

Abstract
The Diels–Alder reactions of a series of silyloxydienes and silylated dienes with acyclic α,β-unsaturated ketones and N-acyloxazolidinones have been investigated. The endo/exo stereochemical outcome is strongly influenced by the substitution pattern of the reactants. High exo selectivity was observed when the termini of the diene and the dienophile involved in the shorter forming bond are both substituted, while the normal endo preference was found otherwise. The exo-selective asymmetric Diels–Alder reactions using Evans’ oxazolidinone chiral auxiliary furnished a high level of π-facial selectivity in the same sense as their well-documented endo-selective counterparts. Computational results of these Diels–Alder reactions were consistent with the experimental endo/exo selectivity in most cases. A twist-asynchronous model accounts for the geometries and energies of the computed transition structures.
Introduction
Precise control of stereoselectivity in the installation of asymmetric centers constitutes a prominent goal in organic synthesis. The Diels–Alder reaction is indispensable for the stereoselective construction of six-membered rings,1 a structural motif widely found in natural products. In its intermolecular variant, a cyclohexene ring with up to 4 stereocenters can be formed in a single step from acyclic precursors, often with high levels of regio- and stereocontrol. A familiar property of this reaction, whether thermal or catalyzed, is the endo diastereoselectivity.2 As a result, the formation of these stereogenic centers on the ring is often regarded as “predictable in a relative sense.”3
In contrast, instances of the Diels–Alder reaction showing exo-selectivity occur less frequently, at least in an intermolecular context. Apart from some isolated cases,4, 5 certain structural motifs on the dienophile are well-known to lead predominantly to exo cycloadducts, including conformationally restricted cyclic s-cis- dienophiles,6 and α-halo α,β-unsaturated carbonyl compounds.7
Synthetic protocols geared toward an exo-selective Diels–Alder reaction usually entail the use of structurally elaborate starting materials that incorporate strategic design features. These features are often anticipated to sterically encumber the endo mode of approach of the reactants. (Scheme 1) Danishefsky has utilized a gem-dimethyl group and a bulky aroyl group on the diene and dienophile respectively to divert the cycloaddition into the desired exo manifold by virtue of their steric bulk. This was applied in the total synthesis of racemic mamanuthaquinone.8 (Eq. 1)
Scheme 1.

Selected Examples of Substrate- and Catalyst-Controlled Exo-Selective Intermolecular Diels–Alder Reactions.
Homochiral α,β-unsaturated N-acylamino Fischer carbenes containing an oxazolidinone or imidazolidinone moiety have been used as exo-selective dienophiles in cycloadditions of cyclopentadiene and other acyclic dienes.9 The apically disposed carbonyl ligands on the octahedrally coordinated metal center furnished the steric hindrance for impeding the endo trajectory. (Eq. 2) Other stoichiometric organometallic reagents have also been reported to give a high exo selectivity.10
An early example of catalyst-controlled exo-selective Diels–Alder reactions was reported by Yamamoto et al. The bulky Lewis acid, aluminum tris(2,6-diphenylphenoxide), was shown to facilitate exo cycloaddition of cyclopentadiene and phenyl vinyl ketone, a pair of reactants which give predominantly an endo cycloadduct otherwise.11 (Eq. 3) Exo-selective cycloadditions of cyclopentadiene or diphenylisobenzofuran and α,β-unsaturated aldehydes have been documented by MacMillan amongst the first highly enantioselective organocatalytic Diels–Alder reactions.12 More recently, Ishihara et al. reported the use of a chiral binaphthyl-substituted diamine in cycloadditions using α-acyloxyacroleins as dienophiles.13 A structurally related, binaphthyl-based diamine was found by Maruoka to be an effective catalyst in the exo-selective cycloadditions with trans-cinnamaldehyde and two other α,β-unsaturated aldehydes.14 The high exo selectivity has been attributed to the steric hindrance of the binaphthyl moiety in the reactive iminium intermediate formed from the dienophile and the catalyst.15 (Eq. 4) Hayashi extended the scope of α,β-unsaturated aldehydes suitable for exo-selective cycloadditions by using a diarylprolinol silyl ether as the organocatalyst.16 In all these reports on organocatalyzed Diels–Alder reactions, instances where a high exo-selectivity is observed involve the use of cyclopentadiene as the diene component.
Examples of exo-selective Diels–Alder reactions are also found in host-guest chemistry. Thus, a trimeric porphyrin host facilitates an exclusively exo cycloaddition of a furan and a maleimide by orienting them in the required disposition.17 The exo orientation can also be fulfilled by hydrogen bonding within the supramolecular complex.18 Monoclonal antibodies that catalyze enantioselective Diels–Alder reactions of N-acylamino-1,3-butadienes and N,N-dimethylacrylamide in an either endo- or exo-specific fashion have also been derived.19
During the study of the preparation of enantioenriched fluorinated carbocycles,20 we synthesized an array of Diels–Alder cycloadducts featuring an allylsilane or a silyl enol ether, starting respectively from all-carbon silylated dienes21 or silyloxy dienes.22, 23 (Scheme 2) We have observed a remarkable change in the endo/exo stereoselectivity of the cycloaddition that depends on the substitution pattern of the starting material. As depicted in Scheme 2, when both R1 and R2 groups are a methyl group (right, Scheme 2), the adducts with four stereocenters are formed with a consistently high exo selectivity. Use of the desmethyl diene and/or dienophile leads to the “normal” endo products predominantly (left, Scheme 2). The observation that such a minor modification of substituents on these common substrates could lead to a dramatic change in endo/exo selectivity is intriguing, not least because it holds obvious implications in the stereodefined synthesis of carbocycles in general. We now detail the synthetic results obtained with this class of substituent-dependent, exo-selective Diels–Alder reactions, as well as computational studies that reveal the origins of these selectivities.
Scheme 2.

Stereochemical Dependence on Substitution Pattern of Starting Materials.
Synthetic Results and Discussion
The dienes 1−2 and dienophiles 3−6 that form the basis of this work are shown in Chart 1. The silylated and silyloxydienes,24 and the dienophiles25 were either readily synthesized by reported methods or were commercially available.
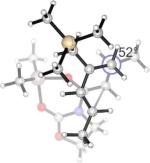
The Diels–Alder reactions involving the α,β-unsaturated N-acyloxazolidinone 3 and their asymmetric variant mediated by Evans’ chiral auxiliary 3*26 are listed in Entries 1−8 of Table 1. These were performed in dichloromethane at −40 °C promoted by 1.4 equivalents of Me2AlCl. As previously reported,20 the asymmetric Diels–Alder reaction of silylated diene 1a with the homochiral 3* delivered endo-7 as the major product with complete Cα-Re facial selectivity.26 (Entry 1) Under otherwise identical conditions, however, diene 1b, which has an extra C1-methyl group, afforded exo-8 as the sole isolated product. (Entry 2) As proved by X-ray crystallography of exo-8, this cycloaddition also arises from the attack on the homochiral dienophile from its Cα-Re face exclusively. The sense of asymmetric induction in this case is thus identical to that observed in their endo-selective counterparts involving 1a and other reported dienes.26 Reaction of diene 1c bearing an additional methyl group at C3 yielded comparable results. (Entry 3) The sense of asymmetric induction in both endo- and exo-selective cycloadditions could be explained by Evans’ model.26 (Figure 2) The oxazolidinone–Lewis acid complex is rigidified by chelation of both carbonyl groups to the aluminum. The benzyl group of the chiral auxiliary effectively shields the Cα-Si face of the dienophile, giving rise to the sense of facial selectivity observed.
Table 1.
