

The crystal structure of iron superoxide dismutase from the cold-adapted organism A. salmonicida has been determined at a resolution of 1.7 Å.
Keywords: superoxide dismutases, sodB, FeSOD, cold adaptation, Aliivibrio salmonicida
Abstract
Superoxide dismutases (SODs) are metalloenzymes that catalyse the dismutation of the superoxide radical anion into O2 and H2O2 in a two-step reaction. The crystal structure of the iron superoxide dismutase from the cold-adapted and fish-pathogenic bacterium Aliivibrio salmonicida (asFeSOD) has been determined and refined to 1.7 Å resolution. The protein has been characterized and compared with the closely related homologous iron superoxide dismutase from the mesophilic Escherichia coli (ecFeSOD) in an attempt to rationalize its environmental adaptation. ecFeSOD shares 75% identity with asFeSOD. Compared with the mesophilic FeSOD, the psychrophilic FeSOD has distinct temperature differences in residual activity and thermostability that do not seem to be related to structural differences such as intramolecular or intermolecular ion bonds, hydrogen bonds or cavity sizes. However, an increased net negative charge on the surface of asFeSOD may explain its lower thermostability compared with ecFeSOD. Activity measurements and differential scanning calorimetry measurements revealed that the psychrophilic asFeSOD had a thermostability that was significantly higher than the optimal growth temperature of the host organism.
1. Introduction
Oxidative stress to cells results from the presence of reactive oxygen species (ROS) such as the superoxide radical anion (O2 ·−), hydrogen peroxide (H2O2) and the hydroxyl radical (OH·). ROS are harmful to the cell and may oxidize biologically important molecules such as lipids, proteins or nucleic acids. Superoxide dismutases (SODs) play a major role in detoxifying harmful oxygen species and are found in organisms ranging from microorganisms to plants and animals, including humans. SODs catalyse the dismutation of superoxide to oxygen and hydrogen peroxide in a two-step reaction in which the metal ion (M = Fe, Mn, Cu or Ni) cycles between the 2+ and 3+ oxidation states (Lavelle et al., 1977 ▶),
Four different SODs have been identified in bacterial strains and they have been divided into four groups depending on their metal cofactor and molecular structure: two highly homologous SODs, manganese-containing and iron-containing enzymes (MnSOD and FeSOD, respectively), a copper–zinc SOD (CuZnSOD) and a nickel-containing SOD (NiSOD). CuZnSODs are commonly distributed in the periplasm of prokaryotes (Steinman & Ely, 1990 ▶; Benov & Fridovich, 1994 ▶), while FeSODs are mainly found in the cytoplasm, MnSODs are found in both the cytoplasm and periplasm of prokaryotes (Hopkin et al., 1992 ▶; Steinman et al., 1994 ▶; Geissdorfer et al., 1997 ▶; Leclere et al., 2001 ▶) and NiSODs have mostly been found in several Streptomyces species (Kim et al., 1996 ▶; Youn et al., 1996 ▶). However, recent structural bioinformatics studies have revealed that the gene encoding NiSOD can be found in many organisms, including both eukaryotes and prokaryotes, and it has been suggested that NiSODs are found in organisms that do not possess FeSODs (Dupont et al., 2008 ▶).
The different groups of SODs are structurally distinct from each other. FeSODs and MnSODs constitute a class of structurally similar metalloenzymes and appear as homodimers and infrequently as homotetramers. They are constructed of helix-rich domains, whereas CuZnSODs are homodimers in which the essential structural motif is a β-barrel (Tainer et al., 1982 ▶) and NiSODs are described as homohexamers consisting of four-helix-bundle subunits (Barondeau et al., 2004 ▶; Wuerges et al., 2004 ▶).
In MnSOD and FeSOD, a metal ion is bound to each monomer. The monomer consists of two domains; two amino-acid residues from each domain contribute to coordination of the metal ion (Stallings et al., 1983 ▶, 1984 ▶). The N-terminal domain is constructed of two long helices that are separated by a shorter and more variable helix, whereas the C-terminal domain is constructed of a three-stranded β-sheet in which the middle strand is antiparallel to the other two. The β-sheet in the C-terminal domain is flanked on both sides by a total of four α-helices. The metal ion in each monomer is coordinated by three His residues, one Asp residue and one water molecule or OH− ion. The interface between the monomers is highly conserved; it is predominantly hydrophobic and generates funnels that allow the substrate to enter the active sites (Lah et al., 1995 ▶). Because the molecular environment around the metal is highly conserved in the various FeSOD and MnSOD structures, some SODs can function with either Mn or Fe (Meier et al., 1982 ▶, 1994 ▶; Gregory & Dapper, 1983 ▶; Amano et al., 1992 ▶), while some SODs are only functional if bound to the original metal despite being able to bind either Mn or Fe (Ose & Fridovich, 1979 ▶; Yamakura & Suzuki, 1980 ▶).
Superoxide dismutases from a diverse group of organisms, including psychrophilic, mesophilic and thermophilic FeSODs, MnSODs and CuZnSODs, have been characterized and shown to possess remarkably high thermostability (Lepock et al., 1990 ▶; Lim et al., 1997 ▶; Knapp et al., 1999 ▶; Kardinahl et al., 2000 ▶; Hunter et al., 2002 ▶; Schafer & Kardinahl, 2003 ▶; Castellano et al., 2006 ▶). This might reflect the important and distinct role of SODs in oxygen tolerance and evolution.
ROS are mainly produced as byproducts of aerobic metabolism. However, Aliivibrio salmonicida is a pathogenic bacterium that causes cold-water vibriosis in Atlantic salmon (Salmo salar), rainbow trout (Oncorhynchus mykiss) and Atlantic cod (Gadus morhua) (Egidius et al., 1986 ▶; Wiik et al., 1989 ▶; Sorum et al., 1990 ▶). Being pathogenic, A. salmonicida has to withstand the defence mechanism of the host, including the oxidative burst from the macrophages in the fish. During the oxidative burst, large quantities of ROS are generated and superoxide dismutases are involved in the first-line defence mechanism against these. In Escherichia coli, suppressed SOD activity leads to several oxygen-dependent phenotypic alterations, such as serious defects in amino-acid biosynthesis (Carlioz & Touati, 1986 ▶), structural instability in the cell envelope (Imlay & Fridovich, 1992 ▶) and an increase in the amount of spontaneous mutagenesis (Farr et al., 1986 ▶).
