

The crystal structure of human protein kinase CK2α2 with a potent indazole-derivative inhibitor has been determined at 3.2 Å resolution.
Keywords: casein kinase 2, CK2α2, CK2α inhibitors, selective kinase inhibitors
Abstract
Casein kinase 2 (CK2) is a serine/threonine kinase that functions as a heterotetramer composed of two catalytic subunits (CK2α1 or CK2α2) and two regulatory subunits (CK2β). The two isozymes CK2α1 and CK2α2 play distinguishable roles in healthy subjects and in patients with diseases such as cancer, respectively. In order to develop novel CK2α1-selective inhibitors, the crystal structure of human CK2α2 (hCK2α2) complexed with a potent CK2α inhibitor which binds to the active site of hCK2α2 was determined and compared with that of human CK2α1. While the two isozymes exhibited a high similarity with regard to the active site, the largest structural difference between the isoforms occurred in the β4–β5 loop responsible for the CK2α–CK2β interface. The top of the N-terminal segment interacted with the β4–β5 loop via a hydrogen bond in hCK2α2 but not in hCK2α1. Thus, the CK2α–CK2β interface is a likely target candidate for the production of selective CK2α1 inhibitors.
1. Introduction
Protein kinase CK2 (previously called casein kinase 2) is a pivotal and ubiquitously expressed member of the protein kinase CMGC subfamily, which includes cyclin-dependent kinases (CDKs), mitogen-activated protein kinases (MAPKs), glycogen synthase kinases (GSKs) and CDK-like kinases (CLKs). The CMGC kinases possess a structurally highly conserved catalytic core and catalyze the transfer of the γ-phosphoryl group of ATP to an appropriate serine or threonine in specific protein substrates. CK2 exists predominantly as a heterotetramer composed of two catalytic subunits (CK2α) and two regulatory subunits (CK2β) (Issinger, 1993 ▶; Guerra, Boldyreff et al., 1999 ▶; Allende & Allende, 1999 ▶; Pinna, 2002 ▶; Litchfield, 2003 ▶). CK2α is a constitutively active protein kinase and is further fully activated by association with CK2β, providing structural stabilization and serving as a docking platform for substrate and other binding partners (Bolanos-Garcia et al., 2006 ▶). In higher animals, CK2α1 and CK2α2 exist as two CK2α isozymes in combination with CK2β to produce three isoforms of the holoenzyme: α12β2, α1α2β2 and α22β2 (Lozeman et al., 1990 ▶).
CK2 plays important roles in transducing signals between extracellular growth factors and nuclear responses during cell division, cellular differentiation and embryogenesis (Guerra & Issinger, 1999 ▶). In mammals, the CK2α2 subunit is highly and exclusively expressed in the brain and testis, supporting the notion that CK2α2 has specific functions in these tissues (Guerra, Siemer et al., 1999 ▶); CK2α2-subunit knockout mice, for example, develop a condition similar to globozoospermia in humans (Xu et al., 1999 ▶). In contrast, the CK2α1 subunit is expressed ubiquitously in the body (Guerra, Siemer et al., 1999 ▶) and has been found in many diseases, particularly cancer, making it an interesting target within the druggable family of eukaryotic protein kinases (Pagano et al., 2006 ▶). Recently, inhibition of CK2α1 by emodin, a potent CK2 inhibitor, has been shown to cure glomerulonephritis in a mouse model (Yamada et al., 2005 ▶). A series of 4,6-disubstituted pyrazine derivatives including CC04820 (Fig. 1 ▶), consisting of a carboxyl group, a pyrrole ring and an indazole ring, were developed as novel CK2α inhibitors and potently blocked the activity of both human CK2α1 (hCKα1) and human CK2α2 (hCKα2) (Suzuki et al., 2008 ▶); CC04820 exhibited no selectivity, with an IC50 of 17 nM for hCK2α1 and an IC50 of 11 nM for hCK2α2. These data are consistent with the fact that CK2α inhibitors provide beneficial effects on nephritis via Ck2α1 inhibition and adverse effects on spermatogenesis via CK2α2 inhibition (Xu et al., 1999 ▶; Yamada et al., 2005 ▶).
