

Abstract
Cellular signal transduction pathways are controlled by specific protein-protein interactions mediated by the binding of short peptides to small modular interaction domains. To gain insights into the specificity of these interactions, the association of phosphotyrosine-containing peptides to Src Homology 2 (SH2) domains is characterized using computations. Molecular dynamics simulations based on high-resolution crystal structures complemented by homology models are used to calculate the absolute binding free energies for 25 SH2-peptides pairs. The calculations are carried out using a potential of mean force free energy simulations method with restraining potentials that was developed previously [Woo and Roux, Proc. Nat. Acad. Sci. USA 102:6825 (2005)]. The method is utilized in conjunction with an implicit solvent representation to reduce the computational cost to characterize the association of five SH2 domains and five peptides. Specificity is ascertained by directly comparing the affinities of a given SH2 domain binding for any of the different peptides. For three of the five SH2 domains, the computational results rank the native peptides, as the most preferred binding motif. For the remaining two SH2 domains, high affinity binding motifs other than the native peptides are identified. The study illustrates how free energy computations can complement experiments in trying to elucidate complex protein-protein interactions networks.
I. INTRODUCTION
Small modular binding domains, such as SH2, SH3, PH or PDZ domains, mediate specific protein-protein interactions that largely define specificity in cellular signal transduction [1–4]. The SH2 domains are a family of small proteins of about 100 amino acids. They are highly conserved, even though they are present in a wide variety of signaling molecules, including protein kinases (Src, Lck), protein phosphatases (Shp2, SHIP2), protein phospholipases (PLCγ1), transcription factors (Stat), signal regulator proteins (SOCS), adaptor proteins (Grb2), scaffold proteins (Shc) et al [5]. Functionally, SH2 domains are predominantly involved in protein tyrosine kinases (PTK) signaling pathways because they specifically recognize phosphotyrosine (pY)-containing motifs within their target proteins. The binding of SH2 domains to specific phosphotyrosine sites recruit specific proteins to various subcellular locations, assemble multi-protein signaling complexes, and regulate enzymes activities [6]. Mutations in SH2 domains or their binding motifs will disrupt the specific interactions between SH2 domains and their binding partner and cause malfunctions in cell signaling which have been linked to a variety of human diseases [7].
The inspiring work by Cantley and co-workers marked the first systematic investigation on the specificity of SH2 domains mediated protein-protein interactions [8, 9]. Using a peptide library, they predicted the preferred binding motifs for 25 SH2 domains and further classified all SH2 domains into 5 groups based on the SH2 domain βD5 residue. Their results indicate that SH2 domains recognize specific residues in the C-terminal pY+1, +2 and +3 positions in a way that varies from one SH2 domain to another. For example, Src kinase SH2 domains preferentially recognize Glu-Glu-Ile, thus their binding motif is designated as pYEEI, whereas Grb2 SH2 domains prefer the pYVNV motif. As more information about SH2 domain bound and ligand-free forms are becoming available, structural studies are shedding light on the observed binding specificity of SH2 domains [10–15]. However, a complete understanding of the origin of specificity requires a detailed study of the thermodynamic basis of the binding of phosphopeptides to SH2 domains.
In contrast to extensive experimental investigations [8, 9, 16–20], few computational studies of SH2 domain binding specificity have been reported to date. Campbell et al analyzed the SH2 domain specificity in terms of the similarity in binding site residues [21]. Sheinerman et al used a continuum electrostatic approach to identify the energetic determinants of specificity [22]. While it was possible to gain significant insight from these previous studies, the scope of such analysis is nonetheless limited because peptide-SH2 absolute binding free energies are not actually calculated and dissected.
In fact, calculations of the absolute binding free energy of a phosphotyrosine peptides to SH2 domain are exceedingly challenging. The difficulties are due, in part, to the large magnitude of the electrostatic interactions involved and the significant flexibility of the peptide ligand. In particular, conventional binding free energy approaches, which are based on a thermodynamic decoupling scheme of the ligand from its environment [23–26], are essentially impractical in this case. Even a statistical imprecision of about 1 percent on the ligand solvation free energy is already larger than the experimental binding free energy. To circumvent those issues, a novel potential of mean force (PMF) free energy simulation method with biasing restraints was previously formulated [27]. Using this method in the context of MD simulations with explicit solvent molecules, it was shown that a rigorously calculated binding affinity of phosphotyrosine peptide pYEEI to the Lck SH2 domain was in very good agreement with experimental data [27]. Although such results are encouraging and suggest that atomic models have the ability to accurately represent molecular interactions, the computational cost of simulations with explicit solvent prohibits a broader exploration of peptide-SH2 specificity. One straightforward strategy to reduce the computational cost, which is tentatively explored here, is to replace the explicit solvent molecules by an implicit solvent approximation.
The goal of the present study is to investigate the ability of computations based on the PMF free energy simulation approach in combination with an implicit solvent representation to account for peptide-SH2 specificity. The method is first validated by comparing with the previous results obtained with explicit solvent simulations, and is then applied to a number of ligands and SH2 domains. Relying on the high sequence similarity and structural conservation of the SH2 domain family, it was possible to model 20 additional bound structures from a set of 5 high resolution crystal structures of SH2 domains with a bound peptide. Using these native and modeled structures, MD simulations with implicit solvent were then performed for a total of 25 pairs, representing all possible interactions between 5 different phosphotyrosine peptides and 5 different SH2 domains. Specificity is ascertained by directly comparing the affinities of a given SH2 domain binding for the five different peptides. For three of the five SH2 domains, the results rank the native peptides as the most preferred binding motif. For the remaining two SH2 domains, high affinity binding motifs other than the native peptides are identified. The present study demonstrates that, computational methods provide a powerful complement to experiments in trying to elucidate complex protein-protein interactions.
II. METHODS
A. Dataset selection
Five different SH2 domains, for which at least one phosphopeptide-bound structure is available, are selected for specificity study: Lck, Grb2, Cbl, p85αN, Stat1 (PDB: 1LKK [12], 1JYR [14], 2CBL [13], 2IUH [11], 1YVL [15]). Each of the five SH2 domains belongs to one of the five SH2 domain groups classified by Cantley and co-workers [8, 9], thus this dataset covers the entire SH2 domain family. The selected SH2 domains and their binding peptide sequences in the bound structures are summarized in Table 1 and detailed below.
TABLE I.
Selected SH2-phosphopeptide complexes for specificity study.
Group-1a SH2 domain
Lck (lymphocyte-specific protein tyrosine kinase) is a member of the Src family nonreceptor protein tyrosine kinase mainly involved in T-cell activation and development. Structurally from N-terminal to C-terminal, Lck contains a myristylation site, a unique domain, followed by an SH3 and an SH2 domain, a tyrosine kinase catalytic domain and a C-terminal tail.
