

The crystal structure of the ligand-binding domain of a receptor tyrosine kinase EphB2, an important mediator of cell-cell communication, has been determined at a resolution of 2 Å. The structure confirms the induced-fit mechanism for the binding of ligands to EphB receptors.
Keywords: EphB2, Eph receptors, ligand binding
Abstract
Eph tyrosine kinase receptors, the largest group of receptor tyrosine kinases, and their ephrin ligands are important mediators of cell–cell communication regulating cell attachment, shape and mobility. Recently, several Eph receptors and ephrins have also been found to play important roles in the progression of cancer. Structural and biophysical studies have established detailed information on the binding and recognition of Eph receptors and ephrins. The initial high-affinity binding of Eph receptors to ephrin occurs through the penetration of an extended G–H loop of the ligand into a hydrophobic channel on the surface of the receptor. Consequently, the G–H loop-binding channel of Eph receptors is the main target in the search for Eph antagonists that could be used in the development of anticancer drugs and several peptides have been shown to specifically bind Eph receptors and compete with the cognate ephrin ligands. However, the molecular details of the conformational changes upon Eph/ephrin binding have remained speculative, since two of the loops were unstructured in the original model of the free EphB2 structure and their conformational changes upon ligand binding could consequently not be analyzed in detail. In this study, the X-ray structure of unbound EphB2 is reported at a considerably higher 2 Å resolution, the conformational changes that the important receptor loops undergo upon ligand binding are described and the consequences that these findings have for the development of Eph antagonists are discussed.
1. Introduction
The Eph family represents the largest class of receptor tyrosine kinases (RTKs), with 16 members (Klein, 2001 ▶; Boyd & Lackmann, 2001 ▶; Lackmann & Boyd, 2008 ▶). Nine membrane-attached ligands, the ephrins, bind to the Eph receptors and activate their tyrosine kinase catalytic domains. The role of these molecules in axon pathfinding is well established (Flanagan & Vanderhaeghen, 1998 ▶). In addition to axon guidance, Eph receptors and ephrins are now known to have important roles in controlling a diverse array of other cell–cell interactions, including those of vascular endothelial cells and specialized epithelia (Kullander & Klein, 2002 ▶; Lackmann & Boyd, 2008 ▶). Both Eph receptors and ephrins are divided into two subclasses, A and B, based on sequence conservation and their binding affinities (Gale et al., 1996 ▶; http://cbweb.med.harvard.edu/eph-nomenclature). The extracellular region (ECD) of Eph receptors contains a highly conserved N-terminal domain (LBD) that is necessary and sufficient for ligand recognition and binding (Labrador et al., 1997 ▶). Our original structure of the EphB2 ligand-binding domain showed a jelly-roll folding topology and our model at 2.9 Å resolution was refined to an R factor of 20.6% (Himanen et al., 1998 ▶). Notably, of the several loops of varying length packing against the β-sheets, two (loops D–E and J–K) were not well ordered, with several residues that could not be located in the electron-density map.
Since Ephs and ephrins are involved not only in early developmental processes, but also in the function of the adult organism (Adams & Klein, 2000 ▶), controlling their signaling could have important medical applications (Dodelet & Pasquale, 2000 ▶; Pasquale, 2008 ▶). Eph–ephrin crystal structures (Himanen et al., 2001 ▶, 2004 ▶; Chrencik et al., 2006 ▶) document that the ligand–receptor interface is dominated by the insertion of an ephrin loop into a hydrophobic channel on the Eph surface, suggesting that Eph–ephrin binding can be disrupted by small molecules or peptides that target this channel. Indeed, phage display technology has identified a series of peptides that specifically bind EphB2, EphB4, EphA2 and EphA4 (Koolpe et al., 2002 ▶, 2005 ▶; Murai et al., 2003 ▶), often effectively competing with the cognate ephrin ligands. The crystal structures of these antagonistic peptides in complex with Eph receptors (Chrencik, Brooun, Recht et al., 2006 ▶; Chrencik et al., 2007 ▶) have confirmed that the peptides bind in the hydrophobic ligand-binding channel of Eph.