In addition to being pathogenic, A. salmonicida is a psychrophilic bacterium, with an optimum growth temperature of 288 K. The relative ease of crystallization of thermophilic proteins has led to several structural studies that have revealed molecular mechanisms of heat adaptation by comparing thermophilic proteins with their mesophilic homologues. Owing to the limited number of available structures, knowledge of the molecular mechanism of cold adaptation is limited. However, some common features of psychrophilic proteins have emerged. For instance, fewer intramolecular and intermolecular interactions are involved in stabilization in psychrophilic proteins compared with thermophilic proteins (Aghajari et al., 1998 ▶; Kim et al., 1999 ▶; Van Petegem et al., 2003 ▶). Another common feature of psychrophilic proteins seems to be a lower stability owing to increased flexibility (Fields, 2001 ▶; D’Amico et al., 2002 ▶). In addition, a net negative charge on the surfaces of cold-adapted enzymes has been observed and implicated as a common feature of psychrophilic enzymes (Smalas et al., 2000 ▶; Siddiqui & Cavicchioli, 2006 ▶).
The crystal structures of FeSODs from mesophilic and thermophilic organisms have previously been solved. However, the three-dimensional structure of FeSOD from A. salmonicida (asFeSOD) reported here is the first crystal structure of a superoxide dismutase from a psychrophilic organism. In an attempt to elucidate the nature of asFeSOD, we have compared its structure, temperature-dependent residual activity and biochemical thermostability with those of FeSOD from the mesophilic E. coli (ecFeSOD).
2. Materials and methods
2.1. Bacterial strains, plasmids and E. coli FeSOD
Genomic DNA from A. salmonicida LFI1238 isolated from cod (G. morhua) was extracted using the standard phenol/chloroform DNA-isolation protocol (Sambrook et al., 1989 ▶). The donor vector pDONR201 and the destination vector pDEST14 from the Gateway system (Invitrogen Life Technologies, Great Britain) were used to clone the sodB gene into an expression strain. ecFeSOD was purchased from Sigma–Aldrich (USA).
2.2. Sequence analysis
The NCBI server (http://www.ncbi.nlm.nih.gov/blast/Blast.cgi) was used for BLAST searches. Alignments were produced using ClustalW (Thompson et al., 2000 ▶).
2.3. Identification of the sodB gene in A. salmonicida
Based on alignment of both the FeSOD amino-acid sequence and nucleotide sequence of various Vibrio species, conserved sequence regions were chosen for the design of degenerate primers. A 300 bp sodB fragment was generated by PCR using 150–1200 ng genomic A. salmonicida DNA as template. PCR was carried out using 5 µl 10× Taq buffer (Promega, USA), 1.5 mM MgCl2, 0.2 mM dNTP mix, 10 µM upstream (5′-CACGGTAARCACCACAACACTTACG-3′) and downstream (5′-ACMARCCAAGTCCAAGAWGAACC-3′) degenerate primers (Invitrogen Life Technologies, Great Britain) and 1.5 U Taq polymerase (Promega, USA) in a final volume of 50 µl. PCR amplification was performed at 367 K for 6 min followed by 30 cycles of 30 s at 367 K, 1 min at 323 K and 2 min at 345 K and then 345 K for 5 min; the reaction mixture was then kept at 277 K. The PCR product was purified using the QIAquick PCR Purification Kit (Qiagen, Germany) according to the manufacturer’s protocol.
2.4. DNA sequencing
DNA sequencing was performed according to the protocol supplied by Applied Biosystems (USA) using the PE Biosystems BigDye Terminator Cycle Sequencing Kit version 3.0/3.1, an ABI 377 Genetic Analyser and ABI Sequence Analysis software version 3.0.
2.5. Southern hybridization
An A. salmonicida BAC library (Molecular Engines Laboratories, France) was used to find the clone harbouring the A. salmonicida sodB gene. Southern hybridization using a 299 bp sodB PCR product as a probe was performed using the DIG High Prime DNA Labelling and Detection Starter Kit II (Roche, Germany). A Lumi-Imager F1 with LumiAnalyst 3.0 software (Boehringer Mannheim, Germany) was used for analysis, according to the manual supplied by the manufacturer. The hybridization temperature was set to 315 K.
2.6. Isolation of the BAC clone harbouring sodB and sequencing of the full-length sodB gene
BAC clones were identified by Southern hybridization. Clones harbouring the sodB gene were grown overnight in Luria–Bertani (LB) liquid medium containing 12.5 µg ml−1 chloramphenicol. BAC DNA harbouring the sodB gene was purified using NucleoBond BAC 100 (Macherey-Nagel, Germany) following the manufacturer’s protocol. To verify that the correct clone had been isolated, the 299 bp sodB fragment was generated by PCR using BAC DNA as template and sequencing was carried out as described above. To obtain the complete sequence of the sodB gene, primer walking was performed on 1–2 µg purified BAC DNA using 5′-CAACTTTGGTTCATCTTGGACTTGG-3′ and 5′-AGCGTCTTGCCTTCAAATTCAGTCC-3′ primers. The full-length sodB gene was amplified by PCR with 5′-TAGAGCACATATTTTTGGA-3′ and 5′-TTATTTTATTGAAAGGGCA-3′ primers using 0.1–1 µg purified BAC DNA and 500 ng A. salmonicida genomic DNA as template. The primers were purchased from Invitrogen Life Technologies and Sigma–Genosys (Great Britain) and sequencing was carried out as described above.
2.7. Cloning
The Gateway system (Invitrogen Life Technologies, Great Britain) was used to clone the genes into the pDONR221 donor vector and pDEST14 destination vector according to the manufacturer’s protocol using the forward and reverse primers 5′-GGGGACAAGTTTGTACAAAAAAGCAGGCTTCGAAGGAGATAGAACCAATGTCATTTGAAT-3′ and 5′-GGGGACCACTTTGTACAAGAAAGCTGGGTCTTACTTAGCTAGGTTTTCTTCTAC-3′, respectively. Entry and expression clones were transformed into competent E. coli DH5α cells. Colony PCR was used to verify that the clones used for further work contained the gene of interest. Plasmids were extracted by alkali lysis and 2-propanol precipitation (Sambrook et al., 1989 ▶) or by using a Wizard Plus SV Miniprep kit (Promega GmbH, Germany). Before expression clones were made, all entry clones were sequenced as described previously using M13 (Sigma–Genosys) primers (5′-GTAAAACGACGGCCAG-3′ and 5′-CAGGAAACAGCTATGAC-3).