Group-1b SH2 domain
Grb2 (growth factor receptor-binding protein 2) is a small adaptor protein with no catalytic activity. Comprising only one SH2 domain surrounded by two SH3 domains, Grb2 direct complex formation with proline-rich regions of other proteins through its two SH3 domains and binds phosphotyrosine sites of other proteins through its SH2 domain.
Group-2 SH2 domain
Cbl (Casitas B-lineage lymphoma) protein is an adaptor that functions as a negative regulator of several signaling pathways initiated from cell surface receptors. The highly conserved amino-terminal region of Cbl is composed of a four-helix bundle domain, an EF-hand calcium-binding domain and an SH2 domain.
Group-3 SH2 domain
Phosphatidylinositol 3-kinase (PI3K) is a heterodimeric enzyme composed of a 110-kDa catalytic subunit (p110) and an 85-kDa regulatory subunit (p85α). The p85α chain contains an SH3 domain followed by two SH2 domains, which regulate the catalytic activity of the enzyme and serve as adapters to couple different phosphoproteins to the catalytic subunit.
Group-4 SH2 domain
Stat1 is a member of the STAT (Signal Transducers and Activator of Transcription) family transcription factor proteins that participate in transcriptional regulation of various cytokine responsive genes. Stat1 is composed of an N-domain, a coiled-coil domain, DNA binding domain, followed by an SH2 domain and transcriptional activation domain.
B. Structure preparation
The bound structures of each SH2 domain in complex with the four peptides other than their native peptide were constructed by a homology modeling protocol. First, two SH2 domains in the native bound structures were superimposed by the combinatorial extension (CE) structure alignment algorithm [28]. Then, the ligand coordinate of each native structure was exchanged to produce two hybrid bound structures. The modeling scheme is shown in Figure 1. This homology modeling scheme exploits the fact that the SH2 domains family is structurally conserved and all known SH2 domain structures show a similar fold [7].
FIG. 1.

homology modeling scheme illustrating how the non-native bound structures are constructed.
C. Binding free energy calculation
A rigorous formulation of absolute binding free energies that is amenable to accurate simulations has been established over several years [23–26]. In those traditional approaches, the ligand is typically decoupled from its environment using thermodynamic integration or free energy perturbation schemes. However, these approaches are not practical for phosphotyrosine peptides binding to SH2 domain because the solvation energies are too large and even a small imprecision is sufficient to render the computation ineffectual. An alternative approach, based on a staged PMF free energy simulations method with restraining potentials was developed to circumvent those difficulties [27]. The thermodynamic cycle illustrating this method is shown in Figure 2. For the sake of completeness, a brief summary of the theoretical formulation is given here and the detailed derivation of this method can be found elsewhere. From classical statistical mechanics, the equilibrium binding constant of a protein-ligand complex can be expressed as the ratio of two integrals [27, 29, 30],
FIG. 2.
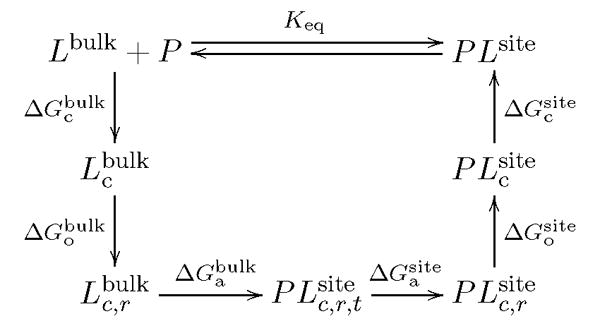
Thermodynamic cycle illustrating the stage method of protein-ligand binding free energy calculation.
| (1) |
where the denominator and the numerator can be viewed as the initial and final state of the binding process: the ligand with its center of mass held at position far away from the protein in the bulk solution and the ligand bound to the protein binding site, U is the total potential energy of the system, (1) and (X) are the degrees of freedom of the ligand and remaining system (solvent or protein), respectively. For computational convenience, a series of intermediate states are inserted into Eq. (1), resulting in the following form for the equilibrium binding constant,
| (2) |
where each term represents a specific stage in the binding free energy computation during which the various restraining potentials uc (conformation), uo (orientation) and ua(axial) are applied and then removed. Those restraining potentials serve to reduce the fluctuations and help the convergence of the free energy computations. However, it is important to note that the final results is rigorously independent of the choice biasing potentials [29, 30]. The free energy change of each stage is computed correspondingly either by MD free energy perturbation (FEP) [31] or by direct numerical integration. The equilibrium binding constant Keq is written as,
| (3) |
(all terms are given in the same order as in Eq. (2) above). Assuming a standard reference state concentration of 1 M (C° = 1/1661 Å−3), the standard binding free energy can be expressed as
| (4) |
The first term and the last term in Eq. (4), corresponding to the free energy changes when applying and releasing a restraining potential on the conformation of the ligand, are calculated by integrating over the PMF as a function of the ligand root mean square deviation (RMSD) with respect to the reference ligand conformation [27, 29, 30]. The difference provides a measure of the free energy cost for the ligand to adopt the bound conformation. The second term is obtained by direct numerical integration over three angles that are defined to represent the orientation of the ligand relative to the protein. The third term is expressed as the product of two integrals S* and I*, representing the surface and radial part of the spherical coordinates describing the position of the ligand relative to the SH2 domain. The angular part S*(dimension of Å2) is calculated by direct integration over two angles,
| (5) |
and I* (dimension of Å) is calculated by integrating over the 1D-PMF as a function of the radial distance between the center-of-mass of the protein and ligand,
| (6) |
(it should be noted that the 1D-PMF is obtained in the presence of the configurational and orientational restraints uc, uo, and ua). To improve the sampling in some regions, the distance between the phosphate of the ligand and phosphate binding residues in SH2 domain is introduced as a second variable and the 2D-PMF W(r1; r2) is first constructed, and then reduced to W(r1) (see supporting information for details). The fourth term and fifth term , corresponding to the free energy changes when the translational and rotational restraint is turned off in the binding site, are calculated by FEP with 10 evenly-spaced sampling windows. Whenever possible, umbrella sampling [32] is used to ensure adequate sampling in protein-ligand configuration space and weighted histogram analysis methods (WHAM) [33] to process the umbrella sampling and FEP data.
