

Preliminary neutron crystallographic data from the serine protease proteinase K have been recorded using the LADI-III diffractometer at the Institut Laue–Langevin. The results illustrate the feasibility of a full neutron structural analysis aimed at further understanding the catalytic mechanism of proteinase K.
Keywords: neutron diffraction, proteinase K, serine protease
Abstract
A preliminary neutron crystallographic study of the proteolytic enzyme proteinase K is presented. Large hydrogenated crystals were prepared in deuterated crystallization buffer using the vapor-diffusion method. Data were collected to a resolution of 2.3 Å on the LADI-III diffractometer at the Institut Laue–Langevin (ILL) in 2.5 d. The results demonstrate the feasibility of a full neutron crystallographic analysis of this structure with the aim of providing relevant information on the location of H atoms, particularly at the active site. This information will contribute to further understanding of the molecular mechanisms underlying the catalytic activity of proteinase K and to an enriched understanding of the subtilisin clan of serine proteases.
1. Introduction
H atoms play a role in many enzyme mechanisms, but X-ray diffraction can only locate H atoms directly when data can be obtained to better than 1.0 Å resolution and when the atoms are sufficiently well ordered (Howard et al., 2004 ▶). In contrast, neutron diffraction can determine the positions of H atoms at much lower resolutions (1.5–2.5 Å; Schoenborn, 1969 ▶). The enzyme used in this study is proteinase K, a member of the subtilisin clan, family S8 of the serine proteases (SPs). Complete amino-acid sequences are known for more than 170 subtilisins and they are found in eubacteria, archaebacteria, eukaryotes and viruses. Neutron diffraction has been used to determine the protonation state of the active site of trypsin (Kossiakoff & Spencer, 1980 ▶). Although the subtilisin clan of SPs and the chymotrypsin clan (of which trypsin is a member) have similar catalytic triads at their active sites, they are evolutionarily distinct (Rawlings & Barrett, 1994 ▶). While trypsin cleaves peptide bonds C-terminal to a basic amino-acid residue, proteinase K preferentially cleaves C-terminal to a hydrophobic or aromatic residue (Keil, 1992 ▶).
The serine protease family of enzymes catalyze proteolysis via a conserved active-site catalytic triad: histidine, aspartic acid and serine. One major question about the serine protease mechanism is whether His69 acts as a true chemical base towards serine or whether the histidine acts as a shuttle that transfers an H atom to the aspartate group. For the trypsin clan of serine proteases, neutron analysis of an inhibitor-bound form of trypsin permitted a definitive answer to this question: the histidine residue is the chemical base (Kossiakoff & Spencer, 1980 ▶, 1981 ▶). The subtilisin clan, of which proteinase K is a member, is evolutionarily distinct from the trypsin clan and the environment around the active site differs, opening the possibility of a different chemical environment for the catalytic triad.
The most potent known proteolytic enzyme, proteinase K is a 28.9 kDa protein that can hydrolyze keratin (Ebeling et al., 1974 ▶). Proteinase K is the main protease obtained from the fungus Tritirachium album. Proteinase K finds utility among molecular biologists for its ability to rapidly digest nucleases that might compromise a nucleic acid preparation, even in the presence of denaturants (such as SDS and urea; Hilz et al., 1975 ▶) or chelating agents. This endopeptidase depends on a Ca2+ ion for stability, but retains much of its protease activity even after incubation with EDTA (Bajorath et al., 1988 ▶). Previous work reported the crystallization of proteinase K under microgravity conditions, yielding the X-ray structure at 0.98 Å resolution (PDB code 1ic6) and allowing visualization of 46% of the H atoms above the 2σ level in F o − F c electron-density maps (Betzel et al., 2001 ▶), including those of the active-site catalytic triad.
The neutron work on subtilisin itself (Kossiakoff et al., 1991 ▶) and the 0.98 Å X-ray structure of proteinase K (Betzel et al., 2001 ▶) both deal with substrate-free forms of these enzymes, so the identity of the chemical base for proteinase K and for the subtilisin class of serine proteases remains an open question, making it desirable to obtain paired apo and inhibitor-bound neutron structures of proteinase K. Our study has the potential to confirm and extend the X-ray study, providing unambiguous coordinate determination for essentially all ordered H and D atoms in the apo proteinase K structure. While our focus is on proteinase K, it is our hope that this work will also enhance our understanding of the subtilisin class of serine proteases.
