

Abstract
Escherichia coli selenophosphate synthetase (SPS, the selD gene product) catalyzes the production of monoselenophosphate, the selenium donor compound required for synthesis of selenocysteine (Sec) and seleno-tRNAs. We report the molecular cloning of human and mouse homologs of the selD gene, designated Sps2, which contains an in-frame TGA codon at a site corresponding to the enzyme’s putative active site. These sequences allow the identification of selD gene homologs in the genomes of the bacterium Haemophilus influenzae and the archaeon Methanococcus jannaschii, which had been previously misinterpreted due to their in-frame TGA codon. Sps2 mRNA levels are elevated in organs previously implicated in the synthesis of selenoproteins and in active sites of blood cell development. In addition, we show that Sps2 mRNA is up-regulated upon activation of T lymphocytes and have mapped the Sps2 gene to mouse chromosome 7. Using the mouse gene isolated from the hematopoietic cell line FDCPmixA4, we devised a construct for protein expression that results in the insertion of a FLAG tag sequence at the N terminus of the SPS2 protein. This strategy allowed us to document the readthrough of the in-frame TGA codon and the incorporation of 75Se into SPS2. These results suggest the existence of an autoregulatory mechanism involving the incorporation of Sec into SPS2 that might be relevant to blood cell biology. This mechanism is likely to have been present in ancient life forms and conserved in a variety of living organisms from all domains of life.
It is now well established that the UGA (TGA) codon directs the incorporation of the amino acid selenocysteine (Sec) into proteins (1). The synthesis of Sec and its incorporation into proteins (1, 2), here referred to as selenoproteins (3), requires the activity of a number of genes first described in bacteria (4, 5). Selenoprotein-encoding cDNAs present stem-loop secondary structures, termed SECIS elements, which are required for the incorporation of Sec at UGA codons (4–6); these mRNA motifs have been shown in bacteria to engage a specific elongation factor (7) that in turn binds selenocystyl-bearing tRNAs complementary to the UGA codon (8).
The synthesis of Sec in bacteria usually requires the initial production of monoselenophosphate (MSP) from selenide and ATP, a reaction catalyzed by selenophosphate synthetase (SPS), the selD gene product (1, 9). MSP (10) is a highly reactive selenium donor compound used to synthesize Sec from seryl-tRNAs and to modify 2-thiouridine tRNAs (11). We have identified novel selD homologs in human, mouse, Haemophilus influenzae, and Methanococcus jannaschii genomes which feature a Sec codon corresponding in position to the enzyme’s predicted active site, differing from E. coli selD (12) and another human selD homolog described by Low et al. (13); we will from here on refer to this latter gene as Sps1 and to the novel human and mouse selD homologs described here as Sps2.
Here we demonstrate the readthrough of a UGA codon and concomitant incorporation of 75Se into mouse SPS2 protein, providing evidence for the existence of an autoregulatory mechanism.
MATERIALS AND METHODS
Molecular Cloning of Mouse and Human Sps2.
For the isolation of mouse Sps2 a cDNA library was constructed and screened as described (14, 15), using as probe the differential display product Clone 1000 (14, 15).
A full-length human Sps2 cDNA was isolated from a cDNA library derived from activated CD8+ T cells (16) by standard methods (17), using a cDNA complementary to Sps2 as a probe. The Sps2-specific cDNA was generated by PCR using CD8+ T-cell cDNA (16) as a template and primers designed from an expressed sequence tag (GenBank accession no. T09327T09327) that represents a 426-bp fragment of the human Sps2 homolog: sense primer, 5′-GTAAAGATGGTGGTCTCCAG-3′; antisense primer, 5′-CTCTCCCGTACTTGGAGGATTTG-3′.
Protein Alignment, Protein Phylogeny, and mRNA Folding Analysis.