D. Computation details
All the computations were performed with a similar protocol. Hydrogen atoms were built to the initial PDB bound structure using the hbuild module of the charmm molecular simulation program [34]. After minimization, the minimized structure was then equilibrated for 1 ns by Langevin dynamics. The resulting structure after equilibration was used as the starting structure for following simulations and the average structure from the 1 ns equilibration was used as the reference conformation. The Langevin simulations were performed at a constant temperature of 300K and a friction of 5 ps−1. The generalized Born with switching (GBSW) implicit solvent [35] was used, with salt concentration set to 150 mM. All computations were carried out with version c33a2 of charmm and the all-atom PARAM27 force field [36].
The lengths of all MD simulations are determined on the basis of a benchmark study of Cbl SH2 domain bound with its native peptide pYTPE. To estimate the error of the computed binding free energy, all full calculations are repeated three times. Standard deviations from the three independent simulations are taken as the uncertainty of the calculations. For the PMF of translating the ligand into SH2 domain binding site in stage 3, simulations were repeated four times because larger uncertainty is observed for this part. The computational cost of each step in binding free energy calculation is tabulated in supporting information (Table S1).
III. RESULTS AND DISCUSSION
A. Constructed structures by homology modeling
The 20 modeled ligand-bound structures and the five native bound structures as described in METHODS are shown in Figure 3, with SH2 domains superimposed. The backbones of the five different peptides generally overlap well and the phosphotyrosine position relative to the SH2 domain is similar, except for the peptide pYTPE from Cbl SH2 domain. MD simulation is expected to relax the peptides and adjust the relative position of the peptides to SH2 domain because the initial configurations are all very similar.
FIG. 3.

native and modeled structures for binding free energy calculations, coloring scheme: red, Cbl+pYTPE; green, p85αN+pYMDM; blue, Grb2+pYVNV; yellow, Lck+pYEEI; purple, Stat1+pYDKP
The major assumptions made in this homology modeling protocol are: (1) a given SH2 domain binds different phosphotyrosine peptides in the same binding site; (2) a given phosphopeptide adopts the same binding mode when binding different SH2 domains; (3) The binding of phosphopeptide does not induce large conformational change in SH2 domain. In order to assess the accuracy of the modeled structures, the three assumptions made in the homology modeling need further validation. To this end, we performed an exhaustive search of all the available SH2 domain structures deposited in PDB up to date. Seven SH2 domains are found to bind more than one distinct peptide and six peptides are found to bind more than one SH2 domain. A superposition of these structures (supporting information Figure 4 and 5) indicates that pY in the peptides generally occupy similar position and the backbone of the three residues at position +1, +2 and +3 C-terminal to pY overlap well. However, for residues beyond +4 position or N-terminal to pY, large deviations are often observed. This high similarity in binding mode is consistent with the fact that the structures of the entire SH2 domain family are highly conserved. Therefore, the major binding sites in SH2 domain are very likely to be conserved, although a second unconventional binding site has been reported for SH2 domains in PLC-γ-C [37] and p85α-N [38].
To estimate the ligand binding induced conformational change in SH2 domain, we computed the Cα RMSD between all SH2 domain apo and holo structures and between different holo structures. Figure 4 shows the normalized distribution of the RMSD values. The RMSD between any pair of structures is below 1.5 Å including the full length. The majority of RMSD is around 0.6 Å for holo-holo and 0.8 angstrom for apo-holo. Because the binding of the relatively short peptide occurs at the surface of SH2 domain, it is unlikely to induce dramatic conformational changes. The major conformational change observed by aligning apo and holo structures is localized in the loop region near the binding surface. For different holo structures, the conformational change is even smaller compared with that between apo and holo structure.
FIG. 4.

The distribution of RMSD between ligand-bound and ligand-free structures of the same SH2 domains (black, green) and RMSD between different ligand-bound structures of the same SH2 domains (red, blue)
B. Binding free energies for Lck SH2 domain bound with pYEEI and mutants
Computed binding free energies from previous explicit solvent simulations [27] and from present GB implicit solvent simulations are compared in Table 2. The results indicate that GB implicit solvent simulations are able to reproduce each of the energetic contributions to the total binding free energy fairly well. Also, the PMF from GB model simulations (Figure 5) displays very similar characteristics compared with explicit solvent simulations. Thus, a change from previous explicit solvent simulations to GB implicit solvent simulations does not severely compromise the accuracy of computed binding free energy, while speed up the calculation by approximately 5 fold.
TABLE II.
Computed binding free energy for Lck SH2 domain-pYEEI and mutants.
| liganda | ΔGtotal | ΔGexp | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| pYEEI | 3.70 | 5.35 | −15.94 | 0.40 | 0.04 | 1.43 | −8.8b | −9.4 | ||||||
| pYEEI | 4.9±0.1 | 5.55 | −16.6±0.8 | 0.35±0.03 | 0.9±0.1 | 1.7±0.1 | −9.1±0.8 | −9.4c | ||||||
| de-Ac | 3.3±0.1 | 5.56 | −15.4±1.3 | 0.37±0.05 | 1.0±0.2 | 0.9±0.1 | −8.9±1.3 | −7.6d | ||||||
| YEEI | 4.1±0.2 | 5.56 | −8.8±1.1 | 0.41±0.07 | 1.8±0.2 | 2.2±0.1 | −3.5±1.1 | −3.6e | ||||||
| p(−1) | 4.2±0.2 | 5.56 | −11.9±1.3 | 0.33±0.08 | 1.6±0.1 | 0.8±0.1 | −4.8±1.3 | −6.9e | ||||||
| pY | 1.6±0.1 | 5.57 | −7.5±1.5 | 0.37±0.04 | 1.8±0.1 | 1.4±0.1 | −3.9±1.5 | −4.7e | ||||||
| EEI | 2.0±0.1 | 5.54 | −5.2±1.0 | 0.44±0.10 | 2.1±0.5 | 1.0±0.2 | −1.2±1.1 | N/A | ||||||
| pYAEI | 3.7±0.1 | 5.55 | −14.9±1.2 | 0.36±0.02 | 1.3±0.2 | 0.9±0.1 | −8.2±1.2 | −8.7e | ||||||
| pYEAI | 4.3±0.1 | 5.54 | −15.4±1.3 | 0.71±0.25 | 1.4±0.2 | 0.9±0.1 | −8.6±1.3 | −8.2e | ||||||
| pYEEA | 4.2±0.1 | 5.55 | −12.6±1.0 | 0.38±0.08 | 1.2±0.2 | 1.5±0.2 | −5.9±1.0 | −7.8e |
See the text for a description of the mutated ligands. All ΔG values are in the unit of kcal/mol. Errors are estimated from the standard deviation of four independent calculations.
Results from previous explicit solvent simulations Ref.[27].
Experimental values are from Ref.[12].
Ref.[57].
Ref.[39].
FIG. 5.