Intermolecular Diels–Alder Reactions Involving Oxazolidinone Dienophiles.
| Entry | Reacting partners | Conditions | Major product | exo/endoa | Yield |
|---|---|---|---|---|---|
| 1b | 1a + 3* | 1.4 equiv. Me2AlCl, CH2Cl2, −40 °C | 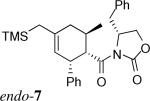 |
1:5 | 50% |
| 2c | 1b + 3* | 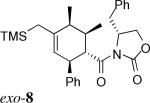 |
> 20:1 | 58% | |
| 3d | 1c + 3* | 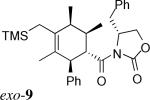 |
> 20:1 | 65% | |
| 4e | 2a + 4 |
 f f
|
1:20 | 78% | |
| 5 | 2a + 3 | 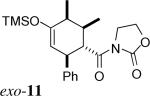 |
> 20:1 | 62% | |
| 6 | 2b + 3 | 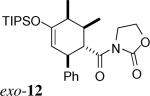 |
> 20:1 | 89% | |
| 7 | 2c + 3 | 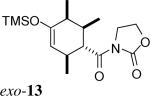 |
> 20:1 | 80% | |
| 8 | 2d + 3* | 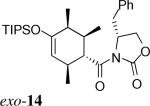 |
> 20:1 | 48% | |
| 9 | 1a + 5 | 0.2−0.3 equiv. Me2AlCl, CH2Cl2, RT | 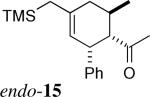 |
1:4 | 15% exo 62% endo |
| 10c | 1b + 6 | 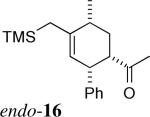 |
1:10 | 4% exo 40% endo | |
| 11c | 1b + 5 | 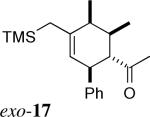 |
11: (1:1.5:0.4)g | 78% exo |
By 400 MHz 1H NMR of the product mixture after aqueous workup.
ref. 20.
1b used as an inseparable isomeric mixture with (Z,E):(E,E) = 3:1. Reaction of only the major isomer was observed unless otherwise stated.
1c used as a mixture with (Z,E):(E,E) = 5:1. Only reaction from the major isomer was observed.
At −100 °C.
Product of protodesilylation of the primary, endo cycloadduct, epimerized at C5, was isolated after chromatographic purification over silica gel.
Four sets of signals were observed by 1H NMR of the crude mixture. Two of the adducts were assigned as exo-17 and its C-5 epimer. The other two, minor adducts were tentatively assigned as the corresponding endo isomers.
Figure 2.
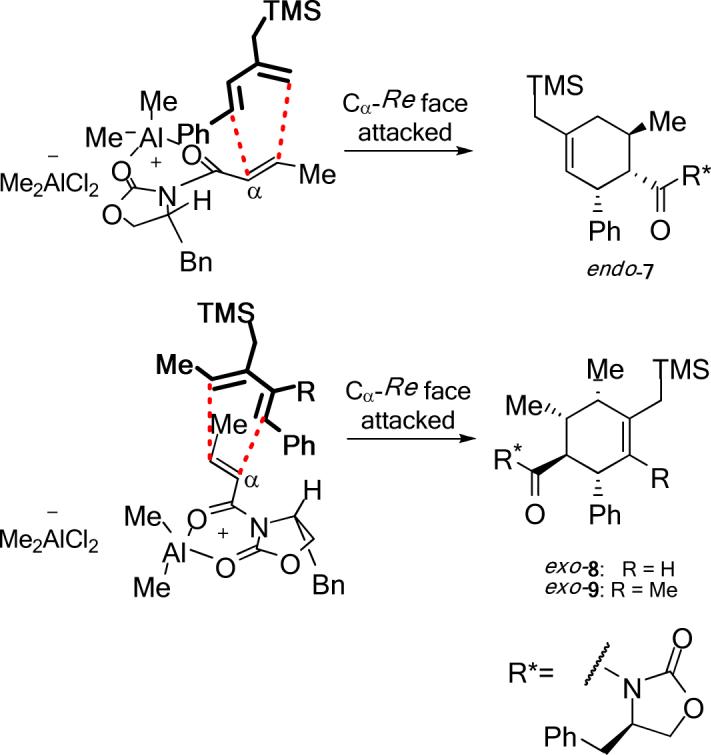
Rationalization of the Sense of Asymmetric Induction in the 1a–1c + 3* Cycloadditions.
The same substituent-dependent variation in endo/exo selectivity was observed in the Diels–Alder reactions of 2-silyloxy-1,3-butadienes with 3 and 4. Thus the cycloaddition of diene 2a and the achiral acryloyl oxazolidinone 4 was highly endo selective, (Entry 4) while the same reaction with the crotonyl oxazolidinone 3 gave the exo cycloadduct 11 as the only product. (Entry 5) The stereoselectivity was not affected to any appreciable extent by changing either the silyl protecting group or the C4 substituent of the diene. The TIPS-protected silyloxydiene 2b and the 1,4-dimethyl silyloxydiene 2c delivered respectively exo-12 (Entry 6) and exo-13 (Entry 7) highly selectively upon cycloaddition with 3. The asymmetric cycloaddition of the silyloxydiene 2d with 3* also yielded the expected product exo-14 in high exo/endo and facial selectivities. (Entry 8) The high exo-selectivity of asymmetric carbo Diels–Alder reactions using the auxiliary of 3* is rare in an intermolecular context.27
α,β-Unsaturated ketones have also been studied with silylated or silyloxy dienes in the presence of catalytic Me2AlCl. (Entries 9−11) The same trend in endo/exo selectivity with substitution pattern was found. Thus predominantly endo cycloaddition occurred when either C1 of the diene (Entry 9) or the β-position of the dienophile (Entry 10) was unsubstituted. For the combination of diene 1b and dienophile 5, each of which contained a terminal methyl group on the reacting double bonds, the reaction was exo-selective. (Entry 11)
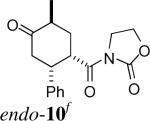
The stereochemistry of the cycloadducts was readily assigned for all cycloadducts based on proton NMR coupling constants (Figure 3), and confirmed in the case of exo-8 by X-ray crystallography. In the exo cycloadducts, the signal due to the methine ring proton (H1, See Figure 1 for numbering) geminal to the carbonyl group appears as a triplet of ca. 10 Hz, consistent with vicinal coupling of H1 to two trans-diaxial protons (H2 and H6). In the endo counterparts with a C6-Me group (endo-720 and 15), this signal exists as a doublet of doublets with coupling constants of about 10 and 6 Hz, which arise, respectively, from one trans-diaxial and one axial-equatorial pair of coupling spins. The couplings of H1 in cycloadducts derived from acryloyl dienophiles (endo-10 and 16) were also consistent with their stereochemistry.
Figure 3.

Key NMR Coupling Constants for Stereochemical Assignments Illustrated for Endo- and Exo-15.
Figure 1.
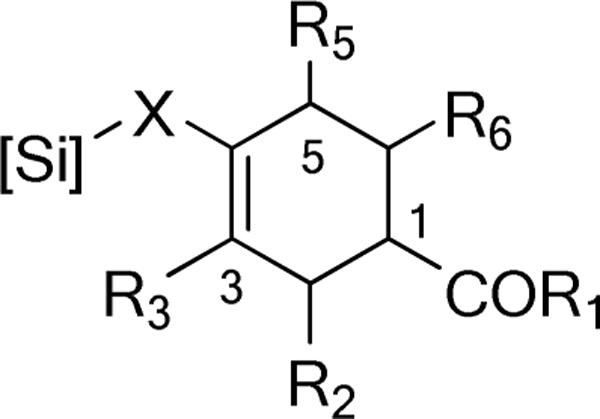
Numbering of Cycloadducts.
Acyclic 2-silyloxy-1,3-butadienes substituted at one or both termini are useful dienes in Diels–Alder reactions toward a number of medicinally relevant targets. Despite their structural simplicity, the stereochemical outcome observed with these substrates and cyclic dienophiles proves to be subtly dependent on the structure of the reactants and the Lewis acid.22, 28 The variation of the observed endo/exo selectivity with substitution pattern was rationalized in terms of steric clash between substituents attached on the approaching diene and dienophile. On the other hand, the use of oxazolidinone-containing dienophiles of the type 3* in R2AlCl-mediated Diels–Alder reactions is well precedented for simple dienes, and the use of 2-silyloxydienes was documented recently.29 We computationally investigated the TSs of the current series of cycloadditions, focusing on how the substitution pattern of the starting material impacts on the stereochemical outcome.