2.8. Expression
Expression was performed using a 6 × 0.5 l Sixfors fermenter (Infors, Switzerland). A 100 ml pre-culture of E. coli BL21 (DE3) transformed with pDEST14 containing the sodB gene was used to inoculate 3 l of 2× LB medium supplemented with 20 mM glucose and 100 µg ml−1 ampicillin. The cells were grown at 310 K until the OD600 reached about 3.0; the temperature was then set to 303 K and the cells were incubated for a further 30 min before expression was induced with 1 mM IPTG. When needed, 4 ml 20% glucose was added to the growth medium of each of the 0.5 l incubators to avoid glucose starvation. The cells were harvested about 8 h after induction by centrifugation at 5000g for 30 min at 277 K, resuspended in extraction buffer (50 mM Tris–HCl pH 7.4) to an OD600 of about 70 and stored at 203 K.
2.9. Protein purification
Before the cells were disrupted by sonication on ice, EDTA-free proteinase inhibitors (Roche) and DNaseI (Sigma–Aldrich) were added to the solution. During sonication using a Vibracell ultrasonic processor VCX 750 (Sonics and Materials, Switzerland), the pulse on/off was set to 9.9 s, the temperature to 288 K, the output to 40% and the timer to 45 min. The recombinant proteins were isolated from cell debris by centrifugation at 25 000g for 80 min at 277 K. asFeSOD was purified using the ÄKTA FPLC and ÄKTA Explorer Purification System and columns obtained from Amersham Pharmacia Biotech (Uppsala, Sweden). All enzyme-containing solutions were kept on ice or at 277 K, apart from during the purification steps, which were performed at room temperature or 277 K. The protein extract was loaded onto a Q-Sepharose Fast Flow (FF) column (1.6/10) equilibrated in buffer A (50 mM Tris–HCl pH 7.5) using a flow rate of 5 ml min−1. The column was washed with two column volumes of buffer A and the asFeSOD enzyme was eluted with a gradient of 0–100% buffer B (buffer A containing 1 M NaCl). Fractions containing asFeSOD were collected and pooled; ammonium sulfate was then added to 1.5 M and applied onto a Phenyl Sepharose (High Substitution) FF column (1.6/10) equilibrated in buffer A containing 1.5 M ammonium sulfate using a flow rate of 5 ml min−1. The column was washed with two column volumes of buffer A containing 1.5 M ammonium sulfate and the asFeSOD enzyme was eluted with a gradient of 0–100% buffer A. Finally, fractions containing asFeSOD were pooled, diluted in buffer A and concentrated using an Amicon Ultra-15 centrifugal filter column with a cutoff of 10 kDa (Millipore, USA).
2.10. Protein concentration
The protein concentration was determined using the BCA protein-assay reagent in a microtitre plate (Pierce, USA) with BSA as standard or by using a NanoDrop spectrophotometer (Thermo Scientific, USA); both methods were performed according to the manufacturer’s protocol. The same method was used for the individual samples during an experiment: the BCA protein assay (Pierce) was used for activity measurements and the NanoDrop spectrophotometer (Thermo Scientific) was used for DSC measurements.
2.11. Superoxide activity measurements
The SOD Assay Kit–WST (Fluka, Sigma–Aldrich) was used to determine the specific activities of asFeSOD and ecFeSOD. The measurements were performed according to the manufacturer’s protocol. WST-1 [2-(4-iodophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium, monosodium salt] is a soluble tetrazolium salt that produces a water-soluble formazan dye upon reduction by a superoxide anion. The rate of the reduction by O2 − is linearly related to xanthine oxidase activity and is inhibited by SOD. Thus, the IC50 (50% inhibition activity of SOD or SOD-like materials) can be determined by a colorimetric method. For the standard curve, solutions of ecFeSOD (Sigma–Aldrich) were prepared at concentrations of 500, 200, 100, 50, 20, 10, 5 and 1 U ml−1 in the SOD Assay Kit–WST dilution buffer before appropriate dilutions of asFeSOD and ecFeSOD were measured. Before the addition of 200 µl WST working solution and 20 µl enzyme working solution (supplied by the manufacturer) to the wells, 20 µl of the asFeSOD or ecFeSOD solutions and three blank solutions (water blank 1, sample blank 2 and water blank 3) were transferred in triplets into the microplate wells. For the blank 2 and 3 solutions, 20 µl dilution buffer was added. The reactions were incubated at 295 K for 20 min and the absorbance was read at 405 nm in a Vmax microplate reader (Molecular Devices, USA). The inhibition rate (SOD activity) was calculated using the equation
and plotted onto an inhibition curve. The asFeSOD and ecFeSOD activity in the samples was determined from the curve in U ml−1 and used for calculations of the specific activity.
2.12. SDS–PAGE
SDS–PAGE was performed using NuPAGE 4–12% bis-tris gels run in MES buffer and stained with Simply Blue Safe Stain, all according to the manufacturer’s protocol (Invitrogen).
2.13. Residual SOD activity at different temperatures
Residual SOD activity at different temperatures was examined by incubating asFeSOD and ecFeSOD in dilution buffer (50 mM Tris–HCl pH 7.5, 100 mM NaCl). Incubations were performed at 273, 283, 295, 303, 313, 323 and 326 K for asFeSOD, and at 273, 283, 295, 303, 313, 323, 333, 343 and 346 K for ecFeSOD. The pH of the buffers was adjusted at the different temperatures. Samples were collected and transferred to ice after 30 min incubation and 2 min pre-incubation at the different temperatures. Residual activity was measured using the SOD Assay Kit–WST from Fluka, Sigma–Aldrich. The highest individual activity measured was set to 100%. Triplets were made of each reaction in the activity measurements in addition to performing the experiment multiple times at the different temperatures.
2.14. Differential scanning calorimetry (DSC)
DSC experiments were performed on a Nano-Differential Scanning Calorimeter III (model CSC6300; Calorimetry Sciences Corp., USA). Native enzyme preparations of asFeSOD and ecFeSOD were filtered using a 0.45 µm Spin-X centrifuge tube filter (Corning, Netherlands) before dialysing overnight at 277 K. Enzyme-containing solutions were dialysed against 1 l dialysis buffer (20 mM HEPES, 100 mM NaCl pH 7.5 for asFeSOD and 30 mM NaH2PO4 pH 7.0, 20 mM NaCl for ecFeSOD) using Slide-A-Lyzer dialysis cassettes from Pierce (2 kDa cutoff). Reference buffers and samples were carefully degassed before loading into the DSC cells and the dialysates were used as blank references in each DSC run. The scans were performed at a constant pressure of 304 kPa in the range 283–348 or 283–383 K with a heating rate of 1 K min−1. Different batches of protein sample were used for both asFeSOD and ecFeSOD, as well as both 20 mM HEPES buffer pH 7.5 and 100 mM NaCl and 30 mM NaH2PO4 buffer with different salt concentrations for ecFeSOD, all of which gave the same result as presented in Fig. 3(b).