Crystal structures have also indicated that the intra-class EphB2–ephrin-B2 (Himanen et al., 2001 ▶) and EphB4–ephrin-B2 (Chrencik, Brooun, Kraus et al., 2006 ▶) dimerization interfaces include two distinct regions, but only one of these is observed in the inter-class EphB2–ephrin-A5 complex (Himanen et al., 2004 ▶), resulting in a smaller overall contact area and decreased binding affinity. The flexibility of the receptor loops that comprise the ligand-binding cavity observed in these structures suggests that the B-class molecules use an ‘induced-fit’ mechanism for recognition. Therefore, it appears to be possible to design a surrogate binding assay for high-throughput inhibitor screens using ligand and receptor constructs that interact solely via the Eph-channel–ephrin-loop interface, provided that the suggested induced-fit model for Eph–ephrin binding is correct. However, the exact nature of the ligand–receptor recognition has remained elusive, since two of the loops were unstructured in the original model of the free EphB2 structure and their potential conformational changes upon ligand binding consequently could not be analyzed. Therefore, we have determined a higher resolution structure of the EphB2 ligand-binding domain at 2 Å resolution.
2. Experimental
The murine EphB2 globular domain (residues 28–210) was expressed using the pET32 vector and Escherichia coli strain AD494(DE3) and purified by affinity and ion-exchange chromatography as described previously (Himanen et al., 1998 ▶). The histidine-containing N-terminal thioredoxin-fusion sequence was removed by thrombin proteolysis. Purified EphB2 was concentrated to 10 mg ml−1 in a buffer containing 10 mM KCl, 2 mM MgCl2 and 10 mM HEPES pH 7.2 and was crystallized in a hanging drop by vapor diffusion at 297 K. Unlike the original crystals, which grew using a reservoir solution containing 15%(w/v) polyethylene glycol (PEG) 4000 and 50 mM sodium acetate pH 4.8, the new crystal forms were obtained using 1.2 M sodium potassium phosphate pH 5.4 as a precipitant. This resulted in much better diffracting crystals that grew in space group P41212 (unit-cell parameters a = 54.03, b = 54.03, c = 157.41 Å) with one molecule in the asymmetric unit.
Data were collected on NSLS beamline X6A at 100 K using 20% PEG 400 as a cryoprotectant. Images were integrated, scaled and merged using HKL-2000 (Otwinowski & Minor, 1997 ▶). Subsequent calculations were performed with the CCP4 program suite (Collaborative Computational Project, Number 4, 1994 ▶). The structure was determined using the molecular-replacement method with the CCP4 program AMoRe (Navaza, 1994 ▶) using PDB entry 1nuk (Himanen et al., 1998 ▶) as a search model. The top solution had a correlation coefficient of 72% prior to refinement in REFMAC5 (Murshudov et al., 1997 ▶). The model was built and the misfit regions trimmed using the program O (Jones et al., 1991 ▶).
3. Results and discussion
Fig. 1 ▶ shows our new EphB2 structure superimposed on the previous unbound structure. The structure has a jelly-roll folding topology and overall dimensions of approximately 50 × 40 × 30 Å. The jelly roll consists of 13 β-strands arranged into two antiparallel β-sheets connected by loops of varying length and two disulfide bonds, forming a compact β-sandwich. The new model was refined at 2.0 Å resolution to an R factor of 19.8% (R free = 26.4%) with an overall temperature factor of 42.5 Å2. The two crystal forms belonged to the same space group and had similar unit-cell parameters (a = 54.03, b = 54.03, c = 157.41 Å for the new crystal form and a = 55.15, b = 55.15, c = 158.9 Å for the old crystal form). In the new structure it was possible to trace the J–K and D–E loops that were not built in the original structure (residues 59–61 and 160–163, respectively). Both loops comprise extensions of the corresponding β-strands (E and K). The temperature factors of the Cα atoms in these regions were between 60 and 80 Å2, indicating moderate flexibility. The D–E loop makes crystal contacts with the same region of a symmetry-related molecule, which may have contributed to stabilizing its conformation. The side chains of Met61 and Val63 in the D–E loop as well as of Arg163 in the J–K loop remained disordered (Fig. 2 ▶ a). The flexible C-terminus is also visible in the improved model. In addition, the N-terminus of a symmetry-related molecule is located inside the central cavity of the receptor, which is formed between the G–H, J–K and D–E loops, close to the binding site of the antagonistic peptide (Chrencik et al., 2007 ▶). The deviation (r.m.s.d.) from the old structure is 0.61 Å as judged by the positions of 167 equivalent Cα atoms. The differences are concentrated in the loops and the N-terminal region. Also, four Pro residues (47, 136, 142 and 176) were not identified as cis-proline in Himanen et al. (1998 ▶) owing to the limited resolution.