Computational Methodology
Geometry optimization and thermodynamic corrections were performed with hybrid density functional theory (B3LYP)30 using the 6−31G(d)31 basis set. MP2 (default, frozen core)/6−31+G(d)//B3LYP/6−31G(d) single-point energies of endo and exo transition states have also been computed and compared with the B3LYP derived values and experimental data where available. Solvation corrections for the experimentally used solvent, dichloromethane, were calculated by the PCM method32 and UAKS radii with single-points at the HF/6−31+G(d,p) level of theory.33
Optimization of transition structure geometries was started from a boat-like input structure expected from the general Diels–Alder reaction. The distance of the shorter forming bond (C5–C6) was first held fixed and the remainder of the structure optimized. This distance constraint was then lifted, and the structure was re-optimized to the transition state.34 All energy minima and transition structures were characterized by frequency analysis.
All calculations were performed with Gaussian 03.35
Computational Results and Discussion
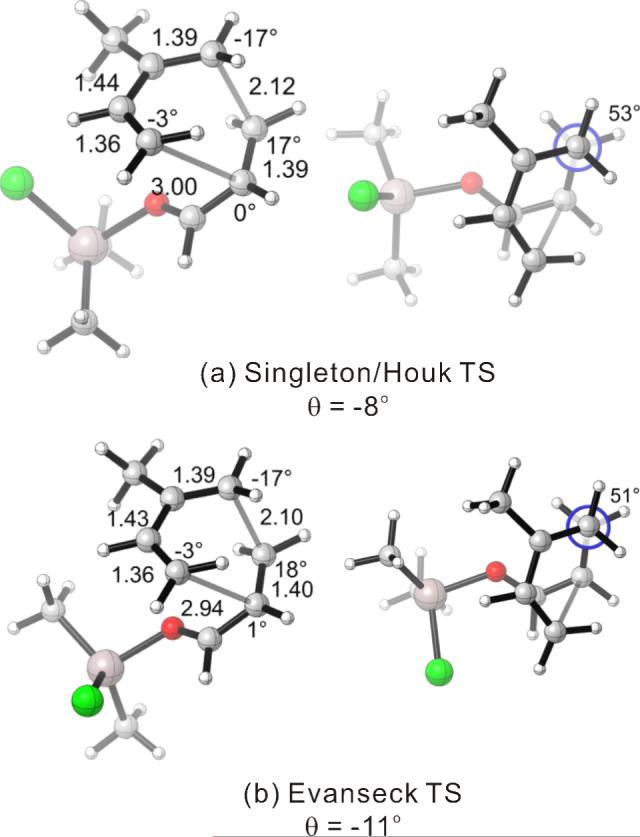
We first computationally examined a Diels–Alder reaction with the oxazolidinone-containing dienophile 3 for which experimental data were available. Dienophile 3 in the presence of Me2AlCl was represented by the bidentate complex A2 in computations. (Figure 4) Chiral complexes of the type A2 have been invoked as the reactive species in R2AlCl-mediated asymmetric Diels–Alder reactions,26 and have been characterized in solution by NMR.36
Figure 4.
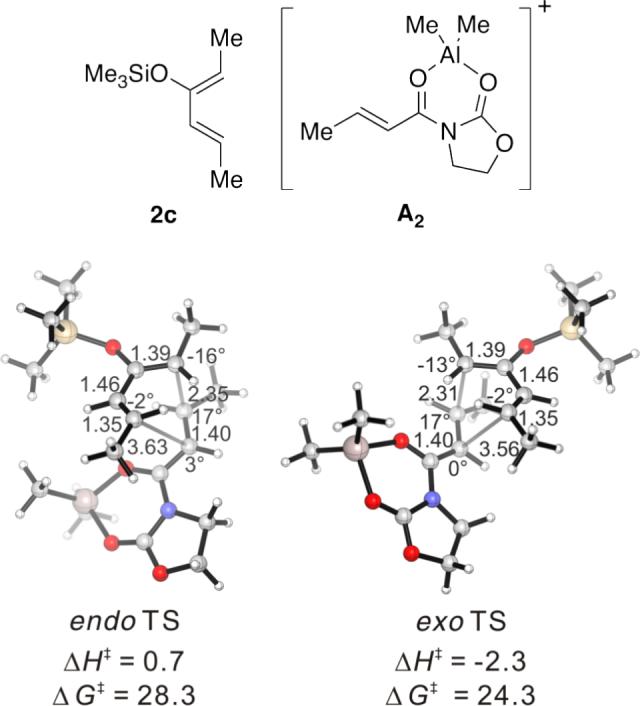
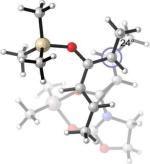
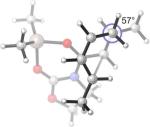
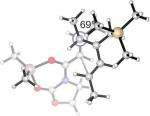
Endo and Exo TSs of the 2c+A2 Cycloadditions. Gas-phase enthalpies of activation computed at the B3LYP/6−31G(d) level. Free energies of activation computed by B3LYP/6−31G(d), corrected for solvent effects by PCM single-points at HF/6−31+G(d,p). Interatomic distances around the forming ring in Å and angles of pyramidalization at the termini of the partially formed bonds are labeled.
The Me2AlCl-mediated cycloaddition of silyloxydiene 2c and 3 was completely exo-selective. (Table 1, Entry 7) The endo and exo TSs of the 2c+A2 addition are presented showing their boat-like geometries in Figure 4. B3LYP and MP2 predict a difference in electronic activation barrier of 2.9 and 2.1 kcal/mol, respectively, both in favor of the exo pathway (See Table 2 below). The difference in free energy of activation between the endo and the exo approaches, after correcting for solvent effects, is 4.0 kcal/mol. (Figure 4) The experimental level and sense of diastereoselectivity was thus consistent with computation. The activation enthalpies refer to the difference between the enthalpy of the TS and the sum of enthalpies of diene and the dienophile complex infinitely separated in the gas phase. The small and negative gas-phase ΔH‡ (and the negative ΔE‡, see below) values are often observed in modeling gas-phase ion/molecule reactions in general.37, 38 In the present case they are consistent with the more favorable electrostatic attractions expected between the diene's electrons and the cationic A2 at the TS, compared with the separate, unbound state.
Table 2.
Activation Barriers and Their Dissection into Distortion and Interaction Energies for Endo and Exo TSs of 2c+A2 (kcal/mol).
| Entries | Method | ΔE‡ | ΔEd‡ | ΔEi‡ | ||
|---|---|---|---|---|---|---|
| diene 2c | dienophile complex A2 | |||||
| 1 | B3LYP/6−31G(d) | endo | −0.1 | 9.5 | 11.9 | −21.4 |
| 2 | exo | −3.0 | 7.9 | 8.8 | −19.7 | |
| 3 | MP2/6−31+G(d) | endo | −11.4 | 9.8 | 12.0 | −33.1 |
| 4 | exo | −13.5 | 8.3 | 8.8 | −30.6 | |
Decomposition of the activation barrier (ΔE‡) into distortion (ΔEd‡) and interaction energies (ΔEi‡)37, 39 yields valuable insight into the differing activation energies of the endo and exo pathways. This analysis has previously been applied in understanding 1,3-dipolar and Diels–Alder cycloadditions.40 The distortion energy is the energy required to distort the diene and the dienophile complex into the geometries they have at the transition state without allowing interaction between them. The activation barrier is then the sum of distortion and interaction energies: ΔE‡ = ΔEd‡ + ΔEi‡, where the interaction energy term encompasses all the stabilizing and repulsive interactions between the diene and dienophile fragments at the transition state. Table 2 lists the activation, transition-state distortion and transition-state interaction energies for the endo and exo Diels–Alder TSs of 2c+A2, computed by B3LYP and MP2 methods.