2.15. Crystallization and data collection
Crystals of asFeSOD were grown by vapour diffusion using the hanging-drop method. The best crystals were grown by mixing 1 µl drops of 6.5 mg ml−1 protein solution with a solution containing 0.1 M Tris–HCl, 1.4 M sodium citrate pH 8.5. The drops were equilibrated at 291 K; crystals appeared within a few days. The crystals tested for diffraction had overall dimensions of about 200 × 200 × 200 µm and were prepared for data collection at cryogenic temperature by rapid transfer into mineral oil before transfer into liquid nitrogen. During data collection, the crystal was maintained at 100 K using a nitrogen cold stream (Oxford Instruments, England). X-ray diffraction data were collected using a MAR345 image-plate detector on the Swiss–Norwegian Beamline (SNBL) at the European Synchrotron Radiation Facility (ESRF), Grenoble, France. The data were indexed, integrated and scaled using the XDS program package (Kabsch, 1993 ▶); the intensities were then converted to structure factors using the CCP4 program TRUNCATE (Collaborative Computational Project, Number 4, 1994 ▶). The crystals were trigonal, with unit-cell parameters a = b = 70.72, c = 170.25 Å, α = β = 90, γ = 120°. Systematic absences in the collected data identified a threefold screw axis along the c axis, with P3121 or P3221 being the only possible choices of space group. This was also indicated by analysis of the intensity data using the Bruker–Nonius program XPREP. The solvent content was estimated to be around 57%, with a Matthews coefficient of 2.87 Å3 Da−1, assuming the presence of two protein molecules in the asymmetric unit. Data-collection statistics are presented in Table 1 ▶.
Table 1. Data-collection and refinement summary.
Values in parentheses are for the highest resolution shell.
| Data collection | |
| Resolution range (Å) | 15–1.70 (1.79–1.70) |
| No. of unique reflections | 55155 |
| Redundancy | 5.8 (5.5) |
| Rmerge† (%) | 11.9 (34.0) |
| Completeness (%) | 99.8 (98.6) |
| Mean I/σ(I) | 9.5 (3.6) |
| Wilson B factor (Å2) | 15.5 |
| Refinement statistics | |
| R value (%) | 20.5 (32.7) |
| Free R value (%) | 23.9 (34.4) |
| Deviations from ideal geometry | |
| Bond lengths (Å) | 0.014 |
| Bond angles (°) | 1.467 |
| ESU‡ (Å) | 0.088 |
| Average B values (Å2) | |
| Protein atoms | 20.6 |
| Fe atoms (2) | 12.0 |
| Water molecules (208) | 26.7 |
| All atoms | 21.0 |
| Ramachandran plot (%) [No. of residues] | |
| Most favoured | 91.4 [307] |
| Additionally allowed | 6.8 [23] |
| Generously allowed§ | 1.2 [4] |
| Disallowed§ | 0.6 [2] |
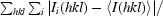
 , where Ii(hkl) is the ith measurement of reflection hkl and 〈I(hkl)〉 is the weighted mean of all measurements of hkl.
, where Ii(hkl) is the ith measurement of reflection hkl and 〈I(hkl)〉 is the weighted mean of all measurements of hkl.
Estimated overall coordinate error from REFMAC5 based on maximum likelihood.
The outliers are Ala85 and Arg169 (generously allowed) and Asn141 (disallowed) in both polypeptide chains. These residues are conserved in ecFeSOD and are also outliers in the Ramachandran plot for that crystal structure, indicating a conserved structural role.
2.16. Structure determination and refinement
The crystal structure of asFeSOD was determined by molecular replacement using MOLREP (Collaborative Computational Project, Number 4, 1994 ▶), in which the deposited structure of ecFeSOD (Lah et al., 1995 ▶) with a sequence identity of 75.3% was used as a starting model. The automated program functions of MOLREP were utilized in order to obtain a model with the best fit to the search model, i.e. the side chains of the ecFeSOD model were stripped in order to fit the sequence of asFeSOD. A high-resolution cutoff of 4 Å was applied to the data. The correct space group was determined to be P3221 by trial and error in MOLREP and one well resolved solution that identified both monomers in the asymmetric unit was found. This solution had a score function of 0.674 and an R factor of 35.8% (the next solution had a score function of 0.373 and an R factor of 50.3%). Automated model building with ARP/wARP (Perrakis et al., 1999 ▶) using all available reflections built 381 of a possible 388 residues (each asFeSOD monomer comprises 194 amino-acid residues). Subsequent refinement was performed by alternating cycles of manual adjustment with O (Jones et al., 1991 ▶) followed by positional refinement with REFMAC5 (Murshudov et al., 1999 ▶). The final R work and R free values were 20.5% and 23.9%, respectively, with acceptable protein stereochemistry. The final model of asFeSOD consists of 386 amino-acid residues in two polypeptide chains comprising residues 2–194 of each monomer. One Fe atom was also identified in each monomer and was added during the course of refinement, as well as a total of 208 water molecules. The data-collection and refinement statistics are summarized in Table 1 ▶.
3. Results and discussion
3.1. Gene identification
The sequence amplified by the degenerate primers encoded part of an open reading frame that displayed a strong similarity to SOD-encoding genes. Homology searches with the deduced amino-acid sequence revealed a high homology with iron-containing SODs. assodB and its flanking regions were found in the total sequence obtained, revealing a gene of 585 bp. The promoter region of assodB showed putative −10, −35 and SD consensus sequences. The presence of a palindromic region followed by a stretch of T residues downstream of the assodB sequence indicated a transcriptional terminator.
3.2. Amino-acid composition of asFeSOD
The assodB open reading frame (ORF) revealed a protein consisting of 194 amino-acid residues with a predicted molecular weight per subunit of 21.4 kDa and a pI of 4.9. Alignment based on the amino-acid sequences of asFeSOD and FeSODs from other Vibrionaceae species as well as that from E. coli showed high homology between the sequences compared (Fig. 1 ▶). asFeSOD revealed highest identity to FeSOD from A. fischeri (93%) and lowest to that from E. coli (75%), while it had sequence identities of 89, 88 and 86% with the FeSODs from Vibrio vulnificus, V. parahaemolyticus and V. cholerae, respectively. The residues discriminating between the iron and manganese SOD proteins (Ala69, Glu70, Trp72 and Ala142; Cortez et al., 1998 ▶), as well as the amino acids involved in metal binding (His27, His74, Asp158 and His162), were conserved in the proteins compared.
Figure 1.