2. Methods
2.1. Crystallization
Proteinase K (Sigma–Aldrich P6556) was used directly without further purification. To reduce hydrogen incoherent scattering (Sears, 1992 ▶), all salt and buffer solutions were prepared in D2O with two exchanges by rotary evaporation. The protein was dissolved to ∼60 mg ml−1 (measured by A 280, E 1% = 14.2) in a buffer consisting of 20 mM HEPES pD 7 (where pD = pH-meter reading + 0.4; Glasoe & Long, 1960 ▶), 1 mM CaCl2 and 0.02% NaN3. D2O-grown crystals of a size suitable for neutron analysis (∼0.8 mm3) were obtained at room temperature using sitting-drop vapor diffusion. Obtaining sufficiently large crystals from deuterated solutions required reoptimization from published conditions. Proteinase K exhibits salting-in behavior that prevented effective macroseeding in our hands, so an autonucleation approach was employed. The well solution consisted of 500 µl 0.07 M sodium citrate–DCl pD 6.5, 1 M NaNO3 (prepared in D2O). Sample drops were prepared by adding 35 µl well solution to 35 µl protein solution. Crystals autonucleated within 1 d and grew to full size within one week. The number of nucleation events per drop was variable, so multiple drops were set up to obtain the crystal for the diffraction experiment. The crystal used for data collection is shown in Fig. 1 ▶. X-ray diffraction tests on a smaller crystal from a duplicate crystallization drop with a sealed-tube copper source showed that the crystals formed in the expected space group P43212 (unit-cell parameters a = b = 68.269, c = 108.343 Å). During crystal mounting in a quartz capillary, solution from the well was added to each side of the crystal to maintain crystal solvation and deuteration. The ends of the capillary were sealed with wax.
Figure 1.

Hydrogenated proteinase K crystals grown in deuterated buffer at pD 6.5 by vapor diffusion. The estimated crystal volume is 0.81 mm3. Visualization was by transmitted polarized white light.
2.2. Neutron and X-ray data collection and processing
Neutron quasi-Laue data were collected at 293 K on the LADI-3 beamline installed on end-station T17 of cold neutron guide H142 at ILL. The data set collected for proteinase K was composed of seven image frames of 8 h exposure each. As is typical for a Laue experiment, the crystal was held stationary at a different ϕ setting for each 8 h exposure; the angular interval between settings was 7°. A portion of the first frame of the quasi-Laue neutron diffraction data is shown in Fig. 2 ▶. While the angular range was 100% complete for this point group (Fig. 3 ▶), the overall completeness for the processed data only reached 70.8%. This issue is addressed further in §3.
Figure 2.

An enlarged view of one quarter of a quasi-Laue neutron diffraction image from proteinase K collected in 8 h on the LADI-III beamline at the Institut Laue–Langevin. The horizontal axis of the figure spans approximately half of the full detector width.
Figure 3.

The stereographic projection of averaged radial completeness demonstrates complete angular coverage of the unique octant (and thus full sampling of the asymmetric unit in reciprocal space), which resulted in an overall data completeness of 70.8%.
The neutron Laue data frames were processed using the Daresbury Laboratory LAUE suite program LAUEGEN, which was modified to account for the cylindrical geometry of the detector (Campbell et al., 1998 ▶). The program LSCALE (Arzt et al., 1999 ▶) was used to determine the wavelength-normalization curve using the intensities of symmetry-equivalent reflections measured at different wavelengths and to apply wavelength-normalization calculations to the observed data. The data were then scaled and merged in SCALA (Collaborative Computational Project, Number 4, 1994 ▶). Data-reduction statistics are summarized in Table 1 ▶.
Table 1. Data-reduction statistics.
Values in parentheses are for the highest resolution shell.