Figure 1.

Structure of the unbound EphB2 of this study (2.0 Å resolution) superimposed on the previously published unbound structure (2.9 Å resolution; Himanen et al., 1998 ▶). The new model is in green and the old model is in red. Clearly traceable J–K and D–E loops that were not built in the original structure can be seen in the new structure. Both loops comprise extensions of the corresponding β-strands (E and K). The flexible N- and C-termini are now also visible. The deviation (r.m.s.d.) from the old structure is 0.61 Å as judged by the positions of 167 equivalent Cα atoms.
Figure 2.

(a) A region of the 2F o − F c electron-density map showing some of the amino-acid residues in the D–E loop with the final model drawn as a stick diagram. The map was calculated before residues 59–61 and 160–163 were built in. The map is contoured at 0.8σ. (b) A sample of the final 2F o − F c electron-density map in the region of the ‘class-specificity loop’ (H–I loop; Himanen et al., 1998 ▶): Asn141 has unfavorable ϕ and ψ angles but its conformation can clearly be determined. The map is contoured at 1σ.
Of particular interest are residues Arg103, Ser101 and Ile108 in the improved model. These residues have been shown to be crucial for the formation of functional Eph–ephrin complexes and possibly for the subclass specificity (Himanen et al., 2001 ▶, 2004 ▶; Chrencik, Brooun, Kraus et al., 2006 ▶). In the electron-density map of the new structure, these residues are clearly seen to protrude towards the central cavity of the receptor. Arg103 is well ordered and forms a van der Waals contact with and a hydrogen bond to the main-chain O atom of Pro27 from the N-terminus of the symmetry-related molecule, while Ser101, which is found at a distance of 3.4 Å from Met165, packs against and stabilizes the J–K loop. In the new structure, the D–E loop is also observed to pack against another D–E loop from a symmetry-related molecule. Finally, the residues in the ‘class-specificity loop’ (Himanen et al., 1998 ▶) are particularly well ordered and can be seen in the electron-density map (Fig. 2 ▶ b).
The ability of EphB2 to bind the cross-class ephrin-A5 with considerably lower affinity than the inter-class ephrin-B2 (Himanen et al., 2004 ▶) makes the ephrin-A5–EphB2 interaction a potential platform for high-throughput screening for binding inhibitors. The G–H loop of ephrin undergoes a distinct conformational rearrangement upon receptor binding and serves as a starting point for the development of structure-based Eph antagonists (Nikolov et al., 2005 ▶, 2007 ▶; Xu et al., 2008 ▶). However, the molecular details of the conformational rearrangements that create a hydrophobic interaction pocket on the surface of the receptor, wrapping around the ligand loop, have thus far been elusive. Fig. 3 ▶ compares the improved model of the unbound EphB2 with the ephrin-A5-bound structure (Himanen et al., 2004 ▶) and reveals a striking movement of the J–K loop upon binding. This movement towards the bound ligand is considerably more dramatic (13 Å) than the movement of the disulfide-stabilized G–H loop (5 Å) that was observed and calculated for the ephrin-B2-bound EphB2 based on the original lower resolution structure (Himanen et al., 2001 ▶). The movement of the D–E loop is also prominent, showing a maximum movement of 9 Å for the EphB2–ephrin-A5 complex. As shown previously in the comparison of the two peptide-bound receptor structures, EphB2–SNEW and EphB4–TNYL (Chrencik, Brooun, Recht et al., 2006 ▶; Chrencik et al., 2007 ▶), differences between various receptor–ligand pairs can be considerable with respect to the movement of the loops upon binding. However, within the B-class Eph receptors, movement of the loops towards the bound ligand appears to be obligatory. In particular, the induced-fit mechanism for the binding of the lower affinity ephrin-A5 ligand to EphB2 shown in this study proves the principle that high-affinity low-molecular-weight compounds can potentially be found that can efficiently inhibit the ligand binding and can thus be developed into Eph antagonists. Once high-resolution structures of unbound and bound EphA receptors have been determined, it will be possible to evaluate whether the dramatic movement of the J–K, G–H and D–E loops is universal throughout the entire Eph-receptor family.
