhibit the release of AA from cellular phospholipids suggests it does not inhibit phospholipase A2. Finally, neither CuDips nor MnTMPyP at concentrations up to 50 μM decreased cell viability as judged by trypan blue exclusion.
DISCUSSION
We demonstrate in the present experiments that peroxynitrite activates the cyclooxygenase activity of PGH synthase by serving as a hydroperoxide substrate for the enzyme’s peroxidase activity. Cyclooxygenase activation was observed with pure peroxynitrite added as a bolus, with peroxynitrite generated in situ from SIN-1, or with peroxynitrite generated by production of NO in the presence of the O2−-generating system xanthine/xanthine oxidase. Activation by in situ generation of peroxynitrite from NO and O2− was completely prevented by SOD. No activation was effected by NO or O2− alone, by the NO decomposition products nitrite or nitrate, or by S-nitrosoglutathione. Thus, the peroxidase-dependent activation of the cyclooxygenase activity of purified or microsomal PGH synthase is specific for peroxynitrite.
Peroxynitrite was shown previously to react with other heme peroxidases including myeloperoxidase, lactoperoxidase, and horseradish peroxidase (56). However, our experiments demonstrate that it does not react with the selenium-containing peroxidase, GSH-Px, under the conditions employed in the present studies. Peroxynitrite reacts directly and rapidly with cysteine (k = 6 × 103 M−1·sec−1) (46) so GSH consumption was observed in the absence of GSH-Px. However, addition of GSH-Px did not increase the rate of GSSG formation. The lack of reactivity of peroxynitrite with GSH-Px may provide a basis for selective activation of the cyclooxygenase activity of PGH synthases by peroxynitrite relative to fatty acid hydroperoxides. Fatty acid hydroperoxides react with GSH-Px with a rate coefficient of 4 × 107 M−1·sec−1 (24). The data in Fig. 2 indicate that gradual generation of peroxynitrite by coupling of NO and O2− can activate cyclooxygenase even in the presence of 8 units GSH-Px and 2.5 mM GSH. Under comparable conditions, no activation is detected following addition of fatty acid hydroperoxides.
Two structurally distinct SOD-mimetic agents, CuDips and MnTMPyP, effectively inhibited PGE2 and PGD2 production by RAW264.7 cells pretreated with LPS/IFN-γ. LPS/IFN-γ treatment was carried out for 7 hr before replacement of the medium with PBS containing the SOD-mimetic agents. By 7 hr, the induction of NO synthase and PGH synthase-2 was near maximal so the effects of the inhibitors were not on the induction of either enzyme. In fact, addition of CuDips prior to treatment of the cells with LPS/IFN-γ did not inhibit induction of NO synthase at any time up to 24 hr (L.M.L. and B.C.C., unpublished results). Tetsuka et al. (57) have reported that treatment of rat mesangial cells for 24 hr with scavengers of reactive oxygen species lowers the level of PGH synthase-2 protein. However, a similar effect does not appear to be responsible for inhibition of PGD2 and PGE2 biosynthesis by O2− in the present experiments. Inhibition was seen after only a 30- to 60-min incubation of RAW264.7 cells with CuDips or MnTMPyP and the inhibitory effect could be abolished by washing the cells to remove the inhibitors prior to the addition of [1-14C]AA. Furthermore, control experiments indicated that neither compound lowered the level of PGH synthase-2 protein under the conditions of these experiments.
Strong inhibition of prostaglandin biosynthesis by CuDips or MnTMPyP was observed from either endogenous or exogenous AA and the release of AA in the presence of CuDips or MnTMPyP appeared comparable to that of untreated cells. This establishes that the SOD-mimetic agents were not acting by inhibiting the release of AA from phospholipid stores. Likewise, neither compound inhibited the cyclooxygenase activity of either PGH synthase-1 or PGH synthase-2 when incubated directly with purified enzyme. Thus, all of the available evidence suggests that CuDips and MnTMPyP reduce prostaglandin biosynthesis by scavenging O2− or peroxynitrite (58, 59). Minimal inhibition of prostaglandin biosynthesis in RAW264.7 cells was observed when SOD was added to the cells. This was not unexpected because the SOD protein does not effectively penetrate intact cells and does not prevent extracellular formation of peroxynitrite by macrophages (28). Salvemini et al. (60) recently reported that recombinant human SOD coupled to polyethyleneneglycol does not lower PGE2 levels in inflammatory of exudates of rat paws injected with carrageenan.