The distortion energy values calculated by either B3LYP or MP2 are very similar in most cases, but MP2 predicts that the interactions at the endo TS are more stabilizing than the exo by a larger margin than B3LYP does. MP2 favors the endo pathway to a greater extent than B3LYP for all cycloadditions. The underestimation of endo/exo selectivity by B3LYP due to problems in describing long-range dispersion interactions has been noted.41, 42
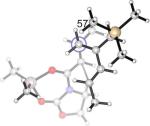
It is noteworthy that the interactions between the diene and dienophile fragments are more stabilizing at the endo TS than at the exo TS, regardless of the computational method used. The net preference of exo selectivity observed in 2c+A2 is therefore the result of more severe reactant distortions that overwhelm the more favorable interaction energies in the endo TS. The geometries of reactants 2c and A2 in their cycloaddition TS geometries provide an illustrative example of reactant distortions. (Table 2) The diene fragment is more heavily deformed at the endo TS by about 1.5 kcal/mol, and the dienophile is distorted twice as much, 3.1 kcal/mol. The origin of the dramatic difference in distortion energies of A2 in the two TSs is most easily visualized by a side-on view of the dienophile complex shown in Figure 5. The methyl group of dienophile A2 is bent out of the olefinic plane more severely in the endo TS (25°, Figure 5a) than in the exo TS (14°, Figure 5b), indicating that it suffers greater repulsion from the vicinal diene methyl in the endo TS.
Figure 5.
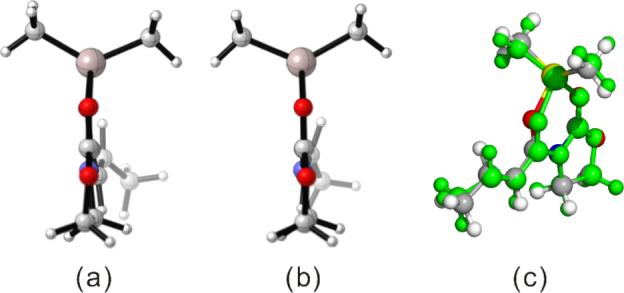
Side View of A2 Dienophile Fragment in (a) the Endo 2c+A2 TS, and (b) the Exo 2c+A2 TS. (c) Overlay of A2 Dienophile Fragments in TSs of 2c+A2 (Green: Endo, Other-Colored Atoms: exo).
A twist-asynchronous model shows how vicinal di-substitution can lead to a high distortion in the endo TS, which is chiefly responsible for the exo selectivity found computationally and experimentally.
In general, Diels–Alder reactions of unsymmetrical dienes and dienophiles proceed through concerted43 asynchronous44 mechanisms.45 Two modes of asynchronicity in Diels–Alder reactions can be distinguished. (Figure 6) The stretch mode refers to the differing lengths of the two forming C–C bonds at the transition state. (Figure 6a) The twist mode refers to the deviation of the diene and the dienophile backbones from being parallel, and can be measured by a twist-asynchronicity parameter, θ, which is the C2−5−6−1 dihedral angle, where C5–C6 is the shorter forming C–C bond. (Figure 6b) The impact of catalyst coordination to stretch-mode asynchronicity of Diels–Alder reactions is well discussed.46 A twist-asynchronous model was first put forward by Brown and Houk to account for substituent-induced selectivity variation in the context of intramolecular Diels–Alder (IMDA) reactions.47 This has seen extensive application in the rationalization of stereoselectivity of the IMDA reactions of other substrates.48
Figure 6.

Schematic Illustration of the Two Modes of Asynchronicity at the Transition State of a Diels–Alder Reaction.
Table 3 lists the endo and the exo TSs for the Diels–Alder reactions of a series of silyloxy dienes and other model dienes with the oxazolidinone-bearing dienophile 3, either in the presence of Me2AlCl (represented by the dienophile complex A2) or in its absence. These are presented as Newman projections along the shorter forming (C5–C6) bond.
Table 3.
Endo (n) and Exo (x) TS Geometries and Energetics for a Range of Silyloxydienes and Other Model Dienes with Dienophile Complex A2 or Dienophile 3, with an Emphasis on Twist-Mode Asynchronicity.
| Entry | Diene | TS geometry | ΔG‡298a | ΔE0‡b | θc | Me(H)–Me dihedral angle | |
|---|---|---|---|---|---|---|---|
| endo | exo | ||||||
| 1 |  |
 |
 |
28.3(n) 24.3(x) | −0.1 (−11.4) (n) −3.0 (−13.5) (x) | −33° (n) +22° (x) | 24°(n) 72°(x) |
| 2 | 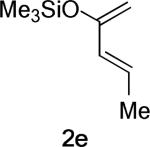 |
 |
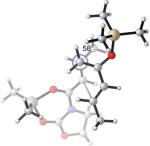 |
24.1(n) 24.0(x) | −2.0 (−13.1) (n) −2.1 (−11.4) (x) | −5°(n) +10°(x) | 53°(n) 58°(x) |
| 3 | 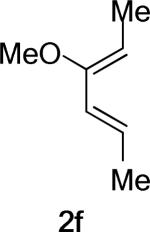 |
 |
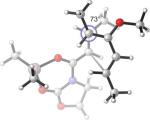 |
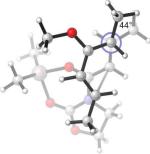
25.9(n) 23.6(x) | 0.4 (−11.0) (n) −1.7 (−11.4) (x) | −13°(n) +23°(x) | 44°(n) 73°(x) |
| 4d | 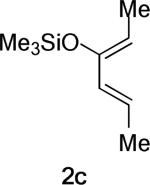 |
 |
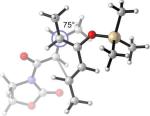 |
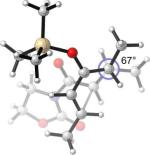
42.7(n) 40.1 (x) | 21.0 (4.5) (n) 20.2 (5.6) (x) | +9°(n) +23°(x) | 67°(n) 75°(x) |
| 5 | 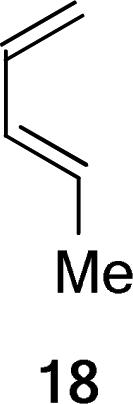 |
 |
 |
28.9(n) 29.6(x) | 6.9 (−3.8) (n) 7.3 (−2.1) (x) | −3°(n) +12°(x) | 57°(n) 58°(x) |
| 6 |  |
 |
 |
34.7(n) 33.3(x) | 10.6 (−4.2) (n) 9.1 (−4.5) (x) | +12°(n) +16°(x) | 71°(n) 64°(x) |
Gas phase Gibbs energy at B3LYP/6−31G(d) level, corrected for solvent effects by PCM single-points at HF/6−31+G(d,p) on B3LYP/6−31G(d) vacuum optimized geometries.
Gas phase uncorrected electronic energy at B3LYP/6−31G(d)//B3LYP/6−31G(d) level. The corresponding energy at MP2/6−31+G(d)//B3LYP/6−31G(d) level in parentheses.
Twist-asynchronicity parameter. See Figure 6b for definition.
In the absence of Me2AlCl (i.e. with dienophile 3).
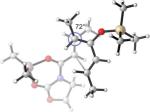
It is clear from Table 3 that the endo series of TSs displays a wider range of twist-mode asynchronicity than the exo TSs. Depending on diene substitution, θ varies from +12° to −33° for the endo TSs and from +10° to +23° for the exo TSs. The endo TS of the exo-selective 2c+A2 cycloaddition is highly twisted, with θ = −33°, and features a very small Me–Me dihedral angle of 23°. (Entry 1) The corresponding exo TS is much less twisted, and places the vicinal Me groups at 72°. The wide difference in Me–Me dihedral angle in the two TSs explains why the dienophile complex experiences a higher distortion at the endo TS.
Replacing the C1-Me group on 2c with hydrogen – resulting in the desmethyl diene 2e – reduces the allylic 1,3-strain across the silyl enol ether and allows TSs containing the s-syn rotamer of 2e to be located. These TSs are more stable than the corresponding TSs containing the s-anti conformer of 2e, and are displayed in Entry 2. The levels of twisting are smaller, leading to similar H–Me dihedral angles in both TSs, concomitant with a much smaller relative stability of the exo TS. MP2 even predicts that the endo TS is more stable. The TS geometries and energies are therefore heavily affected by the C1-Me substituent in this system.