Figure 1.

The structure of the CK2α inhibitor CC04820.
Several crystal structures of maize CK2α1, the hCK2α1 apoenzyme and the hCK2α1 holoenzyme have been determined in states with and without inhibitors (Niefind et al., 1998 ▶, 1999 ▶, 2001 ▶; Ermakova et al., 2003 ▶; Battistutta et al., 2000 ▶, 2001 ▶; De Moliner et al., 2003 ▶; Raaf et al., 2008 ▶). These structures suggest that the extended N-terminal segment fixes the activation segment and αC helix, the conformational plasticity of which is significant for on/off regulation of enzyme activity in CMGC kinases, and thus allows CK2α1 to have constitutive activity.
Although CK2α2 bears a marked resemblance in amino-acid sequence to CK2α1 (Dahmus et al., 1984 ▶), the two isozymes have distinguishable biological functions, as mentioned above. Thus, we determined the first structure of hCK2α2 complexed with CC04820 in order to clarify the structural differences between hCK2α1 and hCK2α2.
2. Materials and methods
2.1. Construction of the expression plasmid
The coding region corresponding to amino-acid residues Met1–Gln334 of human CK2α2 was amplified by the polymerase chain reaction (PCR) and cloned into the vector pGEX-6P-1 (GE Healthcare) at the restriction sites BamHI–EcoRI, providing the construct in a GST-fused form at the N-terminus.
2.2. Protein expression and purification
Escherichia coli strain HMS174 (DE3) cells (Novagen) were transformed with pGEX-hCK2α2. The cells were cultured in 25 ml LB media containing 100 µg ml−1 ampicillin at 310 K on a shaker for 12 h and then transferred to 500 ml LB media and incubated at 310 K on a shaker for 2 h. Protein expression was induced with 0.5 mM isopropyl β-d-1-thiogalactopyranoside (Sigma) at 291 K for 8 h. The extracted supernatant was loaded onto a glutathione-Sepharose 4B column (GE Healthcare). The column was washed with cleavage buffer (20 mM Tris–HCl pH 8.0, 160 mM NaCl, 1 mM EDTA, 0.1% Tween 20 and 1 mM dithiothreitol). The GST-fused protein was cleaved by PreScission protease (GE Healthcare) on the column in cleavage buffer at 277 K for 16 h. The GST-removed hCK2α2 protein, which had a five-residue GST remnant, GPLGS, at the N-terminus, was eluted with PBS buffer containing 1 mM dithiothreitol. The protein was further purified using a MonoQ 4.6/100 column (GE Healthcare) with a linear salt gradient of 0.05–1 M NaCl in 40 column volumes of buffer (25 mM Tris–HCl pH 8.5, 1 mM dithiothreitol) at 277 K using an ÄKTA Explorer system (GE Healthcare). The purified hCK2α2 sample was concentrated without buffer exchange to 10 mg ml−1, as confirmed spectrophotometrically by the Bradford method (Bradford, 1976 ▶). The final concentration of NaCl in this solution was estimated at 250 mM by salt gradient in ion-exchange purification.
2.3. Crystallization
To produce the inhibitor complex, CC04820 powder was suspended in the concentrated protein solution in a sufficient quantity to give a molar ligand:protein ratio of 5:1 and incubated on ice for 4 h. After spinning down undissolved material, crystallization trials were performed by the sitting-drop vapour-diffusion method at 277 K against 60 µl reservoir solution using the commercially available screening kits Crystal Screen HT (Hampton Research) and Wizard (Emerald Biostructures) with the sparse-matrix method (Jancarik & Kim, 1991 ▶). Consequently, thin needle-shaped crystals were obtained by mixing 0.5 µl CK2α2–CC04820 solution with an equal volume of reservoir solution from two conditions from the commercial kit: C12 from Crystal Screen HT (8% PEG 8000, 0.1 M Tris–HCl pH 8.5) and C2 from Wizard (10% PEG 3000, 0.1 M CHES pH 9.5). These conditions were optimized by varying the pH and the PEG concentration. The largest crystals were obtained using a protein:reservoir solution ratio of 1:1 (2:2 µl or 4:4 µl), with the reservoir solution consisting of 8% PEG 8000, 0.1 M Tris–HCl pH 8.0. The crystals grew to maximum dimensions of only 0.08 × 0.005 × 0.005 mm in 2–3 weeks (Fig. 2 ▶).