| Radiation type | X-ray | Neutron |
|---|---|---|
| Source and detector | Cu Kα, MAR345 | Institut Laue–Langevin, LADI-III |
| Wavelength (Å) | 1.54 | 3.25–4.1 |
| Settings or frames | 202 | 7 |
| Time per setting | 7 min | 8 h |
| Space group | P43212 | |
| Unit-cell parameters (Å) | a = b = 68.27, c = 108.34 | |
| Resolution range | 27.09–1.92 (2.02–1.92) | 43.81–2.30 (2.42–2.30) |
| Reflections, measured/unique | 150838/20131 (16912/2738) | 20063/7977 (2004/966) |
| Multiplicity | 7.5 (6.2) | 2.5 (2.1) |
| Completeness | 99.1 (94.5) | 70.8 (59.1) |
| Rmerge† | 0.069 (0.148) | 0.127 (0.197) |
| Rmeas‡ (all I+ and I−) | 0.076 (0.162) | 0.153 (0.246) |
| Rp.i.m.§ (all I+ and I−) | 0.027 (0.060) | 0.084 (0.142) |
| Mean I/σ(I) | 21.9 (16.4) | 7.0 (2.6) |
| Initial phasing Rwork/Rfree | 0.146/0.180 | 0.27/0.28 |
R
merge = 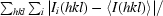
 , where I
i(hkl) is the intensity of the ith observation of reflection (hkl).
, where I
i(hkl) is the intensity of the ith observation of reflection (hkl).
Redundancy-independent merging statistic R
meas (also known as R
r.i.m.) = 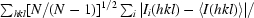 , where N is the redundancy of reflection hkl (Diederichs & Karplus, 1997 ▶; Weiss, 2001 ▶).
, where N is the redundancy of reflection hkl (Diederichs & Karplus, 1997 ▶; Weiss, 2001 ▶).
Precision-indicating merging statistic R
p.i.m. = 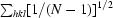
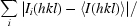 (Weiss, 2001 ▶).
(Weiss, 2001 ▶).
A smaller crystal from a duplicate crystallization condition was used to obtain a room-temperature X-ray data set to a resolution of 1.92 Å using a MAR345 detector on a Cu Kα home X-ray source. The X-ray data were integrated and scaled with the ELVES (Holton & Alber, 2004 ▶) interface to MOSFLM (Leslie et al., 1986 ▶) and SCALA (Collaborative Computational Project, Number 4, 1994 ▶).
2.3. Structure determination
The atomic coordinates of the protein chain from PDB entry 2prk (Betzel et al., 1988 ▶) were used directly for rigid-body X-ray structural refinement. After four cycles of refinement in REFMAC5 (Collaborative Computational Project, Number 4, 1994 ▶), each followed by rebuilding in Coot (Emsley & Cowtan, 2004 ▶), R work and R free stood at 0.146 and 0.180, respectively, indicating successful phasing. H atoms (and D atoms at exchangeable positions) were added to the X-ray model using the phenix.ready_set utility (Adams et al., 2002 ▶), providing the starting atomic coordinates for joint X-ray/neutron refinement. From these coordinates, initial values for neutron R work and R free were obtained from phenix.refine (Adams et al., 2002 ▶; 0.2719 and 0.2808, respectively), indicating successful initial neutron phasing. The way is now clear for full joint refinement and H/D analysis of apo proteinase K, particularly its active site.
3. Results and discussion
For a tetragonal unit cell of point group 422, complete angular data coverage can be obtained with 45° of data, provided that the rotation axis is parallel to the unique c axis and that the a or b axis lies along the direction of the beam at either the beginning or the end of the rotation range (Dauter & Wilson, 2006 ▶). After centering, the unique crystallographic c axis of this proteinase K crystal lay only 0.2° away from the instrument’s spindle axis ϕ, allowing coverage of the complete angular range of the asymmetric unit in reciprocal space with only 45° of data (Fig. 3 ▶). Thus, the high-order space-group symmetry combined with the favorable crystal alignment enabled complete angular coverage of reciprocal space in the short time available for this study, save for the portion eclipsed by the detector’s blind region. However, spatial overlap problems are common in Laue data collection, particularly from large unit-cell crystals (Helliwell, 1992 ▶), and the relatively low overall data completeness (70.8%) reflects this fact. Diffraction data were observed and processed to a resolution of 2.3 Å. A representative neutron Laue diffraction pattern is shown in Fig. 2 ▶. Data completeness is depicted in Fig. 3 ▶.
4. Future work
Full H/D modeling and joint X-ray/neutron refinement of this apo-enzyme crystal structure are under way. It is further anticipated that data will be collected from a soaked or cocrystallized inhibitor-bound transition-state analog during the future ILL scheduling periods, yielding a data set with acceptable completeness to a resolution of ∼2 Å. The final structure refinements will be carried out using both X-ray and neutron data recorded at the same temperature, using the phenix.refine program (Adams et al., 2002 ▶), which enables simultaneous cross-validated energy-weighted refinement against X-ray and neutron data. Such a study will provide direct accurate information about H-atom positions at the active site of proteinase K.