What causes the drastic asynchronous twist in the endo 2c+A2 TS? To assess its origin, we modeled the TSs involving diene 2f, in which a methoxy group replaces the silyloxy group in 2c (Entry 3). The computed free energies of activation indicate that, as a result of this replacement, the exo-endo difference narrows down from 4.0 kcal/mol (Entry 1) to 2.3 kcal/mol (Entry 3). The endo TS is also less twisted, with θ = −13°, placing the vicinal Me groups at a dihedral angle of 44°. The twist-asynchronicity and the resultant Me–Me dihedral angle are practically the same in the exo TSs involving either the methoxy or the silyloxy diene. Since the methoxy and the trimethylsilyloxy groups can be expected to possess similar electronic properties (both dominated by oxygen), steric interactions, plausibly between the trimethylsilyloxy group and the dimethylaluminum portion of A2, are one contributor to the high twist-asynchronicity in endo 2c+A2 TS. Another destabilizing factor is electrostatic repulsion between the oxygenated group and the aluminum-bound dienophile. As the TSs of 2c+3 show, (Entry 4) the asynchronous twist is considerably less severe in the absence of Lewis acid, resulting in the more typical Me–Me dihedral angles of 67° (endo) and 75° (exo) respectively. The exo preference is again diminished; MP2 energies even indicate an endo preference. Electrostatic repulsion between the lone pair on a heterodienophile and the diene is well known to influence stereochemical preferences49 and dictate reaction trajectories50 of hetero-Diels–Alder reactions. Analogous interactions should also operate in the endo 2c+A2 system.
Based on the discussion above, a twist-asynchronous model rationalizing substituent effects on relative endo and exo TS energies in the Diels–Alder reactions of silyloxydienes is presented in Figure 7. In the endo series of TSs (Figure 7a), steric and electrostatic interactions between the diene C2-substituent and the aluminum-complexed dienophile drives the rotation of the latter in the outward direction. This motion is disfavored when both ends of the shorter forming C5–C6 bond are methyl substituted, since the rotation brings them close together and aggravates vicinal repulsion between them. The twist-asynchronicity of these TSs reflects the tradeoff between the two types of repulsion arising from their respective local environments within the TSs. In contrast, the exo TSs are less prone to these destabilizing interactions. As Figure 7b shows, the twisting that would alleviate the steric and electrostatic interactions also brings any vicinal methyl substituents on the C5–C6 forming bond further apart. The exo series of TSs could therefore relax by asynchronous twisting as appropriate. Indeed, when the 2-silyloxy or 2-methoxydiene is methyl substituted at both of their termini, the exo TSs consistently displays a narrow range of Me–Me dihedral angle between 69° and 75°. (Table 3, Entries 1, 3 and 4) This model explains why the relative energies of the endo and the exo TS for a given cycloaddition depend heavily on the substitution pattern, and why the exo TSs are, in most cases, less destabilized than the endo TSs.
Figure 7.

A Twist-Asynchronicity Model Delineating the Origin of Exo Selectivity in Diels–Alder Reactions Involving Oxygenated Dienes.
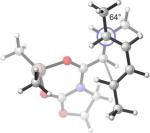
Computations of Diels–Alder TSs of the dienophile complex A2 with a series of structurally simpler dienes give further support to the twist-asynchronicity model. Trans-piperylene (18) was reported by Evans to react with 3* in the presence of Et2AlCl with an endo/exo selectivity superior to 95:5.26 Modeling the TSs of the 18+A2 addition (Table 3, Entry 5), with MP2 single-point energies, reproduced the experimental level and sense of the endo/exo selectivity. The twisting of these TSs is small and comparable to that of the TSs involving the 2-trimethylsilyloxydiene 2e (Entry 2). The substituents around the shorter forming C–C bond are arranged in an approximately staggered manner. The low level of twisting of endo 18+A2 TS could be favorably compared to that of the TSs computed in the dialkylaluminum chloride catalyzed Diels–Alder reaction of isoprene and acrolein. Singleton and Houk,51 and Evanseck52 have shown that the TSs of this reaction, which were validated by experimental and calculated kinetic isotope effects, correspond to the endo approach of the two reactants. These TSs display a low level of asynchronous twist and a nearly staggered arrangement of substituents around the shorter forming C–C bond, similar to the endo 18+A2 TS. (Figure 8) Consequently, the greater stabilization of the endo transition states determine the stereoselectivity.
Figure 8.
The B3LYP/6−31G(d) Endo TSs for the Reaction of Isoprene with Acrolein•AlMe2Cl Obtained by Singleton/Houk and Evanseck.
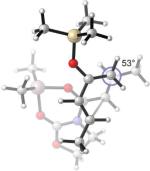
Adding a terminal methyl substituent to trans-piperylene gives 2,4-hexadiene 19. Exo selectivity is now found (Table 3, Entry 6). In the endo TS, the backbones of the diene and the dienophile are skewed, with θ = +12°. The Me–Me repulsion forces the dienophile away from ideal overlap with the remote terminus of the diene. Although the endo TS is now more destabilized (according to B3LYP energies), the magnitude of such destabilization is small (1.4 kcal/mol free energy difference) compared to the cases where the diene is additionally substituted at the 2-position. (2−4 kcal/mol, Table 3, Entries 1 and 3). The “back side” of the endo 19+A2 TS is not hindered by substituents. This allows the dienophile to rotate inward to alleviate the vicinal dimethyl repulsion and the accompanying destabilization.
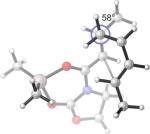
The phenyl substituted silylated dienes 1b and 1a used in the experimental studies were modeled by the methyl analogs 1d and 1e respectively. The free energies of activation of cycloadditions with A2 indicate that the exo preference is very high in the case of diene 1d and much diminished for diene 1e, where the C1-methyl group on the diene is replaced by hydrogen. (Figure 9) This trend is consistent with experimental results of the cycloadditions of 3* with 1a and with 1b (Table 1, Entries 1−2). The moderate endo selectivity in the experimental 1a+3* reaction could not be reproduced by B3LYP energies of the 1e+A2 TSs, although MP2 single-points predict a much higher proportion of the endo product. MP2 methods have been shown, in some cases, to be more accurate in reproducing experimental trends, particularly when weak non-bonding interactions are involved.5, 41, 53
Figure 9.

Endo and Exo TSs of the 1d and 1e+A2 Cycloadditions. Gas-phase enthalpies of activation computed at the B3LYP/6−31G(d) level. Free energies of activation computed by B3LYP/6−31G(d), corrected for solvent effects by PCM single-points at HF/6−31+G(d,p). Interatomic distances around the forming ring in Å and angles of pyramidalization at the termini of the partially formed bonds are labeled.
An analysis of distortion and interaction energies (Table 4) shows that the distortion energies in the endo TS again overwhelm the more favorable interaction energies in the presence of vicinal di-substitution, and account for the exo preference observed. The sum of distortion energies of the diene 1d and dienophile complex A2 is 3.2 kcal/mol higher in the endo TS than in the exo TS, while the corresponding difference involving the desmethyl analog 1e is only about 0.9 kcal/mol favoring the endo pathway.
Table 4.