The multiple protein sequence alignment was carried out by clustal w (18) and then manually refined by superposition of equivalent structural elements predicted by the neural network program phd (19). The phylogenetic tree was calculated by the neighbor-joining method (18) applied to a matrix of difference “distances” derived from the structurally accurate SPS alignments; the reliability of the branching pattern was further tested by performing 5000 bootstrap replications. The folding analysis of the Sps2 mouse and human mRNAs was carried out with the mfold program (20), using other available selenoprotein-encoding nucleotide sequences as positive controls.
Northern Blot Analysis.
Total RNA was obtained from fetal and adult tissues and processed as described (15). Blots were hybridized with the partial cDNA Clone 1000 (see above), which represents the 400 nucleotides at the 3′ end of the entire cDNA Clone 1000-B2, washed and exposed to film as described (15). CD4+ CD8− T lymphocytes derived from peripheral lymph nodes of 8-week-old female BALB/c mice (The Jackson Laboratories) were sorted by flow cytometry and plated at 106 cells per ml in 24-well plates (Becton Dickinson) coated with anti-CD3 (PharMingen) at 10 μg/ml. Cells were harvested after 16 hr of anti-CD3 stimulation. 3T3 ΔB-Raf:ER fibroblasts (21) were made quiescent by overnight incubation in Dulbecco’s modified Eagle’s medium containing 0.5% fetal calf serum (Sigma). Cells were then either untreated or stimulated with 20% (vol/vol) fetal calf serum and incubated for 1 hr at 37°C. The macrophage cell line IG18LA (22) was cultured in RPMI medium 1640 (JRH Biosciences, Lenexa, KS) supplemented with 10% fetal calf serum and stimulated with 10 μg/ml of lipopolysaccharide (E. coli O111:B4; Sigma) for 6 hr. The study of the species distribution was performed by probing a Zooblot (Stratagene)—representing genomic DNA from different species—as described above for Northern analysis with an ≈180-bp DNA sequence obtained from the 5′ untranslated region of mouse Sps2 by digestion of clone 1000 B2 with NarI (Boehringer Mannheim).
Chromosomal Mapping of Mouse Sps2.
Interspecific backcross progeny were generated by mating C57BL/6J × Mus spretus)F1 females and C57BL/6J males as described (23). A total of 205 N2 mice were used to map the Sps2 locus (see Results for details). DNA isolation, restriction enzyme digestion, agarose gel electrophoresis, Southern blot transfer, and hybridization were performed essentially as described (17). All blots were prepared with Hybond-N+ nylon membrane (Amersham). The probe, an ≈180-bp fragment of Clone 1000 B2, corresponding to the 5′ untranslated region of Sps2 obtained by digestion with NarI, was labeled with [α-32P]dCTP using a random priming labeling kit (Stratagene); washing was done to a final stringency of 0.1× standard saline citrate phosphate (SSCP), 0.1% SDS, 65°C. A 6.3-kb fragment was detected in HincII-digested C57BL/6J DNA, and a 7.0-kb fragment M. spretus-specific fragment was followed in the backcross mice.
A description of the probes and restriction fragment length polymorphisms (RFLPs) for the loci linked to Sps2, including interleukin-4 receptor α gene (Il4ra), sialophorin (Spn), fibroblast growth factor 2 (Fgfr2), and O6-methylguanine DNA methyltransferase (Mgmt), has been published previously (24). Recombination distances were calculated as described (25), using the computer program spretus madness. Gene order was determined by minimizing the number of recombination events required to explain the allele distribution patterns.
Generation of Sps2 Constructs for Protein Expression in COS-7 Cells.
Sps2 constructs were generated from the mouse cDNA Clone 1000-B2 by PCR mutagenesis and entirely sequenced as described (15) to introduce a FLAG peptide sequence (IBI) (26) at the N terminus of the SPS2 protein and inserted into the pME18X vector (DNAX) (15): sense primer, 5′-ACTTCTCGAGGCACCATGGACTACAAGGACGACGATGACAAGGCGGAAGCGGCGGCGGCGGGC-3′; antisense primer, 5′-CTAGGTCTAGATTCAAGAACTAGGCTCAGAGGCTGC-3′. PFU enzyme (Stratagene) was used with 22 cycles of PCR as described (15). The Sps2 constructs were inserted into the pME18X vector, using XhoI and XbaI sites incorporated into the 5′ and 3′ primers, respectively.