(A) PMF as a function of ligand rmsd in binding site (solid line) and in bulk solution (dash line), corresponding to the calculations of term and (B) PMF as a function of the radial distance between ligand and protein for Lck-pYEEI from present GB implicit solvent simulations.
To further test the accuracy of the computed binding free energy compared with experimental values, eight mutant peptides of pYEEI are designed. This part serves to evaluate the robustness and reliability of the binding free energy calculations and is motivated by a similar experimental study on Src SH2 domain by Waksman and co-workers [39]. The experiments were done on the SH2 domain of Src while the calculations here are executed with the SH2 domain of Lck. The choice is motivated by the fact that the crystal structure of Lck SH2 domain has higher resolution and previously we have carried out explicit solvent simulations on Lck SH2 domain. Because Lck and Src have very similar binding affinities for a variety of phosphopeptides [40], comparing the computations on SH2 from Lck with experimental data with SH2 from Src is meaningfull. The design of pYEEI mutant peptides aims at dissecting the energetic contributions of each part of the phosphopeptide to the total binding free energy. Specifically, the 8 mutant peptides include the unphosphorylated YEEI, the deacetylated pYEEI, a single phosphotyrosine pTyr, a tripeptide EEI, pYEEI with 1 negative charge on pTyr instead of 2 and three Ala mutants of the Glu-Glu-Ile residue pYAEI, pYEAI, pYEEA. Results from the calculations are listed in Table 2, together with the corresponding experimental binding free energies [39]. A linear fitting of the calculated binding affinities with experimental measurements gives a correlation coefficient of 0.88 and a slope of 1.04, as shown in Figure 6. Another aspect worth noting about present calculations is the free energy cost associated with restricting the conformational degree of freedom of the ligand. The values of ΔΔGc reported in Table 2 indicate that, even for a short peptide ligand, the conformational free energy contribution is on the order of 3 kcal/mol. This energetic cost represents a significant component of the total binding free energy.
FIG. 6.

Comparison of calculated binding free energies with experimental measurements for Lck SH2 domain bound with peptide pYEEI and mutants.
Analysis of the computation confirms several features general to the SH2 domain-phosphopeptide binding. The interaction between the phosphorylated tyrosine and the strictly conserved Arg residue in SH2 domain βB5 position provides the major contribution to the total binding free energy. Nearly one half of the total binding free energy comes from this interaction. The rest three residues C-terminal to pTyr in the peptide provide the other half of the total binding free energy, with the residue at the pTyr+3 position contributing the largest share. This is consistent with a \two-pronged plug engaging a two-holed socket” binding mode [10]. Also, the binding of phosphotyrosine to SH2 domain is dominated by strong electrostatic interaction. When the negative charge on the phosphate group is decreased from 2 to 1, the total binding free energy decreases by almost 3 kcal/mol.
C. Binding specificity for the 5 representative SH2 domains
The absolute binding free energies of the 25 SH2 domain-phosphopeptide pairs were computed (Table 3). In addition, the interaction between each SH2 domain and a phosphotyrosine was also characterized. Only the total binding free energies are given here. The detailed decomposition of the total binding free energy into the contribution from each stage is provided in supporting information. For Lck, Grb2 and Cbl SH2 domains, the calculations correctly ranked the peptide from the original native bound structure as the highest affinity motif. For p85αN and Stat1 SH2 domains, the results suggest that binding motifs other than those in the native bound structures may exist.
TABLE III.
Computed binding free energy for the 25 pair-wise interactions (kcal/mol).
| Lck | Grb2 | Cbl | p85αN | Stat1 | |
|---|---|---|---|---|---|
| pYEEI | −9.1±0.8 | −4.5±1.7 | −4.1±0.9 | −4.6±1.1 | −3.4±1.3 |
| pYVNV | −4.5±1.7 | −10.4±1.7 | −8.3±1.2 | −9.7±1.9 | −10.2±1.6 |
| pYTPE | −1.7±1.8 | −2.7±0.6 | −9.2±1.0 | −4.1±1.4 | −4.0±1.8 |
| pYMDM | −2.3±1.5 | −5.6±2.0 | −7.9±1.4 | −5.1±0.7 | −9.7±0.7 |
| pYDKP | −6.5±1.4 | −6.6±1.2 | −7.2±1.4 | −7.5±1.3 | −8.3±1.2 |
| pY | −3.9±1.5 | −4.2±0.6 | −4.3±1.6 | −4.6±1.4 | −4.7±0.3 |
For Lck SH2 domain, the calculations successfully differentiate the 5 phosphopeptides. The degree of specificity is shown to be high, with a large difference between the binding affinity of the native peptide pYEEI and that of the other peptide. Among the four non-native peptides, pYTPE has the lowest affinity for Lck SH2 domain. This is probably because of the disfavored interaction between the negative charged Glu residue at pY+3 position and the hydrophobic binding pocket in SH2 domain. In fact, among the five SH2 domains pYTPE seems to bind none of the four SH2 domains other than its native Cbl SH2 domain with high affinity.
A number of observations can be made for the results concerning Grb2, Cbl, p85αN and Stat1 SH2 domains. The experimentally determined consensus motif for Grb2 SH2 domain has a favored Asn residue at pY+2 position and varies at pY+1 and +3 position (Table 4). The calculations ranked the native motif pYVNV as the highest binding motif and the computed binding free energies for Grb2 SH2 domain-phosphopeptide interactions agree with experimental data quite well [14, 41]. As to Cbl SH2 domain, previous studies have shown that the two residues at pY-1 and -2 positions N-terminal to pY contribute to binding specificity [42]. However, the calculations show that those two residues may have limited influence on the binding free energy. Even though in present study the residues N-terminal to pY are not included in the simulations, the computed binding free energies for Cbl SH2 domain seem to be in the same range as experimental data [43]. With p85αN SH2 domain, calculated binding free energy for pYMDM is somehow smaller than the experimental result. This may be because residues N-terminal to pY and beyond pY+3 positions provide important contacts with SH2 domain that contribute to the binding free energy. For example, an 11-residues peptide DDGpYMPMSPGV binds with p85αN SH2 domain with an IC50 around 0.7 μM, while the IC50 for the truncated pentapeptide pYMPMS is increased to 60 μM [44]. On the other hand, the peptide pYVNV is ranked as the favored motif for p85αN SH2 domain by the calculation. This suggests that proteins containing the YVNV motif may be potential binding partners for p85αN SH2 domain. In the case of Stat1 SH2 domain, binding affinities seem to be slightly overestimated. For instance, the native peptide pYDKP binds with Stat1 SH2 domain with an estimated binding affinity of −8.7 kcal/mol [17], while the computed binding affinities for peptide pYVNV and pYMDM are both around −10 kcal/mol. Nevertheless, the calculation suggests that the Stat1 SH2 domain may have high binding affinity for the pYVNV or pYMDM motif.