Fig. 6 summarizes the connection of O2−, NO, and prostaglandin biosynthesis in RAW264.7 cells. Treatment of the cells with LPS/IFN-γ induces NO synthase and PGH synthase-2 with comparable time courses (15) (L.M.L. and B.C.C., unpublished results). As the concentration of NO rises, it begins to compete with SOD for O2− generated from cellular sources [e.g., uncoupling of NO synthase, NADPH oxidase, etc. (28)]. This results in the formation of peroxynitrite, which reacts with the peroxidase of PGH synthase to activate its cyclooxygenase activity. Increased activation of PGH synthase results in enhanced conversion of AA to prostaglandins. This mechanism provides an explanation for the enhancement of cellular prostaglandin biosynthesis by NO and a linkage between NO, reactive oxygen species, and lipid mediators of inflammation. Peroxynitrite-mediated cyclooxygenase activation may be particularly important in inflammatory cells which make significant amounts of NO and O2− and contain high levels of PGH synthase. Furthermore, it may provide the basis for a physiological function for peroxynitrite other than as a mediator of toxicity (32).
Figure 6.
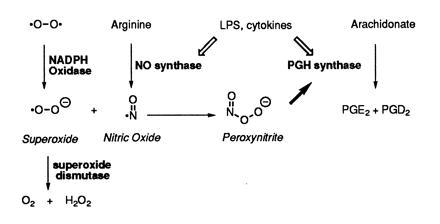
Relation of NO, O2−, and prostaglandin biosynthesis in inflammatory cells.
Acknowledgments
We are grateful to J. Gierse for preparations of purified recombinant human PGH synthase-2, to R. DuBois for RAW264.7 cells and helpful discussions, and to C. Garner for identifying PGD2 as the major metabolite of AA in RAW264.7 cells. This work was supported by research, training, and center grants from the National Institutes of Health (CA47479, DK48831, GM15431, ES07028, ES00267, and CA68485).
Footnotes
Abbreviations: GSH, reduced glutathione; GSSG, oxidized glutathione; GSH-Px, glutathione peroxidase; SOD, Cu,Zn-superoxide dismutase; CuDips, Cu(II) (3,5-diisopropylsalicylate)2; MnTMPyP, Mn(III) tetrakis(1-methyl-4-pyridyl)porphyrin; SNAP, S-nitroso-N-acetyl-penicillamine; SIN-1, 3-morpholinosydnonimine; 15-HPETE, 15-hydroperoxyeicosatetraenoic acid; PGH synthase, prostaglandin endoperoxide synthase; LPS, lipopolysaccharide; IFN-γ, interferon γ; AA, arachidonic acid.
References
- 1.Needleman P, Turk J, Jakschik B A, Morrison A R, Lefkowith J B. Annu Rev Biochem. 1986;55:69–102. doi: 10.1146/annurev.bi.55.070186.000441. [DOI] [PubMed] [Google Scholar]
- 2.Smith W L, Marnett L J. In: Metal Ions in Biological Systems. Sigel H, Sigel A, editors. Vol. 30. Basel: Dekker; 1994. pp. 163–199. [Google Scholar]
- 3.Hamberg M, Svensson J, Wakabayashi T, Samuelsson B. Proc Natl Acad Sci USA. 1974;71:345–349. doi: 10.1073/pnas.71.2.345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nugteren D H, Hazelhof E. Biochim Biophys Acta. 1973;326:448–461. doi: 10.1016/0005-2760(73)90145-8. [DOI] [PubMed] [Google Scholar]
- 5.Vane J, Botting R. FASEB J. 1988;2:89–96. [PubMed] [Google Scholar]
- 6.Jensen N P. Med Chem Res. 1995;5:319–324. [Google Scholar]
- 7.Fu J-Y, Masferrer J L, Seibert K, Raz A, Needleman P. J Biol Chem. 1990;265:16737–16740. [PubMed] [Google Scholar]
- 8.Kujubu D A, Fletcher B S, Varnum B C, Lim R W, Herschman H R. J Biol Chem. 1991;266:12866–12872. [PubMed] [Google Scholar]
- 9.Xie W, Chipman J G, Robertson D L, Erikson R L, Simmons D L. Proc Natl Acad Sci USA. 1991;88:2692–2696. doi: 10.1073/pnas.88.7.2692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.O’Banion M K, Sadowski H B, Winn V, Young D A. J Biol Chem. 1991;266:23261–23267. [PubMed] [Google Scholar]