Figure 2.

Crystals of the hCK2α2–CC04820 complex. The black scale bar is 100 µm in length.
2.4. X-ray crystallographic analysis
After dipping them into Paratone-N oil (Hampton Research), the crystals were frozen in a nitrogen-gas stream at 100 K. Diffraction data were collected at a wavelength of 0.978 Å using synchrotron radiation on Photon Factory beamline BL6A with a Quantum 4R CCD detector (ADSC), using an exposure time of 45 s per image and a crystal-to-detector distance of 230 mm. X-ray diffraction data consisting of 180 images were processed and scaled using the program HKL-2000 (Otwinowski & Minor, 1997 ▶). The selected crystal diffracted to 3.2 Å resolution and was found to belong to an orthorhombic lattice. A complete X-ray diffraction data set was collected using this crystal. The R merge value was high despite the good values of I/σ(I) and redundancy. These values may contain some errors owing to the using of a small crystal in the data-collection procedure. The solvent content was estimated as 41% by the MATTHEWS_COEF program from the CCP4 suite (Collaborative Computational Project, Number 4, 1994 ▶; Matthews, 1968 ▶), assuming the presence of one hCK2α2 molecule in the asymmetric unit.
Molecular-replacement calculations were carried out with the program MOLREP from CCP4 using a homology model derived from the hCK2α1 structure (PDB code 1pjk; Ermakova et al., 2003 ▶). All refinements and electron-density map calculations were performed using the programs DS Modeling and CNX (Accelrys Inc., San Diego, California, USA). Details of data collection and refinement are summarized in Table 1 ▶. The final model includes one hCK2α2 molecule, one CC04820 molecule and 206 water molecules. The five artificial residues at the N-terminus that remained after removal of the GST tag were not visible in the corresponding region of electron-density map. Thus, the residues were presumed to be disordered. With the exception of these residues, the hCK2α2 structure was completely defined by the electron density.
Table 1. Data-collection and structure-refinement statistics.
Values in parentheses are for the highest resolution shell.
| Data collection | |
| Space group | P21212 |
| Unit-cell parameters (Å) | a = 69.81, b = 102.13, c = 46.62 |
| Observations | 55117 (5586) |
| Unique reflections | 5978 (588) |
| Resolution (Å) | 57.64–3.20 (3.31–3.20) |
| Completeness (%) | 100 (100) |
| Rmerge† (%) | 20.0 (41.4) |
| 〈I/σ(I)〉 | 18.6 (8.8) |
| Redundancy | 9.2 (9.5) |
| Refinement statistics | |
| Resolution (Å) | 57.64–3.20 (3.41–3.20) |
| Reflections | 5978 (994) |
| Total atoms | 2946 |
| Rwork‡ (%) | 24.8 (26.8) |
| Rfree§ (%) | 27.1 (32.4) |
| Average B factor (Å2) | 9.14 |
| R.m.s. deviations | |
| Bond lengths (Å) | 0.008 |
| Bond angles (°) | 1.7 |
R
merge = 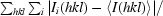
 , where 〈I(hkl)〉 is the mean intensity of the set of i equivalent reflections.
, where 〈I(hkl)〉 is the mean intensity of the set of i equivalent reflections.
R
work = 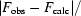
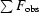 , where F
obs and F
calc are the observed and calculated structure-factor amplitudes, respectively.
, where F
obs and F
calc are the observed and calculated structure-factor amplitudes, respectively.