TABLE IV.
Experimental binding affinity and specificity for the five selected SH2 domains.
| SH2 domain | Experimental binding affinity (kcal/mol) a | Reported consensus motif b | |
|---|---|---|---|
| Lck | AcpYEEI | −9.4 | pY(E/T/Q)(E/D)(I/V/M) |
| AcpYEEIP | −9.5 | ||
| EPQpYEEIPIA | −9.7 | ||
|
| |||
| Grb2 | PSpYVNVQN | −10.6 | pY(Q/Y/V)(N)(Y/Q/F) |
| KPFpYVNV | −9.4 | pY(I/V)(N)(I/L/V) | |
| EEEPQpYEEIPIYL | > −5.9 | ||
|
| |||
| Cbl | GRARAVENQpYSFY | −10.07 | (N)(X)pY(S/T)(X)(X)(P) |
| EDSFLQPpYSSDPT | −8.82 | ||
|
| |||
| p85αN | GESDGGpYMDMSK | −7.1 | pY(M/I/V/E)(X)(M) |
| DGGpYMDMSKDE | −8.2 | ||
| TNEpYMDMKPGV | −8.3 | ||
|
| |||
| Stat1 | PTSFGpYDKPHVL | −8.7 | pY(D/E)(P/R)(R/P/Q) |
| TKASIpYHRPYHR | −5.9 | ||
| VDYEYpYERQHDY | −6.8 | ||
The binding affinities for SH2 domain-phosphotyrosine peptide interactions are reported to be moderate, ranging from 10−5 M to 10−8 M, or −7 to −11 kcal/mol [45]. The difference in affinity between a specific and non-specific interaction is suggested to be relatively small, amounting to less than three orders of magnitude [46, 47]. However, the calculations revealed some binding affinities that are lower than previously reported. One possible explanation of this inconsistency is that experimental characterization of protein-ligand interactions often requires certain degree of binding between the two species of interest. Each kind of experimental technique has its own ideal range of measurement and may not be sensitive to low affinity interactions. For example, the ideal range of Kd values for study by isothermal titration calorimetry (ITC) is 100 μM to 10 nM [45]. In addition, low affinity pairs may not be soluble or stable enough to form complexes in measurable amount. For example, low affinity ligands may be undetected using X-ray crystallography or NMR. In contrast, the binding free energy calculations method is able to assess and quantify all interactions, including those with low affinities that are beyond the range of experimental measurement.
Binding results from a combination of both favorable and unfavorable interactions between SH2 domain and phosphotyrosine peptides. The phosphotyrosine part of the peptide always favors binding with SH2 domain, while residues in the proximate position of pY may stabilize or destabilize binding, depending on which residues are present and which residues comprise the SH2 domain binding site. Assuming that the contribution of individual amino acid are additive, the contribution of pY to total binding free energy amounts to nearly 50% (Table 3) and the remaining three residues C-terminal to pY contribute additional 50% in those high affinity interactions. However, if the three residues C-terminal to pY are completely unfavorable for binding, the net result would be a much lower binding affinity, as shown for some of the SH2-phosphopeptide pairs in present study.
Although the difference between specific and nonspecific interactions is much greater than previously reported, SH2 domains also appear to be promiscuous: multiple phosphopeptide motifs can bind the same SH2 domain and one common phosphopeptide motif can bind several different SH2 domains. For example, the Cbl SH2 domain binds pYTPE, pYMDM, pYVNV with affinities −9.2, −8.3, −7.9 kcal/mol (Table 3), respectively. The phosphopeptide pYVNV can bind four SH2 domains all with relatively high affinities. In fact, previous X-ray crystallography and NMR structural studies also indicate that there exist seven SH2 domains that bind multiple peptides and six common motifs shared by more than one SH2 domains (supporting information).
To reconcile these two seemingly contradictory points of views, it is necessary to make a distinction between “intrinsic” and “effective” specificity of SH2 domain. Here intrinsic specificity refers to the absolute specificity of an isolated SH2 domain that is assumed to function independently, and the effective specificity refers to the specificity exhibited by SH2 domain-containing proteins in vivo. For example, the observation that two different SH2 domain-containing proteins binding to distinct phosphotyrosine sites on the platelet-derived growth factor (PDGF) receptor [48]. Given a particular SH2 domain, whether a phosphopeptide binds the SH2 domain or not mostly depends on the sequence and structure of the peptide. If only the three residues immediately following pY are counted, the tetrapeptide pYXXX has a total of 8000 different combinations. When an isolated SH2 domain exist in the ensemble of all possible peptides, it is very likely that the SH2 domain can bind several peptides with high affinity, given that the major part of binding comes from the pY and some amino acids have very similar structure. Therefore, the intrinsic specificity of SH2 domain, as characterized by peptide library screening [8, 9] or the computational method presented here, is not expected to be very high.
Effective specificity, on the other hand, depends on many factors other than the sequence and structure of the phosphopeptide. First, the spatial and temporal distribution of the interacting species plays an important role in the effective binding specificity [4, 49, 50]. The local concentration of SH2 domains and their binding partners at certain subcellular locations would largely pre-determine whether they interact or not at the time signaling is triggered. Moreover, SH2 domain seldom exists as an isolated single domain in vivo. More often than not, it is part of a big protein that contains multiple domains. The cooperative energetics of multi-domain binding and additional localization of SH2 domains will contribute to a higher effective specificity [51, 52]. Also, the assembling of multi-protein signaling complex can further enhance the specificity by regulating the orientation and position of interacting species [53, 54]. Lastly, it should be pointed out that the effective specificity suggests the interaction is strict. But it does not necessarily imply mutual exclusivity.
While approximately 120 different SH2 domains are encoded in the human genome [7], only 40 of them have structures available in PDB. Based on the analysis of the available SH2 domain structures in PDB, a small database containing 43 phosphopeptide motifs known to bind SH2 domain was built (Table 3 of supporting information). This database will allow a more systematical study on SH2 domain specificity using the method presented here. It should be pointed out that the method itself is of general applicability and can be used to elucidate the interactions mediated by any modular interactions domains whose structures are available.