Acknowledgments
This research was sponsored by the Laboratory Directed Research and Development Program of Oak Ridge National Laboratory, managed by UT-Battelle LLC for the US Department of Energy. We gratefully acknowledge the assistance of Dr Monika Budayova-Spano (University Joseph Fourier/EMBL) and Mr Esko Oksanen (University of Helsinki) with data collection, as well as Dr Suzanne Z. Fisher, Dr Pavel Afonine and Professor Flora Meilleur for helpful discussion.
References
- Adams, P. D., Grosse-Kunstleve, R. W., Hung, L.-W., Ioerger, T. R., McCoy, A. J., Moriarty, N. W., Read, R. J., Sacchettini, J. C., Sauter, N. K. & Terwilliger, T. C. (2002). Acta Cryst. D58, 1948–1954. [DOI] [PubMed]
- Arzt, S., Campbell, J. W., Harding, M. M., Hao, Q. & Helliwell, J. R. (1999). J. Appl. Cryst.32, 554–562.
- Bajorath, J., Hinrichs, W. & Saenger, W. (1988). Eur. J. Biochem.176, 441–447. [DOI] [PubMed]
- Betzel, C., Bellemann, M., Pal, G. P., Bajorath, J., Saenger, W. & Wilson, K. S. (1988). Proteins, 4, 157–164. [DOI] [PubMed]
- Betzel, C., Gourinath, S., Kumar, P., Kaur, P., Perbandt, M., Eschenburg, S. & Singh, T. P. (2001). Biochemistry, 40, 3080–3088. [DOI] [PubMed]
- Campbell, J. W., Hao, Q., Harding, M. M., Nguti, N. D. & Wilkinson, C. (1998). J. Appl. Cryst.31, 496–502.
- Collaborative Computational Project, Number 4 (1994). Acta Cryst. D50, 760–763.
- Dauter, Z. & Wilson, K. S. (2006). International Tables for Crystallography, Vol. F, 1st online ed., edited by M. G. Rossmann & E. Arnold, pp. 183–188. Chester: International Union of Crystallography.
- Diederichs, K. & Karplus, P. A. (1997). Nature Struct. Biol.4, 269–275. [DOI] [PubMed]
- Ebeling, W., Hennrich, N., Klockow, M., Metz, H., Orth, H. D. & Lang, H. (1974). Eur. J. Biochem.47, 91–97. [DOI] [PubMed]
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed]
- Glasoe, P. K. & Long, F. A. (1960). J. Phys. Chem.64, 188–190.
- Helliwell, J. R. (1992). Philos. Trans. Phys. Sci. Eng.340, 221–232.
- Hilz, H., Wiegers, U. & Adamietz, P. (1975). Eur. J. Biochem.56, 103–108. [DOI] [PubMed]
- Holton, J. & Alber, T. (2004). Proc. Natl Acad. Sci. USA, 101, 1537–1542. [DOI] [PMC free article] [PubMed]
- Howard, E. I., Sanishvili, R., Cachau, R. E., Mitschler, A., Chevrier, B., Barth, P., Lamour, V., Van Zandt, M., Sibley, E., Bon, C., Moras, D., Schneider, T. R., Joachimiak, A. & Podjarny, A. (2004). Proteins, 55, 792–804. [DOI] [PubMed]
- Keil, B. (1992). Specificity of Proteolysis. Berlin/New York: Springer-Verlag.
- Kossiakoff, A. A. & Spencer, S. A. (1980). Nature (London), 288, 414–416. [DOI] [PubMed]
- Kossiakoff, A. A. & Spencer, S. A. (1981). Biochemistry, 20, 6462–6474. [DOI] [PubMed]
- Kossiakoff, A. A., Ultsch, M., White, S. & Eigenbrot, C. (1991). Biochemistry, 30, 1211–1221. [DOI] [PubMed]
- Leslie, A. G. W., Brick, P. & Wonacott, A. (1986). Daresbury Lab. Inf. Quart. Protein Crystallogr.18, 33–39.
- Rawlings, N. D. & Barrett, A. J. (1994). Methods Enzymol.244, 19–61. [DOI] [PMC free article] [PubMed]
- Schoenborn, B. P. (1969). Nature (London), 224, 143–146. [DOI] [PubMed]
- Sears, V. F. (1992). Neutron News, 3, 26–37.
- Weiss, M. S. (2001). J. Appl. Cryst.34, 130–135.