Figure 3.

Conformational changes in EphB2 upon binding to ephrin-A5. Free EphB2 (green) is shown superimposed on the structure of ephrin-A5-bound EphB2 (magenta; Himanen et al., 2004 ▶). A striking movement (13 Å) of the J–K receptor loop towards the ligand occurs upon binding. The movement of the D–E loop is also prominent, showing a maximum movement of 9 Å for the receptor in the EphB2–ephrin-A5 complex compared with the free receptor.
While the structural flexibility of the loops participating in B-class Eph–ephrin binding has been suggested to be universally important for receptor–ligand recognition (Ran et al., 2008 ▶), the closing of the hydrophobic cavity on the surface of the receptor in all known complex structures (Himanen et al., 2001 ▶, 2004 ▶; Chrencik, Brooun, Kraus et al., 2006 ▶; Chrencik, Brooun, Recht et al., 2006 ▶; Chrencik et al., 2007 ▶) appears to be characteristic of B-class Eph receptors. Overall, the new structure presented here proves the closing of the receptor cavity after ligand binding, allowing the B-class receptors to use an induced-fit mechanism to recognize and accommodate ligands. To date this model has been speculative, since the two loops indispensable for binding (J–K and D–E) were not visible in the earlier structure of the unbound EphB2 receptor. The finding is of particular interest in light of the search for Eph antagonists and agonists that can potentially be developed into anticancer drugs.
Supplementary Material
PDB reference: EphB2 ligand-binding domain, 3etp, r3etpsf
Table 1. Summary of crystallographic analysis.
Values in parentheses are for the highest resolution shell.
| Resolution (Å) | 50.0–2.0 (2.05–2.0) |
| Wavelength (Å) | 0.9795 |
| Temperature (K) | 100 |
| Completeness (I > 0) (%) | 96.4 (71.6) |
| Total No. of reflections | 135107 (4104) |
| I/σ(I) | 48.0 (3.5) |
| Rmerge† (%) | 4.8 (30.0) |
| Space group | P41212 |
| Unit-cell parameters (Å) | a = 54.03, b = 54.03, c = 157.41 |
| Refinement | |
| Reflections (working/test) | 15158/806 (911/39) |
| No. of residues | 183 |
| Rcryst/Rfree | 19.8/26.3 (26.0/35.7) |
| R.m.s. deviations‡ | |
| Bonds (Å) | 0.022 |
| Angles (°) | 2.085 |
| Average B factor (Å2) | 42.5 |
| Ramachandran analysis, residues in (%) | |
| Favored regions | 95.7 |
| Allowed regions | 3.8 |
| Disallowed regions | 0.5§ |
R
merge = 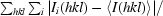
 , where Ii(hkl) is the observed intensity and 〈I(hkl)〉 is the average intensity obtained from multiple observations of symmetry-related reflections.
, where Ii(hkl) is the observed intensity and 〈I(hkl)〉 is the average intensity obtained from multiple observations of symmetry-related reflections.
The r.m.s. deviation in bond lengths and angles are the respective root-mean-square deviations from ideal values.
The only residue found in the disallowed region of the Ramachandran plot is the pre-cis-proline Asn141. Its conformation can clearly be determined, as shown in Fig. 2 ▶(b).
Acknowledgments
We thank the staff at the NSLS synchrotron for the help with data collection. The studies were financially supported by the National Institutes of Health (GM75886; NS38486 to DBN) and the Academy of Finland (to SP).
References
- Adams, R. H. & Klein, R. (2000). Trends Cardiovasc. Med.10, 183–188. [DOI] [PubMed]
- Boyd, A. W. & Lackmann, M. (2001). Sci. STKE, 112, RE20. [DOI] [PubMed]
- Chrencik, J. E., Brooun, A., Kraus, M. L., Recht, M. I., Kolatkar, A. R., Han, G. W., Seifert, J. M., Widmer, H., Auer, M. & Kuhn, P. (2006). J. Biol. Chem.281, 28185–28192. [DOI] [PubMed]
- Chrencik, J. E., Brooun, A., Recht, M. I., Kraus, M. L., Koolpe, M., Kolatkar, A. R., Bruce, R. H., Martiny-Baron, G., Widmer, H., Pasquale, E. B. & Kuhn, P. (2006). Structure, 14, 321–330. [DOI] [PubMed]
- Chrencik, J. E., Brooun, A., Recht, M. I., Nicola, G., Davis, L. K., Abagyan, R., Widmer, H., Pasquale, E. B. & Kuhn, P. (2007). J. Biol. Chem.282, 36505–36513. [DOI] [PMC free article] [PubMed]
- Collaborative Computational Project, Number 4 (1994). Acta Cryst. D50, 760–763.