Activation Barriers and Their Dissection into Distortion and Interaction Energies for Endo and Exo TSs of 1d and 1e+A2 (kcal/mol).
| Entries | Reactants | Method | ΔE‡ | ΔEd‡ | ΔEi‡ | ||
|---|---|---|---|---|---|---|---|
| diene | dienophile complex A2 | ||||||
| 1 | 1d+A2 | B3LYP/6−31G(d) | endo | 2.0 | 10.9 | 14.2 | −23.2 |
| 2 | exo | −0.1 | 9.2 | 12.7 | −22.0 | ||
| 3 | MP2/6−31+G(d) | endo | −14.8 | 10.0 | 14.3 | −39.1 | |
| 4 | exo | −14.1 | 8.4 | 12.7 | −35.2 | ||
| 5 | 1e+A2 | B3LYP/6−31G(d) | endo | −1.4 | 6.0 | 7.6 | −15.0 |
| 6 | exo | −1.3 | 6.3 | 8.2 | −15.8 | ||
| 7 | MP2/6−31+G(d) | endo | −12.9 | 5.9 | 7.6 | −26.3 | |
| 8 | exo | −11.2 | 6.1 | 8.2 | −25.4 | ||
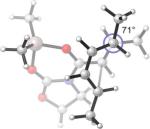
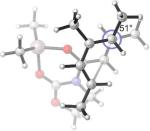
Replacing the oxygen in 2c and 2e with methylene – giving respectively the all-carbon silylated dienes 1d and 1e – leads to considerable alleviation in twisting in both endo and exo TSs with A2. (Table 5, Entries 1−2) These TSs display θ values between −6° and −15°. This is consistent with the reduction of electrostatic repulsion between the diene and dienophile fragments as a result of this replacement. To investigate the influence of the silyl functionality, we computed model TSs of reaction of A2 and 3-methylhexa-2,4-diene (20), in which the silyl group is replaced by a hydrogen. (Table 5, Entry 3) For the 20+A2 addition, a difference in free energies of activation of 1.4 kcal/mol favoring the exo product was computed; this is the same in magnitude and direction as that involving the C2-desmethyl diene 19, (Table 3, Entry 6) but is only half of that observed for the corresponding silylated diene 1d. (Table 5, Entry 1) The C2-Me substituent on the diene does perturb the endo TS geometry, as the vicinal methyl groups are now set at a dihedral angle of 51°, compared to 71° in its absence. The change in the exo TS geometry is negligible. An extra trimethylsilyl group on the diene, however, brings little perturbation on the geometries of either the endo or the exo TS in terms of twist-mode asynchronicity and vicinal Me–Me dihedral angles in 1d+A2. The amplified exo selectivity caused by the presence of the silyl group is likely to be electronic rather than steric in origin, although the precise underpinnings are more subtle in these cases.
Table 5.
Endo (n) and Exo (x) TS Geometries and Energetics for a Range of Substituted Silylated Dienes with Dienophile Complex A2, with an Emphasis on Twist-Mode Asynchronicity.
| Entry | Diene | TS geometry | ΔG‡298a | ΔE0‡b | θc | Me(H)–Me dihedral angle | |
|---|---|---|---|---|---|---|---|
| endo | exo | ||||||
| 1 |  |
 |
 |
30.3(n) 27.4(x) | 2.0 (−14.8) (n) −0.1 (−14.1) (x) | −6°(n) +15°(x) | 47°(n) 69°(x) |
| 2 | 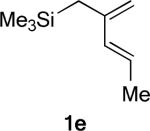 |
 |
 |
23.8(n) 23.0(x) | −1.4 (−12.9) (n) −1.3 (−11.2) (x) | −6°(n) +8°(x) | 52°(n) 57°(x) |
| 3 | 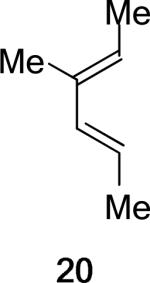 |
 |
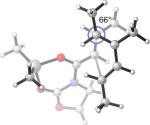 |
31.3(n) 29.9(x) | 6.8 (−9.8) (n) 5.5 (−9.4) (x) | −4°(n) +13°(x) | 51°(n) 66°(x) |
Gas phase Gibbs energies at B3LYP/6−31G(d) level, corrected for solvent effects by PCM single-points at HF/6−31+G(d,p) on B3LYP/6−31G(d) vacuum optimized geometries.
Gas phase uncorrected electronic energy at B3LYP/6−31G(d)//B3LYP/6−31G(d) level. The corresponding MP2/6−31+G(d)//B3LYP/6−31G(d) level in parentheses.
Twist-asynchronicity parameter. See Figure 6b for definition.
Conclusion
A new series of substituent-dependent exo-selective Diels–Alder reactions involving silylated or silyloxy dienes and α,β-unsaturated N-acyloxazolidinone or ketone dienophiles has been studied experimentally and computationally. These reactions demonstrate the maximal stereochemical efficiency of the Diels–Alder reaction by installing four contiguous stereocenters in the cycloadduct with a very favorable exo selectivity and π-facial selectivity. A systematic experimental study identified the structural prerequisites in the starting materials for such a high level of exo preference, namely, simultaneous methyl substitution on the diene and dienophile termini involved in the shorter forming C–C bond. Based on computational investigations of both experimental and other model systems, a twist-mode asynchronicity model was put forward to understand the origin of endo/exo stereocontrol. Specifically, this work showed how the vicinal di-substituent effect could destabilize the highly asynchronous endo TSs, requiring rotations that reduce steric and electrostatic repulsion within the TSs at the expense of reactant distortion. Interaction energies favor the endo pathway, but this is overwhelmed by distortion that relieves gauche-type Me–Me repulsions. This work has moved us closer to a more comprehensive understanding of the factors controlling Diels–Alder endo/exo stereoselectivity. It should also contribute to more effective experimental planning in using this reaction in the stereodefined synthesis of many molecular targets featuring six-membered rings.
Supplementary Material
Chart 1.

Structure of Dienes 1−2 and Dienophiles 3−6.
Acknowledgment
We thank Dr. Andrew Cowley for assistance with X-ray structure determination. This work was funded by the Croucher Foundation, Hong Kong (Y.L.), the Overseas Research Studentship, UK (Y.L.), GlaxoSmithKline (Y.L.), the Engineering and Physical Sciences Research Council, UK (J.M.B.M.), and the National Institute of General Medical Sciences, National Institutes of Health (P.H.-Y.C., K.N.H.). Computational resources were provided by Academic Technology Services, UCLA and the National Service for Computational Chemistry Software, EPSRC.
Footnotes
Supporting Information Available: Complete ref. 35; crystallographic data of exo-8 (.cif); full experimental procedure and data of characterization of all new compounds; Cartesian coordinates, energies, thermal corrections of all optimized reactants and transition structures. These materials are available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- 1.Fringuelli F, Taticchi A. The Diels–Alder Reaction: Selected Practical Methods. Wiley; Chichester: 2002. [Google Scholar]
- 2.a Carey FA, Sundberg RJ. Advanced Organic Chemistry, Part A: Structure and Mechanisms. 5 ed. Springer; New York: 2007. pp. 834–873. [Google Scholar]; b Kürti L, Czakó B. Strategic Applications of Named Reactions in Organic Synthesis. Elsevier; 2005. pp. 140–141. [Google Scholar]
- 3.Nicolaou KC, Snyder SA, Montagnon T, Vassilikogiannakis G. Angew. Chem. Int. Ed. 2002;41:1668. doi: 10.1002/1521-3773(20020517)41:10<1668::aid-anie1668>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 4.Sammis GM, Flamme EM, Xie H, Ho DM, Sorensen EJ. J. Am. Chem. Soc. 2005;127:8612. doi: 10.1021/ja052383c.Pritchard RG, Stoodley RJ, Yuen W-H. Org. Biomol. Chem. 2005;3:162. doi: 10.1039/b410556g.Pellegrinet SC, Spanevello RA. Org. Lett. 2000;2:1073. doi: 10.1021/ol005619k.Kozmin SA, Rawal VH. J. Org. Chem. 1997;62:5252.Fraile JM, García JI, Gracia D, Mayoral JA, Pires E. J. Org. Chem. 1996;61:9479.Node M, Nishide K, Imazato H, Kurosaki R, Inoue T, Ikariya T. Chem. Commun. 1996:2559.Ward DE, Gai Y. Tetrahedron Lett. 1992;33:1851.Lamy-Schelkens H, Giomi D, Ghosez L. Tetrahedron Lett. 1989;30:5887. Exo-selective catalytic asymmetric Diels–Alder reactions of Danishefsky-type dienes and α,β-unsaturated N-acyloxazolidinones have also been disclosed very recently: Sudo Y, Shirasaki D, Harada S, Nishida A. J. Am. Chem. Soc. 2008;130:12588. doi: 10.1021/ja804430n. For a report on an exo-selective Diels–Alder reaction that also includes a mechanistic discussion: Ge M, Stoltz BM, Corey EJ. Org. Lett. 2000;2:1927. doi: 10.1021/ol0060026.