The R free value was calculated with a random 5% subset of all reflections that were excluded from the refinement.
3. Results and discussion
3.1. Overall folding of hCK2α2
hCK2α2 had the fold typical of the CMGC kinases, with an extension of the N-terminal segment (Fig. 3 ▶ a). The protein folding of hCK2α2 involving the N-terminal lobe and the C-terminal lobe was highly similar to that of hCK2α1 (Fig. 5a). The inhibitor bound to the ATP-binding region in the cleft between the N- and C-terminal lobes of hCK2α2.
Figure 3.

Structural overview of hCK2α2 binding the potent inhibitor CC04820. (a) Attachment of the N-terminal segment to the activation segment and to the αC helix. Close-ups are shown of the interactions of (b) the aromatic cluster, (c) the hydrogen bonds between the N-terminal segment and the activation segment and (d) the hydrogen bonds between the activation segment and the αC helix.
The N-terminal segment of hCK2α2 was fixed to its body by an aromatic cluster and several hydrogen bonds in a similar manner to that observed for hCK2α1. At the interface between the N-terminal segment and the activation segment, an aromatic cluster containing Tyr24, Trp25 and Tyr27 on one side and Phe182 and His184 on the other was particularly obvious (Fig. 3 ▶ b). The backbone N atom of Ala10 made a hydrogen bond to Tyr183 Oη and Asn17 Nδ2 made a hydrogen bond to the backbone O atom of Tyr183 (Fig. 3 ▶ c). Besides these interactions, the activation segment interacted with the αC helix via three hydrogen bonds. Lys78 Nζ formed a hydrogen bond to the backbone O atom of Gly178 and Arg81 Nη1 made a hydrogen bond to the backbone O atom of Gly178 and the backbone O atom of Ala180 (Fig. 3 ▶ d). All these residues in the N-terminal segment are highly conserved among CK2α from different species (Niefind et al., 1998 ▶; Ermakova et al., 2003 ▶). The crystal structures of hCK2α2 showed that the activation segment and the αC helix were fixed in the active conformation by close contacts to the N-terminal segment as in hCK2α1 and mCK2α1. Overall, no significant differences were detected in the folding of the two isozymes.
3.2. Interaction of CC04820 with the ATP-binding region of hCK2α2
CC04820 bound tightly to the ATP-binding region as judged by the clear electron-density map in the corresponding region. The inhibitor was surrounded by several hydrophobic amino acids. The pyrazine moiety interacted with Val67 in the β3 strand. The pyrrole ring interacted with Ile175 in the activation segment. The indazole ring was sandwiched between Leu46 in the glycine-rich loop between the β1 and β2 strands and Met164 in the extra β-strand adjusting the activation segment. Furthermore, three hydrogen bonds were found between hCK2α2 and the inhibitor. The carboxyl group made hydrogen bonds to Lys69 Nζ as the active essential residue and the backbone N atom of Asp176 in the activation segment, while one of the N atoms of the pyrazine ring made hydrogen bonds to the backbone O atom of Ile117 on the hinge region connecting the N- and C-lobes (Fig. 4 ▶ a).
Figure 4.

Stereoview of the ATP-binding region. (a) Illustration with the inhibitor. Hydrogen bonds are shown as dotted lines. The N atoms are shown in blue and the O atoms are shown in red. (b) Superposition of the hinge region of hCK2α2 (orange) on that of hCK2α1 (grey). The different residues in the hinge region are highlighted for hCK2α2 (green) and hCK2α1 (blue).