Alignment of FeSODs. Full-length alignment of the FeSOD protein product from A. salmonicida (asFeSOD) with the products of other FeSOD genes from E. coli (ecFeSOD), A. fischeri (afFeSOD), V. vulnificus (vvFeSOD), V. parahaemolyticus (vpFeSOD) and V. cholerae (vcFeSOD). Metal-coordinating residues are marked with a green triangle. Secondary-structure elements from the crystal structure of asFeSOD are indicated above the alignment.
3.3. Expression and purification of asFeSOD
assodB was cloned into three different vectors and was expressed in two different E. coli expression strains at three different temperatures. The native construct expressed in E. coli BL21 (DE3) at 303 K gave the highest amount of soluble asFeSOD protein. About 30 mg protein was purified to apparent homogeneity from 20 ml soluble extract with an OD600 of 70 (Fig. 2 ▶). In order to elucidate the nature of asFeSOD and ecFeSOD in solution, gel filtration of asFeSOD and ecFeSOD was performed. Both proteins appeared to be dimers in solution (data not shown).
Figure 2.

SDS–PAGE of purified asFeSOD. From the left, approximately 5, 10 and 490 µg purified asFeSOD. Mark12 Unstained Standard (Invitrogen) is shown on the right. Purified asFeSOD has a monomer size of 21.4 kDa.
3.4. Residual activity of asFeSOD and ecFeSOD at different temperatures
In order to find the temperature range over which asFeSOD can operate, the enzyme was incubated at different temperatures at pH 7.5 and compared with ecFeSOD treated in the same way. asFeSOD showed the lower temperature optimum as well as a higher residual activity at lower temperature than ecFeSOD. asFeSOD had a relatively broad temperature-optimum profile (ranging from 273 to 303 K) compared with ecFeSOD, which has a narrower temperature profile ranging from 303 to 323 K (Fig. 3 ▶ a). A similar broad range of high activity has previously been observed for psychrophilic FeSOD from Pseudoalteromonas haloplanktis (Castellano et al., 2006 ▶) and has been suggested to be an alternative feature of cold adaptation of proteins (Georlette et al., 2004 ▶). In addition, asFeSOD and ecFeSOD lost more than 50% residual activity at 330 and 350 K, respectively (Fig. 3 ▶ a). Both enzymes showed high activity at temperatures higher than the optimum growth temperature of their host organism. The SOD–WST assay is indirect and the assay temperature had to be altered from 310 K to room temperature in order to perform the temperature/activity measurements. Therefore, the data presented here should be used as relative temperature profiles for asFeSOD and ecFeSOD, rather than their specific optimum temperatures.
Figure 3.

Activity and stability measurements for asFeSOD and ecFeSOD. (a) Enzymes were incubated for 30 min at temperatures ranging from 273 to 328 K for asFeSOD and from 273 to 346 K for ecFeSOD before residual activity was measured by a SOD–WST assay. The highest residual activity measured was set to 100%. (b) Thermal unfolding of asFeSOD and ecFeSOD measured by DSC at a scan rate of 1 K min−1 (1 kcal = 4.184 kJ). The thermograms are baseline-subtracted and normalized for protein concentration.
3.5. Thermostability of asFeSOD and ecFeSOD
Compared with their mesophilic counterparts, a common feature of cold-adapted enzymes that have been studied so far is their low thermal stability. This was also seen in this study, in which we measured thermal stability by differential scanning calorimetry (DSC). The thermogram for asFeSOD showed one single peak with a T m of 335 K (Fig. 3 ▶ b). The transition for asFeSOD is sharp and symmetric. For psychrophilic proteins that display heat-labile and heat-stable domains, it appears that the active site is localized in or close to the heat-labile domain (Bentahir et al., 2000 ▶; Lonhienne et al., 2001 ▶; Watanabe et al., 2005 ▶). Psychrophilic enzymes have been shown to be inactivated by temperatures below the unfolding temperature of the protein, which is supported by this study. This supports the theory of ‘localized flexibility’, which assumes that the low stability of the active site or of the structures bearing the active site is a main determinant of activity at low temperature (Fields & Somero, 1998 ▶).
For ecFeSOD, two peaks were observed that were separated by a temperature interval of about 20 K. The T m for asFeSOD was 335 K, while it was 338 K for the first transition and 358 K for the second transition of ecFeSOD (Fig. 3 ▶ b). The ecFeSOD transition is divided into two distinct peaks: a heat-labile and a heat-stable domain. The inactivation of mesophilic or thermophilic enzymes strongly corresponds to the loss of the structural conformation of the protein (D’Amico et al., 2003 ▶). In this study, the activity of the mesophilic ecFeSOD remains stable up to 333 K before drastically decreasing (Fig. 3 ▶ a), corresponding to the unfolding thermogram measured by DSC (Fig. 3 ▶ b). Also, in contrast to the cooperative unfolding of psychrophilic enzymes, mesophilic and thermophilic homologues seem to unfold in two or three transitions (either observable or indicated by deconvolution of the heat capacity), probably owing to the independent unfolding of structural units of distinct stability (D’Amico et al., 2001 ▶; Feller, 2003 ▶), although this is not always the case (Zecchinon et al., 2005 ▶). However, as was shown in a calorimetric study involving metal interactions in E. coli MnSOD (Mizuno et al., 2004 ▶), the DSC thermogram of native MnSOD shows two distinct peaks, as we have observed for ecFeSOD in this study, and the peak at the lower temperature is smaller in size than the peak at the higher temperature. Mizuno et al. (2004 ▶) found that the different peaks demonstrated the different oxidation states (Mn2+ and Mn3+) of the bound metals, where MnSOD bound to Mn3+ gave the most stable conformation. As far as we know, apart from our study no DSC data have been obtained to date for psychrophilic or mesophilic iron superoxide dismutases. Since the metal-binding properties for MnSOD and FeSOD are quite analogous and the thermograms of ecMnSOD and ecFeSOD revealed similar shapes, it is reasonable to conclude that the different peaks in the thermogram of ecFeSOD also originate from reduced or oxidized forms of iron bound to ecFeSOD.
3.6. asFeSOD structure
3.6.1. Overall structure
The crystal structure of asFeSOD was solved by molecular replacement using the previously published crystal structure of ecFeSOD as a starting model. The structure was then refined to an R work of 20.5% (R free = 23.9%). Crystals grew in the trigonal space group P3221 and contained two protein monomers in the asymmetric unit. Apart from the N-terminal methionine, all amino acids were visible in electron density and were included in refinement. Superimposing the two monomers in the asymmetric unit gave root-mean-square (r.m.s.) deviations of 0.31 Å for Cα atoms and of 0.61 Å for all atoms. The main differences between the two monomers were observed in the ten N-terminal residues, for which the r.m.s. deviations were around 1.2 Å because of differences in crystal packing between the two monomers (monomer A formed crystal contacts with a symmetry-related molecule).