A number of aspects regarding the present computational approach remain to be explored. In particular, the binding free energy calculation methodology used here assumes that the conformational changes in the SH2 domain induced by peptide binding are relatively modest and can be sampled directly by brute force MD. However, although the conformational changes are generally small, as shown by aligning the SH2 domain apo and holo crystal structures, the possibility of larger deformation in some cases may not be excluded a priori. This could be one of the reasons why the computed binding free energies for Stat1 SH2 domain are systematically greater than the experimental data. While the usage of a tetrapeptide in present study simpliffies the modeling and simulations, it may also fall short of modeling accurately the complex binding interactions because peptide residues N-terminal to pY or beyond pY+3 position may further stabilize binding or disfavor binding for some SH2 domains. Furthermore, the extent to which a short peptide composed of 4 to 10 residues adopts the same conformation as it exists as part of a protein in vivo is unclear. Thus, interactions between short phosphopeptides and SH2 domain may not realistically represent the SH2 domain mediated protein-protein interactions. Nevertheless, SH2 domain-phosphopeptide still comprises a good model system to study SH2 domain mediated protein-protein interactions, considering that their binding surface area is relatively small and residues far away from pY contribute little to binding.
IV. CONCLUSION
An efficient PMF free energy simulation strategy with restraining potentials was employed to study the specificity of phosphotyrosine peptides-SH2 domains interactions. The binding free energy calculations successfully reproduced the specific binding motifs for most of the selected SH2 domains. The results suggest that the magnitude of SH2 domain-phosphopeptide interactions may have greater range than previously reported. In addition, the interactions between a single isolated SH2 domain and isolated phosphotyrosine peptides are not so specific to be mutually exclusive. Some of the SH2 domains, if not all, are able to simultaneously bind multiple peptides with equally high affinity, provided all those peptides are present with the same concentration. Also, several peptide motifs are shared by multiple SH2 domains. The present results support the notion that the intrinsic specificity of modular domain mediated interaction alone is not suficient to achieve the high specificity in cell signaling. Other factors such as localization are likely to play an important role.
Specific protein-protein interactions play key roles in almost every cellular process. Even though proteomic or micro-array techniques can screen large amount of protein-protein interactions in a high-throughput manner, the molecular details of the interactions are often missing. Therefore, structure-based computational methods that are able to characterize protein-protein interactions and identify new interacting partners at the molecular level are of great interest. For experimental and computational techniques that have been developed to elucidate protein-protein interactions, see a recent review by Shoemaker et al [55, 56]. As more structures of modular domains are increasingly being solved, further studies on specific modular domain interactions will help understand the origin of cell signaling specificity.
Supplementary Material
Acknowledgments
We are grateful to Jose Faraldo-Gomez, Yuqing Deng and Richard Jones for helpful discussions. Part of this work was carried out on the Tungsten cluster of the National Center for Supercomputing Applications (NCSA). This work was supported by grant CA093577 from the NIH.
References
- 1.Cohen GB, Ren R, Baltimore D. Modular binding domains in signal transduction proteins. Cell. 1995;80:237–48. doi: 10.1016/0092-8674(95)90406-9. [DOI] [PubMed] [Google Scholar]
- 2.Pawson T. Protein modules and signalling networks. Nature. 1995;373:573–80. doi: 10.1038/373573a0. [DOI] [PubMed] [Google Scholar]
- 3.Pawson T, Nash P. Protein-protein interactions define specificity in signal transduction. Genes Dev. 2000;14:1027–47. [PubMed] [Google Scholar]
- 4.Pawson T. Specificity in signal transduction: from phosphotyrosine-sh2 domain interactions to complex cellular systems. Cell. 2004;116:191–203. doi: 10.1016/s0092-8674(03)01077-8. [DOI] [PubMed] [Google Scholar]
- 5.Schlessinger J, Lemmon MA. Sh2 and ptb domains in tyrosine kinase signaling. Sci STKE. 2003;RE12:2003. doi: 10.1126/stke.2003.191.re12. [DOI] [PubMed] [Google Scholar]
- 6.Pawson T, Nash P. Assembly of cell regulatory systems through protein interaction domains. Science. 2003;300:445–52. doi: 10.1126/science.1083653. [DOI] [PubMed] [Google Scholar]
- 7.Liu BA, Jablonowski K, Raina M, Arce M, Pawson T, Nash PD. The human and mouse complement of sh2 domain proteins-establishing the boundaries of phosphotyrosine signaling. Mol Cell. 2006;22:851–68. doi: 10.1016/j.molcel.2006.06.001. [DOI] [PubMed] [Google Scholar]
- 8.Songyang Z, Shoelson SE, Chaudhuri M, Gish G, Pawson T, Haser WG, King F, Roberts T, Ratnofsky S, Lechleider RJ, et al. Cell. 1993;72:767–78. doi: 10.1016/0092-8674(93)90404-e. [DOI] [PubMed] [Google Scholar]
- 9.Songyang Z, Shoelson SE, McGlade J, Olivier P, Pawson T, Bustelo XR, Barbacid M, Sabe H, Hanafusa H, Yi T, et al. Specific motifs recognized by the sh2 domains of csk, 3bp2, fps/fes, grb-2, hcp, shc, syk, and vav. Mol Cell Biol. 1994;14:2777–85. doi: 10.1128/mcb.14.4.2777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Waksman G, Shoelson SE, Pant N, Cowburn D, Kuriyan J. Binding of a high affinity phosphotyrosyl peptide to the src sh2 domain: crystal structures of the complexed and peptide-free forms. Cell. 1993;72:779–90. doi: 10.1016/0092-8674(93)90405-f. [DOI] [PubMed] [Google Scholar]