- 5.Boren B, Hirschi JS, Reibenspies JH, Tallant MD, Singleton DA, Sulikowski GA. J. Org. Chem. 2003;68:8991. doi: 10.1021/jo034462h. [DOI] [PubMed] [Google Scholar]
- 6.a Cernak TA, Gleason JL. J. Org. Chem. 2008;73:102. doi: 10.1021/jo701866g. [DOI] [PubMed] [Google Scholar]; b Qi J, Roush WR. Org. Lett. 2006;8:2795. doi: 10.1021/ol0609208. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Roush WR, Barda DA, Limberakis C, Kunz RK. Tetrahedron. 2002;58:6433. doi: 10.1021/ol025772+. [DOI] [PubMed] [Google Scholar]; d Takeda K, Imaoka I, Yoshii E. Tetrahedron. 1994;50:10839. [Google Scholar]; e Pyne SG, Safaei-G J, Hockless DCR, Skelton BW, Sobolev AN, White AH. Tetrahedron. 1994;50:941. [Google Scholar]; f Roush WR, Brown BB. J. Org. Chem. 1992;57:3380. [Google Scholar]; g Adam W, Albert R, Hasemann L, Nava Salgado VO, Nestler B, Peters E-M, Peters K, Prechtl F, von Schnering HG. J. Org. Chem. 1991;56:5782. [Google Scholar]
- 7.Corey EJ, Loh T-P. J. Am. Chem. Soc. 1991;113:8966. [Google Scholar]
- 8.Yoon T, Danishefsky SJ, de Gala S. Angew. Chem. Int. Ed. Engl. 1994;33:853. [Google Scholar]
- 9.Powers TS, Jiang W, Su J, Wulff WD, Waltermire BE, Rheingold AL. J. Am. Chem. Soc. 1997;119:6438. [Google Scholar]
- 10.a Barluenga J, Canteli R-M, Flórez J, García-Granda S, Gutiérrez-Rodríguez A, Martín E. J. Am. Chem. Soc. 1998;120:2514. [Google Scholar]; b Richardson BM, Welker ME. J. Org. Chem. 1997;62:1299. [Google Scholar]; c Wright MW, Smalley TL, Welker ME, Rheingold AL. J. Am. Chem. Soc. 1994;116:6777. [Google Scholar]; d Sabat M, Reynolds KA, Finn MG. Organometallics. 1994;13:2084. [Google Scholar]; e Gilbertson SR, Zhao X, Dawson DP, Marshall KL. J. Am. Chem. Soc. 1993;115:8517. [Google Scholar]
- 11.Maruoka K, Imoto H, Yamamoto H. J. Am. Chem. Soc. 1994;116:12115. [Google Scholar]
- 12.Ahrendt KA, Borths CJ, MacMillan DWC. J. Am. Chem. Soc. 2000;122:4243. [Google Scholar]
- 13.Ishihara K, Nakano K. J. Am. Chem. Soc. 2005;127:10504. doi: 10.1021/ja053368a. [DOI] [PubMed] [Google Scholar]
- 14.Kano T, Tanaka Y, Maruoka K. Org. Lett. 2006;8:2687. doi: 10.1021/ol060621i. [DOI] [PubMed] [Google Scholar]
- 15.Kano T, Tanaka Y, Maruoka K. Chem. Asian J. 2007;2:1161. doi: 10.1002/asia.200700122. [DOI] [PubMed] [Google Scholar]
- 16.Gotoh H, Hayashi Y. Org. Lett. 2007;9:2859. doi: 10.1021/ol071009+. [DOI] [PubMed] [Google Scholar]
- 17.Walter CJ, Anderson HL, Sanders JKM. J. Chem. Soc., Chem. Commun. 1993:458. [Google Scholar]
- 18.Pearson RJ, Kassianidis E, Philp D. Tetrahedron Lett. 2004;45:4777. [Google Scholar]
- 19.a Cannizzaro CE, Ashley JA, Janda KD, Houk KN. J. Am. Chem. Soc. 2003;125:2489. doi: 10.1021/ja020879d. [DOI] [PubMed] [Google Scholar]; b Heine A, Stura EA, Yli-Kauhaluoma JT, Gao C, Deng Q, Beno BR, Houk KN, Janda KD, Wilson IA. Science. 1998;279:1934. doi: 10.1126/science.279.5358.1934. [DOI] [PubMed] [Google Scholar]; c Yli-Kauhaluoma JT, Ashley JA, Lo C-H, Tucker L, Wolfe MM, Janda KD. J. Am. Chem. Soc. 1995;117:7041. [Google Scholar]; d Gouverneur VE, Houk KN, de Pascual-Teresa B, Beno B, Janda KD, Lerner RA. Science. 1993;262:204. doi: 10.1126/science.8211138. [DOI] [PubMed] [Google Scholar]
- 20.Lam Y.-h., Bobbio C, Cooper IR, Gouverneur V. Angew. Chem. Int. Ed. 2007;46:5106. doi: 10.1002/anie.200701365. [DOI] [PubMed] [Google Scholar]
- 21.For related Diels–Alder reactions of all-carbon silylated dienes, see: Ryu DH, Corey EJ. J. Am. Chem. Soc. 2003;125:6388. doi: 10.1021/ja035393r.Vedejs E, Duncan SM. J. Org. Chem. 2000;65:6073. doi: 10.1021/jo000533q.Organ MG, Winkle DD. J. Org. Chem. 1997;62:1881.Corey EJ, Letavic MA. J. Am. Chem. Soc. 1995;117:9616.Krafft GA, Garcia EA, Guram A, O'Shaughnessy B, Xu X. Tetrahedron Lett. 1986;27:2691.Sakurai H, Hosomi A, Saito M, Sasaki K, Iguchi H, Sasaki J-I, Araki Y. Tetrahedron. 1983;39:883.Trost BM, Shimizu M. J. Am. Chem. Soc. 1982;104:4299.Ojima I, Yatabe M, Fuchikami T. J. Org. Chem. 1982;47:2051.
- 22.a Frankowski KJ, Golden JE, Zeng Y, Lei Y, Aubé J. J. Am. Chem. Soc. 2008;130:6018. doi: 10.1021/ja800574m. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Alonso D, Caballero E, Medarde M, Tomé F. Tetrahedron Lett. 2007;48:907. [Google Scholar]; c Caballero E, Alonso D, Peláez R, Álvarez C, Puebla P, Sanz F, Medarde M, Tomé F. Tetrahedron. 2005;61:6871. [Google Scholar]; d Caballero E, Alonso D, Peláez R, Alvarez C, Puebla P, Sanz F, Medarde M, Tomé F. Tetrahedron Lett. 2004;45:1631. [Google Scholar]
- 23.a Nakashima D, Yamamoto H. J. Am. Chem. Soc. 2006;128:9626. doi: 10.1021/ja062508t. [DOI] [PubMed] [Google Scholar]; b Ruijter E, Schültingkemper H, Wessjohann LA. J. Org. Chem. 2005;70:2820. doi: 10.1021/jo0488311. [DOI] [PubMed] [Google Scholar]; c Boezio AA, Jarvo ER, Lawrence BM, Jacobsen EN. Angew. Chem. Int. Ed. 2005;44:6046. doi: 10.1002/anie.200502178. [DOI] [PubMed] [Google Scholar]; d Chaplin JH, Edwards AJ, Flynn BL. Org. Biomol. Chem. 2003;1:1842. doi: 10.1039/b302522e. [DOI] [PubMed] [Google Scholar]; e Kraus GA, Hon YS, Sy J, Raggon J. J. Org. Chem. 1988;53:1397. [Google Scholar]; f Bell VL, Holmes AB, Hsu S-Y, Mock GA, Raphael RA. J. Chem. Soc., Perkin Trans. 1986;1:1507. [Google Scholar]
- 24.Narayanan BA, Bunnelle WH. Tetrahedron Lett. 1987;28:6261.Hayashi T, Katsuro Y, Kumada M. Tetrahedron Lett. 1980;21:3915.Yamamoto Y, Yamamoto H. Angew. Chem. Int. Ed. 2005;44:7082. doi: 10.1002/anie.200501345.Kanemasa S, Kumegawa M, Wada E, Nomura M. Bull. Chem. Soc. Jpn. 1991;64:2990. See also refs. 20 and 23a.