3.6.2. Active-site entrance and geometry
asFeSOD occurs as a dimer in solution in which the active site of one monomer is joined to the active site of the other monomer. Interestingly, the active-site entrance is connected by the formation of a tunnel through the dimer interface (Fig. 4 ▶ c), which can be found in all FeSOD and MnSOD crystal structures. Six water molecules are found within this tunnel, in addition to 6–8 water molecules in each of the entrances to the active site. The Fe atom in asFeSOD is five-coordinated in a trigonal bipyramidal configuration (Fig. 4 ▶ b). The coordinated residues in asFeSOD (the corresponding ecFeSOD residues are given in parentheses) are, as described previously, His27 (His26), His74 (His73), Asp158 (Asp156) and His162 (His160); the fifth ligand in each protein is a water molecule. Comparing the active-site geometry of asFeSOD and ecFeSOD reveals a few substitutions within a 8 Å radius of the Fe atom in the two enzymes (Gly122 in ecFeSOD is substituted by Ser in asFeSOD, Val159 in ecFeSOD is substituted by Leu in asFeSOD and Tyr28 in ecFeSOD is substituted by His in asFeSOD). None of these substitutions are located between the Fe atoms in the two monomers and there are no obvious differences in either the primary or the secondary coordination shells of the Fe atoms. During the refinement, both Fe atoms were refined with full occupancy. On comparing the resulting B factors for the Fe atoms with those of their coordinating residues, all atoms were found to have similar values, indicating that the metal sites are fully occupied in the asFeSOD structure. From a similar comparison using the deposited coordinates of ecFeSOD (PDB code 1isa), the iron sites appeared to be equally well occupied in that structure. In addition, an X-ray absorption near-edge structure (XANES) scan of a crystal of asFeSOD clearly showed the presence of iron in the sample (data not shown). With all channels open on the fluorescence detector, no detectable amounts of manganese could be observed.
Figure 4.

Structural features of asFeSOD. (a) Cartoon representation of the crystallized dimer of FeSOD from A. salmonicida (asFeSOD). α-Helices are coloured red, β-strands are coloured blue and loop regions are coloured green. Fe atoms are indicated as orange spheres. (b) Close-up view of the active site of asFeSOD, illustrating the Fe coordination. The Fe atom is shown as an orange sphere, the coordinated water molecule as a red sphere and the protein ligands are shown in ball-and-stick representation. Secondary-structure elements are coloured as in (a). (c) Slabbed view of the molecular surface of the dimer of asFeSOD, illustrating the tunnel that runs through the dimer interface and connects the two active sites. The monomers are shown in cartoon representation and are coloured red and green. The view on the right is rotated 90° around x compared with that on the left.
3.6.3. Overall stability and comparison to ecFeSOD
Overall comparison of asFeSOD and ecFeSOD based on their three-dimensional structures revealed few differences. ecFeSOD has a deletion at amino-acid position 150 compared with most other bacterial FeSODs, which have a Gly residue in this position. This residue lies in the β2–β3 loop and points away from the dimer interface. The loop is exposed to water and is not close to the active site or involved in substrate binding.
The superposition of a dimer of asFeSOD with a dimer of ecFeSOD gives an r.m.s. deviation of 0.59 Å for main-chain atoms, while the r.m.s. deviation is 0.55 Å for main-chain atoms in each of the asFeSOD and ecFeSOD monomers. There are no substitutions between the monomers of asFeSOD and ecFeSOD within a radius of 4 Å of the dimer interface. When this radius is increased to 6 Å, there are two amino-acid substitutions compared with ecFeSOD: these are Asn38Lys and Lys117Asn. The distance between Asn38 and Lys117 (and Lys38 and Asn117) is relatively short in both structures and as a consequence these substitutions appear to be complementary.
Comparison of salt bridges within a radius of 4 Å in one dimer of asFeSOD and ecFeSOD shows that both structures have 13 salt bridges; each has three which are unique to that structure. The unique salt bridges that are globally stabilizing (with more than five residues between the contributors) are distant from the dimer interface and the active site. The same is true for salt bridges that are shorter than 6 Å; asFeSOD has 24 salt bridges, of which ten are unique, and ecFeSOD has 23 salt bridges, of which nine are unique. None of these findings appear to explain the higher thermostability of ecFeSOD compared with asFeSOD.
In addition, comparison of the hydrogen bonds along the dimer interface revealed no differences. All donor–acceptor pairs are present in both structures. The total number of hydrogen bonds in asFeSOD is 382, whereas there are 371 in ecFeSOD. The numbers of unique hydrogen bonds are 71 and 60 for asFeSOD and ecFeSOD, respectively.
A decreased number of cavities (Russell et al., 1994 ▶; Delboni et al., 1995 ▶), tight packing of proteins by shorter loops (Russell et al., 1994 ▶), an increased hydrophobic effect in the protein core or subunit–subunit interfaces (Russell et al., 1994 ▶; Delboni et al., 1995 ▶; Korolev et al., 1995 ▶), decreased flexibility owing to an increase in the number of proline residues (Delboni et al., 1995 ▶) and increased charged–neutral hydrogen bonds (Tanner et al., 1996 ▶) or ion pairs (Perutz & Raidt, 1975 ▶; Perutz, 1978 ▶) may act as stabilizing elements for protein structures. However, analysis of the asFeSOD and ecFeSOD protein structures focusing on the features described above did not reveal any significant differences between them and cannot explain the lower thermostability of asFeSOD compared with ecFeSOD
3.6.4. Surface electrostatics and its contribution to thermal stabilization
Visualization of the electrostatic surface potential of asFeSOD indicates an increased net negative charge on the surface compared with ecFeSOD (pI of 4.9 and 5.6, respectively), while the overall hydrophobic surface-exposed areas in the two enzymes are quite similar (data not shown). Previously, an increase in the net negative charge of the surface of several cold-adapted enzymes has been described and has been suggested to be a characteristic of extremophilic enzymes (Siddiqui & Cavicchioli, 2006 ▶, and references therein). Charged surfaces make proteins more soluble as their interaction with the solvent improves. On the other hand, anionic surfaces can make proteins more unstable owing to increased charge–charge repulsions. This may explain the lower stability observed for asFeSOD compared with ecFeSOD.