- 11.Nolte RT, Eck MJ, Schlessinger J, Shoelson SE, Harrison SC. Crystal structure of the pi 3-kinase p85 amino-terminal sh2 domain and its phosphopeptide complexes. Nat Struct Biol. 1996;3:364–74. doi: 10.1038/nsb0496-364. [DOI] [PubMed] [Google Scholar]
- 12.Tong L, Warren TC, King J, Betageri R, Rose J, Jakes S. Crystal structures of the human p56lck sh2 domain in complex with two short phosphotyrosyl peptides at 1.0 a and 1.8 a resolution. J Mol Biol. 1996;256:601–10. doi: 10.1006/jmbi.1996.0112. [DOI] [PubMed] [Google Scholar]
- 13.Meng W, Sawasdikosol S, Burakoff SJ, Eck MJ. Structure of the amino-terminal domain of cbl complexed to its binding site on zap-70 kinase. Nature. 1999;398:84–90. doi: 10.1038/18050. [DOI] [PubMed] [Google Scholar]
- 14.Nioche P, Liu WQ, Broutin I, Charbonnier F, Latreille MT, Vidal M, Roques B, Garbay C, Ducruix A. Crystal structures of the sh2 domain of grb2: highlight on the binding of a new high-affinity inhibitor. J Mol Biol. 2002;315:1167–77. doi: 10.1006/jmbi.2001.5299. [DOI] [PubMed] [Google Scholar]
- 15.Mao X, Ren Z, Parker GN, Sondermann H, Pastorello MA, Wang W, McMurray JS, Demeler B, Darnell JEJ, Chen X. Structural bases of unphosphorylated stat1 association and receptor binding. Mol Cell. 2005;17:761–71. doi: 10.1016/j.molcel.2005.02.021. [DOI] [PubMed] [Google Scholar]
- 16.Dente L, Vetriani C, Zucconi A, Pelicci G, Lanfrancone L, Pelicci PG, Cesareni G. Modified phage peptide libraries as a tool to study specificity of phosphorylation and recognition of tyrosine containing peptides. J Mol Biol. 1997;269:694–703. doi: 10.1006/jmbi.1997.1073. [DOI] [PubMed] [Google Scholar]
- 17.Wiederkehr-Adam M, Ernst P, Muller K, Bieck E, Gombert FO, Ottl J, Graff P, Gross-muller F, Heim MH. Characterization of phosphopeptide motifs specific for the src homology 2 domains of signal transducer and activator of transcription 1 (stat1) and stat3. J Biol Chem. 2003;278:16117–28. doi: 10.1074/jbc.M300261200. [DOI] [PubMed] [Google Scholar]
- 18.Rodriguez M, Li SS, Harper JW, Songyang Z. An oriented peptide array library (opal) strategy to study protein-protein interactions. J Biol Chem. 2004;279:8802–7. doi: 10.1074/jbc.M311886200. [DOI] [PubMed] [Google Scholar]
- 19.Sweeney MC, Wavreille AS, Park J, Butchar JP, Tridandapani S, Pei D. Decoding protein-protein interactions through combinatorial chemistry: sequence specificity of shp-1, shp-2, and ship sh2 domains. Biochemistry. 2005;44:14932–47. doi: 10.1021/bi051408h. [DOI] [PubMed] [Google Scholar]
- 20.Jones RB, Gordus A, Krall JA, MacBeath G. A quantitative protein interaction network for the erbb receptors using protein microarrays. Nature. 2006;439:168–74. doi: 10.1038/nature04177. [DOI] [PubMed] [Google Scholar]
- 21.Campbell SJ, Jackson RM. Diversity in the sh2 domain family phosphotyrosyl peptide binding site. Protein Eng. 2003;16:217–27. doi: 10.1093/proeng/gzg025. [DOI] [PubMed] [Google Scholar]
- 22.Sheinerman FB, Al-Lazikani B, Honig B. Sequence, structure and energetic determinants of phosphopeptide selectivity of sh2 domains. J Mol Biol. 2003;334:823–41. doi: 10.1016/j.jmb.2003.09.075. [DOI] [PubMed] [Google Scholar]
- 23.Roux B, Nina M, Pomes R, Smith JC. Thermodynamic stability of water molecules in the bacteriorhodopsin proton channel: a molecular dynamics free energy perturbation study. Biophys J. 1996;71:670–81. doi: 10.1016/S0006-3495(96)79267-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gilson MK, Given JA, Bush BL, McCammon JA. The statistical-thermodynamic basis for computation of binding affinities: a critical review. Biophys J. 1997;72:1047–69. doi: 10.1016/S0006-3495(97)78756-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hermans J, Wang L. Inclusion of loss of translational and rotational freedom in theoretical estimates of free energies of binding. application to a complex of benzene and mutant t4 lysozyme. J Am Chem Soc. 1997;119:2707–2714. [Google Scholar]
- 26.Boresch S, Tettinger F, Leitgeb M, Karplus M. Absolute binding free energies: A quantitative approach for their calculation. J Phys Chem B. 2003;107:9535–9551. [Google Scholar]
- 27.Woo HJ, Roux B. Calculation of absolute protein-ligand binding free energy from computer simulations. Proc Natl Acad Sci U S A. 2005;102:6825–30. doi: 10.1073/pnas.0409005102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shindyalov IN, Bourne PE. Protein structure alignment by incremental combinatorial extension (ce) of the optimal path. Protein Eng. 1998;11:739–47. doi: 10.1093/protein/11.9.739. [DOI] [PubMed] [Google Scholar]
- 29.Deng Y, Roux B. Calculation of standard binding free energies: Aromatic molecules in the t4 lysozyme l99a mutant. J Chem Theory Comput. 2006;2:1255–73. doi: 10.1021/ct060037v. [DOI] [PubMed] [Google Scholar]
- 30.Wang J, Deng Y, Roux B. Absolute binding free energy calculations using molecular dynamics simulations with restraining potentials. Biophys J. 2006;91:2798–814. doi: 10.1529/biophysj.106.084301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kollman P. Free energy calculations: Applications to chemical and biochemical phenomena. Chem Rev. 1993;93:2395–2417. [Google Scholar]
- 32.Torrie GM, Valleau JP. Nonphysical sampling distributions in monte carlo free-energy estimation: Umbrella sampling. J Comp Phys. 1977;23:187–199. [Google Scholar]
- 33.Kumar S, Bouzida D, Swendsen R, Kollman P, Rosenberg J. The weighted histogram analysis method for free-energy calculations on biomolecules. i. the method. J Comp Chem. 1992;13:1011–1021. [Google Scholar]
- 34.Brooks B, Bruccoleri R, Olafson B, States D, Swaminathan S, Karplus M. Charmm: A program for macromolecular energy minimization and dynamics calculations. J Comput Chem. 1983;4:187–217. [Google Scholar]
- 35.Im W, Lee MS, Brooks CL. Generalized born model with a simple smoothing function. J Comput Chem. 2003;24:1691–702. doi: 10.1002/jcc.10321. [DOI] [PubMed] [Google Scholar]