The interaction in the hCK2α2–CC04820 complex (Fig. 4 ▶ a) is likely to be similar to that in hCK2α1–CC04820, as hCK2α2 was estimated to resemble hCK2α1 with respect to the overall folding, especially in the active site. However, two residues in the hinge region outside of the active site differed between the two isozymes (hCK2α2, Tyr116–Ile117; hCK2α1, His115–Val116). In hCK2α2, the space between the inhibitor and the hinge region composed of Tyr116–Asn119 was slightly smaller than that in hCK2α1 (Fig. 4 ▶ b). This space in hCK2α2 was packed with the side chain of Ile117, which was larger than that of Val116. The observation, together with the Tyr116/His115 difference, indicates that the hinge region is likely to be effective in the acquisition of selectivity, for which the difference in this space is exploited, and suggest that it may be possible to produce a novel inhibitor utilizing the structural difference in this region.
3.3. Structural variations at the CK2α–CK2β interface
hCK2α2 has a unique conformation of the β4–β5 loop (Fig. 5 ▶ a), which corresponds to the CK2α–CK2β interface in the CK2α2β2 heterotetramer (Litchfield, 2003 ▶), compared with apo-hCK2α1 (Ermakova et al., 2003 ▶), holo-hCK2α1 (Niefind et al., 2001 ▶) and mCK2α1 (Niefind et al., 1998 ▶). The three CK2α1 structures are very similar with respect to the β4–β5 loop. The distances between the Cα atoms of the top residue in the β4–β5 loop (Val105 in hCK2α1, Val106 in hCK2α2 and Val105 in mCK2α1) and the Cα atoms of the top residue in the N-terminal segment (Asp37 in hCK2α1, Asp38 in hCK2α2 and Asp37 in mCK2α1) were 8.5 Å in hCK2α2, 12.7 Å in apo-hCK2α1, 12.8 Å in holo-hCK2α1 and 13.7 Å in mCK2α1. The N-terminal segment was very much closer to the β4–β5 loop in hCK2α2.
Figure 5.

Superposition of the Cα traces of hCK2α2 (green) and hCK2α1 (orange; PDB file 1pjk). (a) The largest structural difference is indicated by the dotted circle. (b) A close-up of the interaction is shown from the hydrogen bonds between the N-terminal region and the β4 and β5 loop responsible for α/β association. The N atoms are shown in blue and the O atoms in red.
Consequently, the formation of a hydrogen bond between the backbone O atom of Gly35 in the N-terminal segment and the backbone N atom of Lys103 in the β4 strand was only observed in hCK2α2. Furthermore, two hydrogen bonds were found in the connecting region between the β4 and β5 strands. The Nζ atom of Lys108 on the β5 strand made hydrogen bonds to the backbone O atom of Pro105 and Val106 on the β4 strand (Fig. 5 ▶ b). This observation suggests that the β4–β5 loop in hCK2α2 is more rigid than those in the other three CK2α1 structures.
The structural difference in the CK2α–CK2β interface between the two isozymes probably allows hCK2α2-selective or hCK2α1-selective inhibitors to be produced; nonselective CK2α-inhibitor binding to the CK2α–CK2β interface region has been reported (Raaf et al., 2008 ▶).
In conclusion, we have determined the crystal structure of hCK2α2 complexed with a potent CK2α inhibitor (CC04820). Structural comparison of the two isozymes is likely to allow the production of selective CK2α1 or CK2α2 inhibitors by utilizing the hinge region and particularly the CK2α–CK2β interface.
Supplementary Material
PDB reference: human protein kinase CK2α2, 3e3b, r3e3bsf
Acknowledgments
These studies were supported by the Program of Fundamental Studies in Health Science of the National Institute of Biomedical Innovation (NIBIO). The inhibitor CC04820 used in the present study was provided by TOREY Industries Inc. The synchrotron-radiation experiments were performed at Photon Factory with the approval of the Japan Synchrotron Radiation Research Institute. We thank the staff for their help in data collection at the BL-6A station.
References
- Allende, C. C. & Allende, J. E. (1999). J. Cell. Biochem.30, 129–136.