4. Conclusion
This is the first crystal structure of a cold-adapted iron-containing superoxide dismutase and the first DSC study of psychrophilic and mesophilic bacterial FeSODs that describes their different thermogram profiles and melting temperatures. Biochemical studies involving measurement of the residual activity of asFeSOD and ecFeSOD at different temperatures, as well as differential scanning measurements, indicated that there were distinct differences between the two enzymes related to temperature and stability. asFeSOD was inactivated at temperatures about 20 K lower than those observed for ecFeSOD and showed a thermogram with a lower T m than for ecFeSOD. Superoxide dismutases have previously been shown to be highly thermostable and asFeSOD also falls into this category.
Superoxide dismutases are diffusion-controlled nearly ‘perfect’ enzymes with optimally constructed active sites, making it unlikely that there would be large differences in or near the active site. Comparison of asFeSOD and ecFeSOD revealed minor differences at the sequence level and in the three-dimensional structure of the monomer and dimer. However, the main differences in amino-acid composition between them seem to be positioned at the surface of the enzymes. Also, examination of the surface potential showed an increased net negative charge of asFeSOD, potentially making the protein more unstable, which may explain the lower stability of asFeSOD compared with ecFeSOD.
Supplementary Material
PDB reference: asFeSOD, 2w7w, r2w7wsf
Acknowledgments
This study was supported by the Norwegian Research Council. The Norwegian Structural Biology Centre (NorStruct) is supported by a grant from the National Program in Functional Genomics (FUGE) of the Research Council of Norway. Provision of synchrotron beamtime at the ESRF and at BESSY is gratefully acknowledged.
References
- Aghajari, N., Feller, G., Gerday, C. & Haser, R. (1998). Structure, 6, 1503–1516. [DOI] [PubMed]
- Amano, A., Shizukuishi, S., Tsunemitsu, A. & Tsunasawa, S. (1992). Oral Microbiol. Immunol.7, 368–371. [DOI] [PubMed]
- Barondeau, D. P., Kassmann, C. J., Bruns, C. K., Tainer, J. A. & Getzoff, E. D. (2004). Biochemistry, 43, 8038–8047. [DOI] [PubMed]
- Benov, L. T. & Fridovich, I. (1994). J. Biol. Chem.269, 25310–25314. [PubMed]
- Bentahir, M., Feller, G., Aittaleb, M., Lamotte-Brasseur, J., Himri, T., Chessa, J. P. & Gerday, C. (2000). J. Biol. Chem.275, 11147–11153. [DOI] [PubMed]
- Carlioz, A. & Touati, D. (1986). EMBO J.5, 623–630. [DOI] [PMC free article] [PubMed]
- Castellano, I., Di Maro, A., Ruocco, M. R., Chambery, A., Parente, A., Di Martino, M. T., Parlato, G., Masullo, M. & De Vendittis, E. (2006). Biochimie, 88, 1377–1389. [DOI] [PubMed]
- Collaborative Computational Project, Number 4 (1994). Acta Cryst. D50, 760–763.
- Cortez, N., Carrillo, N., Pasternak, C., Balzer, A. & Klug, G. (1998). J. Bacteriol.180, 5413–5420. [DOI] [PMC free article] [PubMed]
- D’Amico, S., Claverie, P., Collins, T., Georlette, D., Gratia, E., Hoyoux, A., Meuwis, M. A., Feller, G. & Gerday, C. (2002). Philos. Trans. R. Soc. Lond. B Biol. Sci.357, 917–925. [DOI] [PMC free article] [PubMed]
- D’Amico, S., Gerday, C. & Feller, G. (2001). J. Biol. Chem.276, 25791–25796. [DOI] [PubMed]
- D’Amico, S., Marx, J. C., Gerday, C. & Feller, G. (2003). J. Biol. Chem.278, 7891–7896. [DOI] [PubMed]
- Delboni, L. F., Mande, S. C., Rentier-Delrue, F., Mainfroid, V., Turley, S., Vellieux, F. M., Martial, J. A. & Hol, W. G. (1995). Protein Sci.4, 2594–2604. [DOI] [PMC free article] [PubMed]
- Dupont, C. L., Neupane, K., Shearer, J. & Palenik, B. (2008). Environ. Microbiol.10, 1831–1843. [DOI] [PubMed]
- Egidius, E., Wilk, R., Anderson, K., Hoff, A. & Hjeltnes, B. (1986). Int. J. Syst. Bacteriol.36, 518–520.
- Farr, S. B., D’Ari, R. & Touati, D. (1986). Proc. Natl Acad. Sci. USA, 83, 8268–8272. [DOI] [PMC free article] [PubMed]
- Feller, G. (2003). Cell. Mol. Life Sci.60, 648–662. [DOI] [PMC free article] [PubMed]
- Fields, P. A. (2001). Comp. Biochem. Physiol. A Mol. Integr. Physiol.129, 417–431. [DOI] [PubMed]
- Fields, P. A. & Somero, G. N. (1998). Proc. Natl Acad. Sci. USA, 95, 11476–11481. [DOI] [PMC free article] [PubMed]
- Geissdorfer, W., Ratajczak, A. & Hillen, W. (1997). Gene, 186, 305–308. [DOI] [PubMed]
- Georlette, D., Blaise, V., Collins, T., D’Amico, S., Gratia, E., Hoyoux, A., Marx, J. C., Sonan, G., Feller, G. & Gerday, C. (2004). FEMS Microbiol. Rev.28, 25–42. [DOI] [PubMed]
- Gregory, E. M. & Dapper, C. H. (1983). Arch. Biochem. Biophys.220, 293–300. [DOI] [PubMed]
- Hopkin, K. A., Papazian, M. A. & Steinman, H. M. (1992). J. Biol. Chem.267, 24253–24258. [PubMed]
- Hunter, T., Bannister, J. V. & Hunter, G. J. (2002). Eur. J. Biochem.269, 5137–5148. [DOI] [PubMed]
- Imlay, J. A. & Fridovich, I. (1992). J. Bacteriol.174, 953–961. [DOI] [PMC free article] [PubMed]
- Jones, T. A., Zou, J.-Y., Cowan, S. W. & Kjeldgaard, M. (1991). Acta Cryst. A47, 110–119. [DOI] [PubMed]
- Kabsch, W. (1993). J. Appl. Cryst.26, 795–800.