Protein Expression of Sps2 in COS-7 Cells.
COS-7 cells were maintained and transfected as described (15). After transfection, cells grew in media with 10% serum (15) containing 75Se (as H275SeO3; University of Missouri Research Reactor Facility) at 0.1 mCi/liter (1 mCi = 37 MBq). Northern blotting was performed on transfected COS-7 cells collected at the moment of harvesting for protein studies to assess the efficiency of transfection, as described below but using the full-length clone 1000-B2 cDNA. pME18X vector without insert was used as control. Cells were lysed as described (15). Cell lysates were immunopurified by using anti-FLAG M2 affinity gel (IBI), eluted, precipitated, electrophoresed, transferred, and immunoprobed with anti-FLAG M2 antibody (IBI), all procedures as described (15). Polyvinylidene difluoride (PVDF) membranes containing electrotransferred 75Se-labeled immunoprecipitates were exposed to film (Kodak).
RESULTS
Identification of a Novel Homolog of the E. coli selD Gene.
In the course of identifying genes that are preferentially expressed in active centers of embryonic hematopoiesis (14, 15), we isolated a cDNA, designated 1000-B2, that encodes a protein with significant similarity to the E. coli selD gene product (12). The sequence of the mouse cDNA, as well as its human counterpart, unexpectedly revealed an in-frame TGA codon that aligned precisely with the position of a cysteine residue critical for the activity of the E. coli SPS (27) (Cys-17; Fig. 1 a and b), suggesting that the mammalian enzymes could contain Sec in their putative active sites. Two bacterial selD homologs that contain a TGA at the same relative position were found in the genomes of both H. influenzae and M. jannaschii by sequence data base comparisons. These novel counterparts of the E. coli selD gene were, in both cases, misinterpreted [H. influenzae genome locus HI0200 (28) and M. jannaschii genome locus MJ1591 (29)] due to their in-frame TGA (stop) codons. Different from the situation in humans, these two genes are the sole selD homologs (HiSPS and MjSPS) for H. influenzae and M. jannaschii. It is not possible at this time to exclude the existence of a (second) SPS equivalent in E. coli, though this appears unlikely (30); a definitive answer awaits the complete sequencing of the E. coli genome. On the other hand, a rigorous screen of the entire genome sequences of the yeast Saccharomyces cerevisiae and the bacterium Mycoplasma genitalium did not discern any selD equivalent (31, 32).
Figure 1.
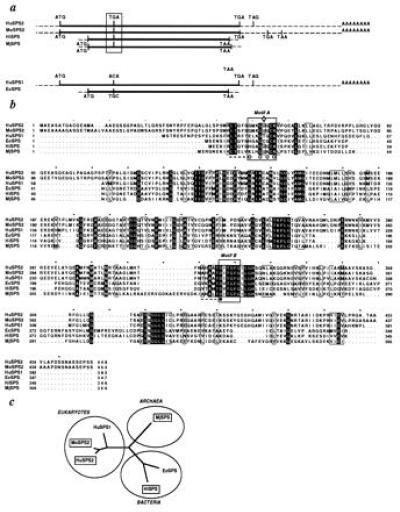
Identification of a novel selD homolog. (a) Schematic representation of the nucleotide sequences of mouse and human Sps2 (MoSPS2 and HuSPS2, respectively) shows the relative positions of the in-frame TGA codon as well as other TGA codons likely used as termination signals (see also Fig. 4). Thick lines denote the effective open reading frame (ORF). Human SPS1 (HuSPS1) and the E. coli SPS (EcSPS) sequences feature different codons opposite the Sps2 TGA. (b) Protein sequence alignment of the available SPS homologs. Reverse-lettered residues are identical among the six sequences; boxed amino acids are chemically conserved; dots are gaps in the alignment. Large boxes highlight the Walker A- and B-like motifs characteristic of nucleotide-binding folds; a Gly-rich active site pattern (where ⋄ is Sec—U in one-letter amino acid code (4)—Cys or Thr); Gly residues are marked by (○), and an invariant Asp (•) starts a predicted turn. Dashed lines represent predicted hydrophobic β-strands (19). (c) Dendrogram showing the divergence of SPS forms. Boxes around the SPS2, M. jannaschii, and H. influenzae molecules denote their special character as selenoenzymes. SPS1 and EcSPS have likely mutated this codon to Thr and Cys, respectively.