- Dodelet, V. C. & Pasquale, E. B. (2000). Oncogene, 19, 5614–5619. [DOI] [PubMed]
- Flanagan, J. G. & Vanderhaeghen, P. (1998). Annu. Rev. Neurosci.21, 309–345. [DOI] [PubMed]
- Gale, N. W., Holland, S. J., Valenzuela, D. M., Flenniken, A., Pan, L., Ryan, T. E., Henkemeyer, M., Strebhardt, K., Hirai, H., Wilkinson, D. G., Pawson, T., Davis, S. & Yankopoulos, G. D. (1996). Neuron, 17, 9–19. [DOI] [PubMed]
- Himanen, J. P., Chumley, M. J., Lackmann, M., Li, C., Barton, W. A., Jeffrey, P. D., Vearing, C., Geleick, D., Feldheim, D. A., Boyd, A. W., Henkemeyer, M. & Nikolov, D. B. (2004). Nature Neurosci.7, 501–509. [DOI] [PubMed]
- Himanen, J. P., Henkemeyer, M. & Nikolov, D. B. (1998). Nature (London), 396, 486–491. [DOI] [PubMed]
- Himanen, J. P., Rajashankar, K. R., Lackmann, M., Cowan, C. A., Henkemeyer, M. & Nikolov, D. B. (2001). Nature (London), 414, 933–938. [DOI] [PubMed]
- Jones, T. A., Zou, J.-Y., Cowan, S. W. & Kjeldgaard, M. (1991). Acta Cryst. A47, 110–119. [DOI] [PubMed]
- Klein, R. (2001). Curr. Opin. Cell Biol.13, 196–203. [DOI] [PubMed]
- Koolpe, M., Burgess, R., Dail, M. & Pasquale, E. B. (2005). J. Biol. Chem.280, 17301–17311. [DOI] [PubMed]
- Koolpe, M., Dail, M. & Pasquale, E. B. (2002). J. Biol. Chem.277, 46974–46979. [DOI] [PubMed]
- Kullander, K. & Klein, R. (2002). Nature Rev. Mol. Cell Biol.3, 475–486. [DOI] [PubMed]
- Labrador, J. P., Brambilla, R. & Klein, R. (1997). EMBO J.16, 3889–3897. [DOI] [PMC free article] [PubMed]
- Lackmann, M. & Boyd, A. W. (2008). Sci. Signal.1, re2. [DOI] [PubMed]
- Murai, K. K., Nguyen, L. N., Koolpe, M., McLennan, R., Krull, C. E. & Pasquale, E. B. (2003). Mol. Cell. Neurosci.24, 1000–1011. [DOI] [PubMed]
- Murshudov, G. N., Vagin, A. A. & Dodson, E. J. (1997). Acta Cryst. D53, 240–255. [DOI] [PubMed]
- Navaza, J. (1994). Acta Cryst. A50, 157–163.
- Nikolov, D. B., Li, C., Barton, W. & Himanen, J. P. (2005). Biochemistry, 44, 10947–10953. [DOI] [PubMed]
- Nikolov, D., Li, C., Jeffrey, P. & Himanen, J. P. (2007). Protein Sci.16, 996–1000. [DOI] [PMC free article] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol.276, 307–326. [DOI] [PubMed]
- Pasquale, E. B. (2008). Cell, 133, 38–52. [DOI] [PubMed]
- Ran, X., Qin, H., Liu, J., Fan, J. S., Shi, J. & Song, J. (2008). Proteins, 72, 1019–1029. [DOI] [PubMed]
- Xu, K., Rajashankar, K. R., Chan, Y.-P., Himanen, J. P., Broder, C. C. & Nikolov, D. B. (2008). Proc. Natl Acad. Sci. USA, 105, 9953–9958. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: EphB2 ligand-binding domain, 3etp, r3etpsf
PDB reference: EphB2 ligand-binding domain, 3etp, r3etpsf