- Battistutta, R., De Moliner, E., Sarno, S., Zanotti, G. & Pinna, L. A. (2001). Protein Sci.10, 2200–2206. [DOI] [PMC free article] [PubMed]
- Battistutta, R., Sarno, S., De Moliner, E., Papinutto, E., Zanotti, G. & Pinna, L. A. (2000). J. Biol. Chem.275, 29618–29622. [DOI] [PubMed]
- Bolanos-Garcia, V. M., Fernandez-Recio, J., Allende, J. E. & Blundell, T. L. (2006). Trends. Biochem. Sci.31, 654–661. [DOI] [PubMed]
- Bradford, M. M. (1976). Anal. Biochem.717, 1448–1454.
- Collaborative Computational Project, Number 4 (1994). Acta Cryst. D50, 760–763.
- Dahmus, G. K., Clover, C. V., Brutlag, D. L. & Dahmus, M. E. (1984). J. Biol. Chem.259, 9001–9006. [PubMed]
- De Moliner, E., Moro, S., Sarno, S., Zagotto, G., Zanotti, G., Pinna, L. A. & Battistutta, R. (2003). J. Biol. Chem.278, 1831–1836. [DOI] [PubMed]
- Ermakova, I., Boldyreff, B., Issinger, O.-G. & Niefind, K. (2003). J. Mol. Biol.330, 925–934. [DOI] [PubMed]
- Guerra, B., Boldyreff, B., Sarno, S., Cesaro, L., Issinger, O.-G. & Pinna, L. A. (1999). Pharmacol. Ther.82, 303–313. [DOI] [PubMed]
- Guerra, B. & Issinger, O.-G. (1999). Electrophoresis, 20, 391–408. [DOI] [PubMed]
- Guerra, B., Siemer, S., Boldyreff, B. & Issinger, O.-G. (1999). FEBS Lett.462, 353–357. [DOI] [PubMed]
- Issinger, O.-G. (1993). Pharmacol. Ther.59, 1–30. [DOI] [PubMed]
- Jancarik, J. & Kim, S.-H. (1991). J. Appl. Cryst.24, 409–411.
- Litchfield, D. W. (2003). Biochem. J.369, 1–15. [DOI] [PMC free article] [PubMed]
- Lozeman, F. J., Litchfield, D. W., Pienning, C., Takio, K., Walsh, K. A. & Krebs, E. D. (1990). Biochemistry, 29, 8436–8447. [DOI] [PubMed]
- Matthews, B. W. (1968). J. Mol. Biol.33, 491–497. [DOI] [PubMed]
- Niefind, K., Guerra, B., Ermakowa, I. & Issinger, O.-G. (2001). EMBO J.20, 5320–5331. [DOI] [PMC free article] [PubMed]
- Niefind, K., Guerra, B., Pinna, L. A., Issinger, O.-G. & Schomburg, D. (1998). EMBO J.17, 2451–2462. [DOI] [PMC free article] [PubMed]
- Niefind, K., Pütter, M., Guerra, B., Issinger, O.-G. & Schomburg, D. (1999). Nature Struct. Biol.6, 1100–1103. [DOI] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol.276, 307–326. [DOI] [PubMed]
- Pagano, M. A., Cesaro, L., Meggio, F. & Pinna, L. A. (2006). Biochem. Soc. Trans.34, 1303–1306. [DOI] [PubMed]
- Pinna, L. A. (2002). J. Cell Sci.115, 32873–33878.
- Raaf, J., Brunstein, E., Issinger, O.-G. & Niefind, K. (2008). Chem. Biol.15, 111–117. [DOI] [PubMed]
- Suzuki, Y., Cluzeau, J., Hara, T., Hirasawa, A., Tsujimoto, G., Oishi, S., Ohno, H. & Fujii, N. (2008). Arch. Pharm.341, 554–561. [DOI] [PubMed]
- Xu, X., Toselli, P. A., Russell, L. D. & Seldin, D. C. (1999). Nature Genet.23, 118–121. [DOI] [PubMed]
- Yamada, M. et al. (2005). Proc. Natl Acad. Sci. USA, 102, 7736–7741.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: human protein kinase CK2α2, 3e3b, r3e3bsf
PDB reference: human protein kinase CK2α2, 3e3b, r3e3bsf