The Walker A- and B-like motifs (33) characteristic of α/β nucleotide-binding folds are evident in two well-conserved stretches of an alignment of the available SPS homologs depicted in Fig. 1b: a Gly-rich GX2G⋄GCK active site pattern (where ⋄ is Sec, Cys, or Thr) and an invariant Asp starting a predicted β/α turn are, respectively, reminiscent of the “P-loop” motif A and a conserved, acidic motif B (34); both motifs follow predicted (19) hydrophobic β-strands (see Fig. 1b). The detection of an ATP-binding sequence pattern in SPS enzymes is consistent with a known ATP hydrolysis requirement for selenophosphate formation from selenide and ATP (9). Other conserved amino acid regions in the global SPS alignment likely indicate additional candidate residues involved in binding and catalysis (34).
Sps2 mRNA Is Differentially Expressed During Development and in Adult Tissues.
Northern blot analysis shows that Sps2 is preferentially expressed in tissues where selenoproteins are produced—liver, kidney, and testis (35–37)—followed by sites of blood cell development—yolk sac, bone marrow, spleen, and thymus (Fig. 2A). As shown in Fig. 2B, Sps2 mRNA levels are up-regulated upon activation of CD4+ CD8− T lymphocytes. This seems to be specific for T cells, since neither macrophages nor fibroblasts showed induction of Sps2 upon activation.
Figure 2.
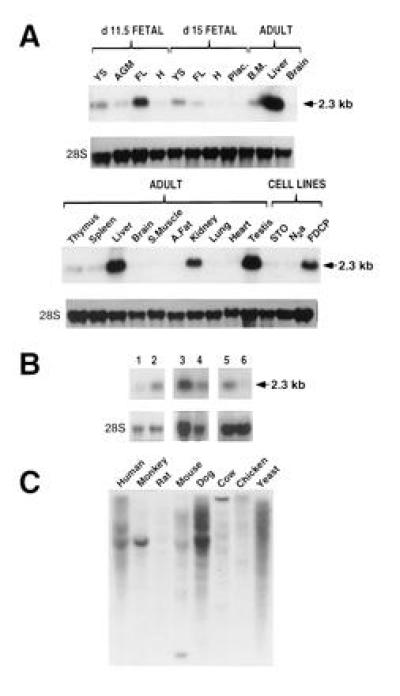
Northern blot and species distribution analyses of Sps2. (A) Sps2 is preferentially expressed in tissues where known selenoproteins are produced and in sites of blood cell development. YS, yolk sac; AGM, AGM region (see text); FL, fetal liver; H, head of the embryo; Plac, placenta; BM, bone marrow; S.Muscle, skeletal muscle; A.Fat, perivisceral abdominal fat; FDCP, hematopoietic FDCPmixA4 cell line; STO, fibroblast cell line STO (15); N2a, neuronal cell line N2a (15); 28S, ribosomal RNA. (B) Sps2 is a T-cell activation gene. Lanes: 1, unstimulated CD4+ CD8− T cells; 2, CD4+ CD8− T cells stimulated with anti-CD3; 3, unstimulated macrophages; 4, macrophages stimulated with lipopolysaccharide; 5, serum-deprived fibroblasts; 6, serum-stimulated fibroblasts. (C) SPS is highly conserved among a variety of animal organisms as shown by probing a Zooblot with the 5′ untranslated region (UTR) of mouse Sps2.