- 25.a Evans DA, Chapman KT, Bisaha J. J. Am. Chem. Soc. 1984;106:4261. [Google Scholar]; b Thom C, Kocienski P. Synthesis. 1992:582. [Google Scholar]; c Kanemasa S, Kanai T. J. Am. Chem. Soc. 2000;122:10710. [Google Scholar]
- 26.Evans DA, Chapman KT, Bisaha J. J. Am. Chem. Soc. 1988;110:1238. [Google Scholar]
- 27.For a recent example of the use of this chiral auxiliary in an exo-selective asymmetric hetero Diels–Alder reaction: Evans DA, Scheidt KA, Downey CW. Org. Lett. 2001;3:3009. doi: 10.1021/ol016420q.
- 28.a Zeng Y, Aubé J. J. Am. Chem. Soc. 2005;127:15712. doi: 10.1021/ja055629m. [DOI] [PubMed] [Google Scholar]; b Zeng Y, Reddy DS, Hirt E, Aubé J. Org. Lett. 2004;6:4993. doi: 10.1021/ol047809r. [DOI] [PubMed] [Google Scholar]
- 29.Armstrong A, Davies NGM, Martin NG, Rutherford AP. Tetrahedron Lett. 2003;44:3915. [Google Scholar]
- 30.a Becke AD. J. Chem. Phys. 1993;98:1372. [Google Scholar]; b Becke AD. J. Chem. Phys. 1993;98:5648. [Google Scholar]; c Lee C, Yang W, Parr RG. Phys. Rev. B. 1988;37:785. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]
- 31.a Hehre WJ, Ditchfield R, Pople JA. J. Chem. Phys. 1972;56:2257. [Google Scholar]; b Hariharan PC, Pople JA. Theor. Chim. Acta. 1973;28:213. [Google Scholar]
- 32.Tomasi J, Persico M. Chem. Rev. 1994;94:2027. [Google Scholar]
- 33.Takano Y, Houk KN. J. Chem. Theory Comput. 2005;1:70. doi: 10.1021/ct049977a. [DOI] [PubMed] [Google Scholar]
- 34.Birney DM, Houk KN. J. Am. Chem. Soc. 1990;112:4127. [Google Scholar]
- 35.Frisch MJ, et al. Gaussian 03. Gaussian, Inc.; Wallingford, CT: 2004. Rev. C.02 & D.01. [Google Scholar]
- 36.Castellino S, Dwight WJ. J. Am. Chem. Soc. 1993;115:2986. [Google Scholar]
- 37.Bickelhaupt FM. J. Comput. Chem. 1999;20:114. [Google Scholar]
- 38.DeChancie J, Acevedo O, Evanseck JD. J. Am. Chem. Soc. 2004;126:6043. doi: 10.1021/ja037702j. [DOI] [PubMed] [Google Scholar]
- 39.Nagase S, Morokuma K. J. Am. Chem. Soc. 1978;100:1666. [Google Scholar]
- 40.a Ess DH, Houk KN. J. Am. Chem. Soc. 2008;130:10187. doi: 10.1021/ja800009z. [DOI] [PubMed] [Google Scholar]; b Ess DH, Houk KN. J. Am. Chem. Soc. 2007;129:10646. doi: 10.1021/ja0734086. [DOI] [PubMed] [Google Scholar]; c Ess DH, Jones GO, Houk KN. Org. Lett. 2008;10:1633. doi: 10.1021/ol8003657. [DOI] [PubMed] [Google Scholar]; d García JI, Martínez-Merino V, Mayoral JA, Salvatella L. J. Am. Chem. Soc. 1998;120:2415. doi: 10.1021/ja003695c. [DOI] [PubMed] [Google Scholar]; e Sbai A, Branchadell V, Ortuño RM, Oliva A. J. Org. Chem. 1997;62:3049. doi: 10.1021/jo960447j. [DOI] [PubMed] [Google Scholar]
- 41.Wannere CS, Paul A, Herges R, Houk KN, Schaefer HF, III, Schleyer P. v. R. J. Comput. Chem. 2007;28:344. doi: 10.1002/jcc.20532. [DOI] [PubMed] [Google Scholar]
- 42.Tsuzuki S, Lüthi HP. J. Chem. Phys. 2001;114:3949. [Google Scholar]
- 43.a Goldstein E, Beno B, Houk KN. J. Am. Chem. Soc. 1996;118:6036. [Google Scholar]; b Storer JW, Raimondi L, Houk KN. J. Am. Chem. Soc. 1994;116:9675. [Google Scholar]; c Houk KN, Lin YT, Brown FK. J. Am. Chem. Soc. 1986;108:554. doi: 10.1021/ja00263a059. [DOI] [PubMed] [Google Scholar]
- 44.a Singleton DA, Schulmeier BE, Hang C, Thomas AA, Leung S-W, Merrigan SR. Tetrahedron. 2001;57:5149. [Google Scholar]; b Beno BR, Houk KN, Singleton DA. J. Am. Chem. Soc. 1996;118:9984. [Google Scholar]
- 45.Houk KN, Gonzalez J, Li Y. Acc. Chem. Res. 1995;28:81. For an up-to-date discussion of Diels–Alder mechanisms, see also: Bachrach SM. Computational Organic Chemistry. John Wiley & Sons, Inc.; 2007. p. 128-133.
- 46.García JI, Mayoral JA, Salvatella L. J. Am. Chem. Soc. 1996;118:11680. See also refs 34, 40d, and 40e.
- 47.a Brown FK, Houk KN. Tetrahedron Lett. 1985;26:2297. [Google Scholar]; b Wu T-C, Houk KN. Tetrahedron Lett. 1985;26:2293. [Google Scholar]
- 48.a Tripoli R, Cayzer TN, Willis AC, Sherburn MS, Paddon-Row MN. Org. Biomol. Chem. 2007;5:2606. doi: 10.1039/b708324f. [DOI] [PubMed] [Google Scholar]; b Paddon-Row MN, Moran D, Jones GA, Sherburn MS. J. Org. Chem. 2005;70:10841. doi: 10.1021/jo051973q. [DOI] [PubMed] [Google Scholar]; c Cayzer TN, Paddon-Row MN, Moran D, Payne AD, Sherburn MS, Turner P. J. Org. Chem. 2005;70:5561. doi: 10.1021/jo0505829. [DOI] [PubMed] [Google Scholar]; d Lilly MJ, Paddon-Row MN, Sherburn MS, Turner CI. Chem. Commun. 2000:2213. [Google Scholar]
- 49.Iafe RG, Houk KN. J. Org. Chem. 2008;73:2679. doi: 10.1021/jo702576r. [DOI] [PubMed] [Google Scholar]
- 50.a Leach AG, Houk KN. J. Org. Chem. 2001;66:5192. doi: 10.1021/jo0104126. [DOI] [PubMed] [Google Scholar]; b McCarrick MA, Wu YD, Houk KN. J. Org. Chem. 1993;58:3330. [Google Scholar]
- 51.Singleton DA, Merrigan SR, Beno BR, Houk KN. Tetrahedron Lett. 1999;40:5817. [Google Scholar]
- 52.Acevedo O, Evanseck JD. Org. Lett. 2003;5:649. doi: 10.1021/ol027408g. [DOI] [PubMed] [Google Scholar]
- 53.a Dinadayalane TC, Vijaya R, Smitha A, Sastry GN. J. Phys. Chem. A. 2002;106:1627. [Google Scholar]; b Baki S, Maddaluno J, Derdour A, Chaquin P. Eur. J. Org. Chem. 2008:3200. [Google Scholar]; c Bakalova SM, Santos AG. J. Org. Chem. 2004;69:8475. doi: 10.1021/jo049298s. [DOI] [PubMed] [Google Scholar]; d Bakalova SM, Kaneti J. J. Phys. Chem. A. ASAP, DOI: 10.1021/jp803701y. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.