- 36.MacKerell AJ, Bashford D, Bellot M, Dunbrack R, Evanseck J, Field M, Fischer S, Gao J, Guo H, Ha DJMS, Kuchnir L, Kuczera K, Lau F, Mattos C, Michnick S, Ngo T, Nguyen D, Prodhom B, Reiher W, III, Roux B, Schlenkrich M, Smith J, Stote R, Straub J, Watanabe M, Wiorkiewicz-Kuczera J, Karplus M. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J Phys Chems B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 37.Groesch TD, Zhou F, Mattila S, Geahlen RL, Post CB. Structural basis for the requirement of two phosphotyrosine residues in signaling mediated by syk tyrosine kinase. J Mol Biol. 2006;356:1222–36. doi: 10.1016/j.jmb.2005.11.095. [DOI] [PubMed] [Google Scholar]
- 38.Weber T, Schaffhausen B, Liu Y, Gunther UL. Nmr structure of the n-sh2 of the p85 subunit of phosphoinositide 3-kinase complexed to a doubly phosphorylated peptide reveals a second phosphotyrosine binding site. Biochemistry. 2000;39:15860–9. doi: 10.1021/bi001474d. [DOI] [PubMed] [Google Scholar]
- 39.Bradshaw JM, Mitaxov V, Waksman G. Investigation of phosphotyrosine recognition by the sh2 domain of the src kinase. J Mol Biol. 1999;293:971–85. doi: 10.1006/jmbi.1999.3190. [DOI] [PubMed] [Google Scholar]
- 40.Payne G, Shoelson SE, Gish GD, Pawson T, Walsh CT. Kinetics of p56lck and p60src src homology 2 domain binding to tyrosine-phosphorylated peptides determined by a competition assay or surface plasmon resonance. Proc Natl Acad Sci U S A. 1993;90:4902–6. doi: 10.1073/pnas.90.11.4902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rahuel J, Gay B, Erdmann D, Strauss A, Garcia-Echeverria C, Furet P, Caravatti G, Fretz H, Schoepfer J, Grutter MG. Structural basis for specificity of grb2-sh2 revealed by a novel ligand binding mode. Nat Struct Biol. 1996;3:586–9. doi: 10.1038/nsb0796-586. [DOI] [PubMed] [Google Scholar]
- 42.Lupher MLJ, Songyang Z, Shoelson SE, Cantley LC, Band H. The cbl phosphotyrosine-binding domain selects a d(n/d)xpy motif and binds to the tyr292 negative regulatory phosphorylation site of zap-70. J Biol Chem. 1997;272:33140–4. doi: 10.1074/jbc.272.52.33140. [DOI] [PubMed] [Google Scholar]
- 43.Hu J, Hubbard SR. Structural characterization of a novel cbl phosphotyrosine recognition motif in the aps family of adapter proteins. J Biol Chem. 2005;280:18943–9. doi: 10.1074/jbc.M414157200. [DOI] [PubMed] [Google Scholar]
- 44.Piccione E, Case RD, Domchek SM, Hu P, Chaudhuri M, Backer JM, Schlessinger J, Shoelson SE. Phosphatidylinositol 3-kinase p85 sh2 domain specificity defined by direct phosphopeptide/sh2 domain binding. Biochemistry. 1993;32:3197–202. doi: 10.1021/bi00064a001. [DOI] [PubMed] [Google Scholar]
- 45.Ladbury JE, Lemmon MA, Zhou M, Green J, Botfield MC, Schlessinger J. Measurement of the binding of tyrosyl phosphopeptides to sh2 domains: a reappraisal. Proc Natl Acad Sci U S A. 1995;92:3199–203. doi: 10.1073/pnas.92.8.3199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ladbury JE, Arold S. Searching for specificity in sh domains. Chem Biol. 2000;7:R3–8. doi: 10.1016/s1074-5521(00)00067-3. [DOI] [PubMed] [Google Scholar]
- 47.O’Rourke L, Ladbury JE. Specificity is complex and time consuming: mutual exclusivity in tyrosine kinase-mediated signaling. Acc Chem Res. 2003;36:410–6. doi: 10.1021/ar020167s. [DOI] [PubMed] [Google Scholar]
- 48.Fantl WJ, Escobedo JA, Martin GA, Turck CW, del Rosario M, McCormick F, Williams LT. Distinct phosphotyrosines on a growth factor receptor bind to specific molecules that mediate different signaling pathways. Cell. 1992;69:413–23. doi: 10.1016/0092-8674(92)90444-h. [DOI] [PubMed] [Google Scholar]
- 49.Saltiel AR, Pessin JE. Insulin signaling pathways in time and space. Trends Cell Biol. 2002;12:65–71. doi: 10.1016/s0962-8924(01)02207-3. [DOI] [PubMed] [Google Scholar]
- 50.Kholodenko BN. Cell-signalling dynamics in time and space. Nat Rev Mol Cell Biol. 2006;7:165–76. doi: 10.1038/nrm1838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ottinger EA, Botfield MC, Shoelson SE. Tandem sh2 domains confer high specificity in tyrosine kinase signaling. J Biol Chem. 1998;273:729–35. doi: 10.1074/jbc.273.2.729. [DOI] [PubMed] [Google Scholar]
- 52.Bar-Sagi D, Rotin D, Batzer A, Mandiyan V, Schlessinger J. Sh3 domains direct cellular localization of signaling molecules. Cell. 1993;74:83–91. doi: 10.1016/0092-8674(93)90296-3. [DOI] [PubMed] [Google Scholar]
- 53.Smith FD, Scott JD. Signaling complexes: junctions on the intracellular information super highway. Curr Biol. 2002;12:R32–40. doi: 10.1016/s0960-9822(01)00646-7. [DOI] [PubMed] [Google Scholar]
- 54.Cho W. Building signaling complexes at the membrane. Sci STKE. 2006;pe7:2006. doi: 10.1126/stke.3212006pe7. [DOI] [PubMed] [Google Scholar]
- 55.Shoemaker BA, Panchenko AR. Deciphering protein-protein interactions. part i. Experimental techniques and databases. PLoS Comput Biol. 2007;3:e42. doi: 10.1371/journal.pcbi.0030042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shoemaker BA, Panchenko AR. Deciphering protein-protein interactions. part ii. computational methods to predict protein and domain interaction partners. PLoS Comput Biol. 2007;3:e43. doi: 10.1371/journal.pcbi.0030043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cousins-Wasti RC, Ingraham RH, Morelock MM, Grygon CA. Determination of affinities for lck sh2 binding peptides using a sensitive fluorescence assay: comparison between the pyeeip and pyqpqp consensus sequences reveals context-dependent binding specificity. Biochemistry. 1996;35:16746–52. doi: 10.1021/bi9620868. [DOI] [PubMed] [Google Scholar]
- 58.Tong L, Warren TC, Lukas S, Schembri-King J, Betageri R, Proudfoot JR, Jakes S. Carboxymethyl-phenylalanine as a replacement for phosphotyrosine in sh2 domain binding. J Biol Chem. 1998;273:20238–42. doi: 10.1074/jbc.273.32.20238. [DOI] [PubMed] [Google Scholar]
- 59.Panayotou G, Gish G, End P, Truong O, Gout I, Dhand R, Fry MJ, Hiles I, Pawson T, Waterfield MD. Interactions between sh2 domains and tyrosine-phosphorylated platelet-derived growth factor beta-receptor sequences: analysis of kinetic parameters by a novel biosensor-based approach. Mol Cell Biol. 1993;13:3567–76. doi: 10.1128/mcb.13.6.3567. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.