- 11.Masferrer J L, Reddy S T, Zweifel B S, Seibert K, Needleman P, Gilbert R S, Herschman H R. J Pharmacol Exp Ther. 1994;270:1340–1344. [PubMed] [Google Scholar]
- 12.Sautebin L, Ialenti A, Ianaro A, Di Rosa M. Br J Pharmacol. 1995;114:323–328. doi: 10.1111/j.1476-5381.1995.tb13230.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Salvemini D, Settle S L, Masferrer J L, Seibert K, Currie M G, Needleman P. Br J Pharmacol. 1995;114:1171–1178. doi: 10.1111/j.1476-5381.1995.tb13330.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Salvemini D, Seibert K, Masferrer J L, Misko T P, Currie M G, Needleman P. J Clin Invest. 1994;93:1940–1947. doi: 10.1172/JCI117185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Corbett J A, Kwon G, Turk J, McDaniel M L. Biochemistry. 1993;32:13767–13770. doi: 10.1021/bi00213a002. [DOI] [PubMed] [Google Scholar]
- 16.Salvemini D, Misko T P, Masferrer J L, Seibert K, Currie M G, Needleman P. Proc Natl Acad Sci USA. 1993;90:7240–7244. doi: 10.1073/pnas.90.15.7240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Di Rosa M, Ialenti A, Ianaro A, Sautebin L. Prostaglandins Leukotrienes Essent Fatty Acids. 1996;54:229–238. doi: 10.1016/s0952-3278(96)90053-8. [DOI] [PubMed] [Google Scholar]
- 18.Tsai A-L, Wei C, Kulmacz R J. Arch Biochem Biophys. 1994;313:367–372. doi: 10.1006/abbi.1994.1400. [DOI] [PubMed] [Google Scholar]
- 19.Hajjar D P, Lander H M, Pearce S F A, Upmacis R K, Pomerantz K B. J Am Chem Soc. 1995;117:3340–3346. [Google Scholar]
- 20.Curtis, J. F., Reddy, N. G., Mason, R. P., Kalyanaraman, B. & Eling, T. E. (1996) Arch. Biochem. Biophys., in press. [DOI] [PubMed]
- 21.Karthein R, Dietz R, Nastainczyk W, Ruf H H. Eur J Biochem. 1988;171:313–320. doi: 10.1111/j.1432-1033.1988.tb13792.x. [DOI] [PubMed] [Google Scholar]
- 22.Smith W L, Eling T E, Kulmacz R J, Marnett L J, Tsai A. Biochemistry. 1992;31:3–7. doi: 10.1021/bi00116a001. [DOI] [PubMed] [Google Scholar]
- 23.Tsai A-L, Kulmacz R J, Palmer G. J Biol Chem. 1995;270:10503–10508. doi: 10.1074/jbc.270.18.10503. [DOI] [PubMed] [Google Scholar]
- 24.Ursini F, Bindoli A. Chem Phys Lipids. 1987;44:255–276. doi: 10.1016/0009-3084(87)90053-3. [DOI] [PubMed] [Google Scholar]
- 25.Marshall P J, Kulmacz R J, Lands W E M. J Biol Chem. 1987;262:3510–3517. [PubMed] [Google Scholar]
- 26.Huie R E, Padmaja S. Free Radical Res Commun. 1993;18:195–199. doi: 10.3109/10715769309145868. [DOI] [PubMed] [Google Scholar]
- 27.Blough N V, Zafiriou O C. Inorg Chem. 1985;24:3504–3505. [Google Scholar]
- 28.Ischiropoulos H, Zhu L, Beckman J S. Arch Biochem Biophys. 1992;298:446–451. doi: 10.1016/0003-9861(92)90433-w. [DOI] [PubMed] [Google Scholar]
- 29.Koppenol W H, Moreno J J, Pryor W A, Ischiropoulos H, Beckman J S. Chem Res Toxicol. 1992;5:834–842. doi: 10.1021/tx00030a017. [DOI] [PubMed] [Google Scholar]
- 30.Wizemann T M, Gardner C R, Laskin J D, Quinones S, Durham S K, Goller N L, Ohnishi S T, Laskin D L. J Leukocyte Biol. 1994;56:759–768. doi: 10.1002/jlb.56.6.759. [DOI] [PubMed] [Google Scholar]
- 31.Hausladen A, Fridovich I. J Biol Chem. 1994;269:29405–29408. [PubMed] [Google Scholar]
- 32.Beckman J S. Chem Res Toxicol. 1996;9:836–844. doi: 10.1021/tx9501445. [DOI] [PubMed] [Google Scholar]
- 33.Funk M O, Isaacs R, Porter N A. Lipids. 1976;11:113–117. doi: 10.1007/BF02532660. [DOI] [PubMed] [Google Scholar]
- 34.Marnett L J, Siedlik P H, Ochs R C, Pagels W D, Das M, Honn K V, Warnock R H, Tainer B E, Eling T E. Mol Pharmacol. 1984;26:328–335. [PubMed] [Google Scholar]
- 35.Odenwaller R, Chen Y-N P, Marnett L J. Methods Enzymol. 1990;187:479–485. doi: 10.1016/0076-6879(90)87054-7. [DOI] [PubMed] [Google Scholar]