- Kardinahl, S., Anemuller, S. & Schafer, G. (2000). Biol. Chem.381, 1089–1101. [DOI] [PubMed]
- Kim, F. J., Kim, H. P., Hah, Y. C. & Roe, J. H. (1996). Eur. J. Biochem.241, 178–185. [DOI] [PubMed]
- Kim, S. Y., Hwang, K. Y., Kim, S. H., Sung, H. C., Han, Y. S. & Cho, Y. (1999). J. Biol. Chem.274, 11761–11767. [DOI] [PubMed]
- Knapp, S., Kardinahl, S., Hellgren, N., Tibbelin, G., Schafer, G. & Ladenstein, R. (1999). J. Mol. Biol.285, 689–702. [DOI] [PubMed]
- Korolev, S., Nayal, M., Barnes, W. M., Di Cera, E. & Waksman, G. (1995). Proc. Natl Acad. Sci. USA, 92, 9264–9268. [DOI] [PMC free article] [PubMed]
- Lah, M. S., Dixon, M. M., Pattridge, K. A., Stallings, W. C., Fee, J. A. & Ludwig, M. L. (1995). Biochemistry, 34, 1646–1660. [DOI] [PubMed]
- Lavelle, F., McAdam, M. E., Fielden, E. M. & Roberts, P. B. (1977). Biochem. J.161, 3–11. [DOI] [PMC free article] [PubMed]
- Leclere, V., Chotteau-Lelievre, A., Gancel, F., Imbert, M. & Blondeau, R. (2001). Microbiology, 147, 3105–3111. [DOI] [PubMed]
- Lepock, J. R., Frey, H. E. & Hallewell, R. A. (1990). J. Biol. Chem.265, 21612–21618. [PubMed]
- Lim, J. H., Yu, Y. G., Han, Y. S., Cho, S., Ahn, B. Y., Kim, S. H. & Cho, Y. (1997). J. Mol. Biol.270, 259–274. [DOI] [PubMed]
- Lonhienne, T., Zoidakis, J., Vorgias, C. E., Feller, G., Gerday, C. & Bouriotis, V. (2001). J. Mol. Biol.310, 291–297. [DOI] [PubMed]
- Meier, B., Barra, D., Bossa, F., Calabrese, L. & Rotilio, G. (1982). J. Biol. Chem.257, 13977–13980. [PubMed]
- Meier, B., Sehn, A. P., Schinina, M. E. & Barra, D. (1994). Eur. J. Biochem.219, 463–468. [DOI] [PubMed]
- Mizuno, K., Whittaker, M. M., Bachinger, H. P. & Whittaker, J. W. (2004). J. Biol. Chem.279, 27339–27344. [DOI] [PubMed]
- Murshudov, G. N., Vagin, A. A., Lebedev, A., Wilson, K. S. & Dodson, E. J. (1999). Acta Cryst. D55, 247–255. [DOI] [PubMed]
- Ose, D. E. & Fridovich, I. (1979). Arch. Biochem. Biophys.194, 360–364. [DOI] [PubMed]
- Perrakis, A., Morris, R. & Lamzin, V. S. (1999). Nature Struct. Biol.6, 458–463. [DOI] [PubMed]
- Perutz, M. F. (1978). Science, 201, 1187–1191. [DOI] [PubMed]
- Perutz, M. F. & Raidt, H. (1975). Nature (London), 255, 256–259. [DOI] [PubMed]
- Russell, R. J., Hough, D. W., Danson, M. J. & Taylor, G. L. (1994). Structure, 2, 1157–1167. [DOI] [PubMed]
- Sambrook, J., Fritsch, E. F. & Maniatis, T. (1989). Molecular Cloning: A Laboratory Manual. Cold Spring Harbor, New York: Cold Spring Harbor Laboratory.
- Schafer, G. & Kardinahl, S. (2003). Biochem. Soc. Trans.31, 1330–1334. [DOI] [PubMed]
- Siddiqui, K. S. & Cavicchioli, R. (2006). Annu. Rev. Biochem.75, 403–433. [DOI] [PubMed]
- Smalas, A. O., Leiros, H. K., Os, V. & Willassen, N. P. (2000). Biotechnol. Annu. Rev.6, 1–57. [DOI] [PubMed]
- Sorum, H., Hvaal, A. B., Heum, M., Daae, F. L. & Wiik, R. (1990). Appl. Environ. Microbiol.56, 1033–1037. [DOI] [PMC free article] [PubMed]
- Stallings, W. C., Pattridge, K. A., Strong, R. K. & Ludwig, M. L. (1984). J. Biol. Chem.259, 10695–10699. [PubMed]
- Stallings, W. C., Powers, T. B., Pattridge, K. A., Fee, J. A. & Ludwig, M. L. (1983). Proc. Natl Acad. Sci. USA, 80, 3884–3888. [DOI] [PMC free article] [PubMed]
- Steinman, H. M. & Ely, B. (1990). J. Bacteriol.172, 2901–2910. [DOI] [PMC free article] [PubMed]
- Steinman, H. M., Weinstein, L. & Brenowitz, M. (1994). J. Biol. Chem.269, 28629–28634. [PubMed]
- Tainer, J. A., Getzoff, E. D., Beem, K. M., Richardson, J. S. & Richardson, D. C. (1982). J. Mol. Biol.160, 181–217. [DOI] [PubMed]
- Tanner, J. J., Hecht, R. M. & Krause, K. L. (1996). Biochemistry, 35, 2597–2609. [DOI] [PubMed]
- Thompson, J. D., Plewniak, F., Thierry, J. & Poch, O. (2000). Nucleic Acids Res.28, 2919–2926. [DOI] [PMC free article] [PubMed]
- Van Petegem, F., Collins, T., Meuwis, M. A., Gerday, C., Feller, G. & Van Beeumen, J. (2003). J. Biol. Chem.278, 7531–7539. [DOI] [PubMed]
- Watanabe, S., Yasutake, Y., Tanaka, I. & Takada, Y. (2005). Microbiology, 151, 1083–1094. [DOI] [PubMed]
- Wiik, R., Andersen, K., Daae, F. L. & Hoff, K. A. (1989). Appl. Environ. Microbiol.55, 819–825. [DOI] [PMC free article] [PubMed]
- Wuerges, J., Lee, J. W., Yim, Y. I., Yim, H. S., Kang, S. O. & Djinovic Carugo, K. (2004). Proc. Natl Acad. Sci. USA, 101, 8569–8574. [DOI] [PMC free article] [PubMed]
- Yamakura, F. & Suzuki, K. (1980). J. Biochem.88, 191–196. [PubMed]
- Youn, H. D., Kim, E. J., Roe, J. H., Hah, Y. C. & Kang, S. O. (1996). Biochem. J.318, 889–896. [DOI] [PMC free article] [PubMed]
- Zecchinon, L., Oriol, A., Netzel, U., Svennberg, J., Gerardin-Otthiers, N. & Feller, G. (2005). J. Biol. Chem.280, 41307–41314. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: asFeSOD, 2w7w, r2w7wsf
PDB reference: asFeSOD, 2w7w, r2w7wsf