- 36.Gierse J K, Hauser S D, Creely D P, Kobolt C, Rangwala S H, Isakson P C, Seibert K. Biochem J. 1994;304:1–9. doi: 10.1042/bj3050479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Beckman J S, Chen J, Ischiropoulos H, Crow J P. Methods Enzymol. 1994;233:229–240. doi: 10.1016/s0076-6879(94)33026-3. [DOI] [PubMed] [Google Scholar]
- 38.Pryor W A, Cueto R, Jin X, Koppenol W H, Ngu-Schwemlein M, Squadrito G L, Uppu P L, Uppu R M. Free Radical Biol Med. 1995;18:75–83. doi: 10.1016/0891-5849(94)00105-s. [DOI] [PubMed] [Google Scholar]
- 39.Putter J. In: Methods of Enzymatic Analysis. Bergmeyer H U, editor. Weinheim, Germany: Verlag Chemie; 1975. pp. 685–690. [Google Scholar]
- 40.Markey C M, Alward A, Weller P E, Marnett L J. J Biol Chem. 1987;262:6266–6279. [PubMed] [Google Scholar]
- 41.Ple P, Marnett L J. J Biol Chem. 1989;264:13983–13993. [PubMed] [Google Scholar]
- 42.DuBois R N, Awad J, Morrow J, Roberts L J, Bishop P R. J Clin Invest. 1994;93:493–498. doi: 10.1172/JCI116998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wendelborn D F, Morrow J D, Roberts L J. Methods Enzymol. 1990;187:51–62. doi: 10.1016/0076-6879(90)87008-q. [DOI] [PubMed] [Google Scholar]
- 44.Hevel J M, Marletta M A. Methods Enzymol. 1994;233:250–258. doi: 10.1016/s0076-6879(94)33028-x. [DOI] [PubMed] [Google Scholar]
- 45.Paglia D E, Valentine W N. J Lab Clin Med. 1967;70:158–169. [PubMed] [Google Scholar]
- 46.Radi R, Beckman J S, Bush K M, Freeman B A. J Biol Chem. 1991;266:4244–4250. [PubMed] [Google Scholar]
- 47.Hogg N, Darley-Usmar V M, Wilson M T, Moncada S. Biochem J. 1992;281:419–424. doi: 10.1042/bj2810419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Augusto O, Gatti R M, Radi R. Arch Biochem Biophys. 1994;310:118–125. doi: 10.1006/abbi.1994.1147. [DOI] [PubMed] [Google Scholar]
- 49.Kuiper J, Zijlstra F J, Kamps J A A M, van Berkel T J C. Biochim Biophys Acta. 1988;959:143–152. doi: 10.1016/0005-2760(88)90025-2. [DOI] [PubMed] [Google Scholar]
- 50.Urade Y, Ujihara M, Hariguchi Y, Ikai K, Hayaishi O. J Immunol. 1989;143:2982–2987. [PubMed] [Google Scholar]
- 51.Garvey E P, Oplinger J A, Tanoury G J, Sherman P A, Fowler M, Marshall S, Harmon M F, Paith J E, Furfine E S. J Biol Chem. 1994;269:26669–26676. [PubMed] [Google Scholar]
- 52.Kensler T W, Trush M A. Biochem Pharmacol. 1983;32:3485–3487. doi: 10.1016/0006-2952(83)90382-9. [DOI] [PubMed] [Google Scholar]
- 53.Kensler T W, Bush D M, Kozumbo W J. Science. 1983;221:75–77. doi: 10.1126/science.6857269. [DOI] [PubMed] [Google Scholar]
- 54.Dworski R, Sheller J R, Wicersham N E, Oates J A, Brigham K L, Roberts L J, Fittzgerald G A. Am Rev Respir Dis. 1989;139:46–51. doi: 10.1164/ajrccm/139.1.46. [DOI] [PubMed] [Google Scholar]
- 55.Faulkner K M, Liochev S I, Fridovich I. J Biol Chem. 1994;269:23471–23476. [PubMed] [Google Scholar]
- 56.Floris R, Piersma S R, Yang G, Jones P, Wever R. Eur J Biochem. 1993;215:767–775. doi: 10.1111/j.1432-1033.1993.tb18091.x. [DOI] [PubMed] [Google Scholar]
- 57.Tetsuka T, Baier L D, Morrison A R. J Biol Chem. 1996;271:11689–11693. doi: 10.1074/jbc.271.20.11689. [DOI] [PubMed] [Google Scholar]
- 58.Groves J T, Marla S S. J Am Chem Soc. 1995;117:9578–9579. [Google Scholar]
- 59.Szabo C, Day B J, Salzman A L. FEBS Lett. 1996;381:82–86. doi: 10.1016/0014-5793(96)00087-7. [DOI] [PubMed] [Google Scholar]
- 60.Salvemini D, Wang Z Q, Wyatt P S, Bourdon D M, Marino M H, Manning P T, Currie M G. Br J Pharmacol. 1996;118:829–838. doi: 10.1111/j.1476-5381.1996.tb15475.x. [DOI] [PMC free article] [PubMed] [Google Scholar]