A study of the species conservation of the Sps2 message was performed by probing a genomic DNA blot with a 180-bp fragment representing the 5′ UTR of mouse Sps2. The result is shown in Fig. 2C, and it indicates a widespread distribution and well-conserved nucleotide sequence of Sps2 among mammals. We have used this portion of the Sps2 gene, as the ORFs of Sps1 and Sps2 show a significant degree of identity and due the observation that the 3′ UTRs of homologous genes can vary significantly between species.
Mapping of Mouse Sps2 to Chromosome 7.
To try to identify potential natural mutants of Sps2 we attempted to map the Sps2 gene. The mouse chromosomal location of Sps2 was determined by interspecific backcross analysis using progeny derived from matings of [(C57BL/6J × M. spretus)F1 × C57BL/6J] mice (see Materials and Methods). The 7.0-kb M. spretus fragment (see Materials and Methods) was used to follow the segregation of the Sps2 locus in backcross mice. The mapping results indicated that Sps2 is located in the distal region of mouse chromosome 7, linked to Il4ra, Fgfr2, Spn, and Mgmt. Although 146 mice were analyzed for every marker and are shown in the segregation analysis (Fig. 3), up to 188 mice were typed for some pairs of markers. Each locus was analyzed in pairwise combinations for recombination frequencies, using the additional data. The ratios of the total number of mice exhibiting recombinant chromosomes to the total number of mice analyzed for each pair of loci and the most likely gene order are centromere–Il4ra– 2/188 –Spn– 0/170 –Sps2– 0/171 –Fgfr2– 8/159 –Mgmt. The recombination frequencies [expressed as genetic distances in centimorgans (cM) ± the standard error] are –Il4ra– 1.1 ± 0.8 –[Spn, Sps2, Fgfr2]– 5.0 ± 1.7 –Mgmt. No recombinations were detected between Spn and Sps2 in 170 animals typed in common, suggesting that the two loci are within 1.7 cM of each other (upper 95% confidence limit). No recombinants were detected between Sps2 and Fgfr2 in 171 animals typed in common, suggesting the two loci are within 1.7 cM of each other (upper 95% confidence level).
Figure 3.
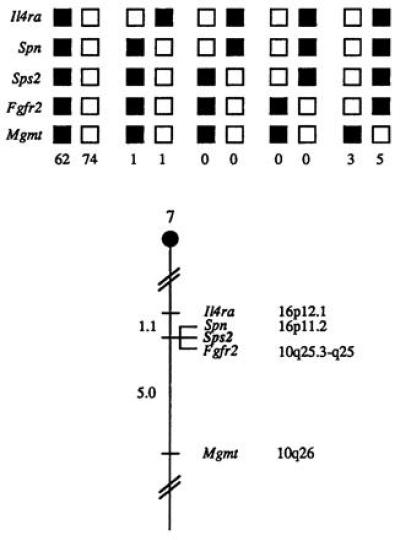
Sps2 maps in the distal region of mouse chromosome 7. Sps2 was placed on mouse chromosome 7 by interspecific backcross analysis. The segregation patterns of Sps2 and flanking genes in 146 backcross animals that were typed for all loci are shown at the top of the figure. Each column represents the chromosome identified in the backcross progeny that was inherited from the (C57BL/6J × M. spretus)F1 parent. The shaded boxes represent the presence of a C57BL/6J × M. spretus)F1 parent. The number of offspring inheriting each type of chromosome is listed at the bottom of each column. A partial chromosome 7 linkage map showing the location of Sps2 in relation to linked genes is shown at the bottom. Recombination distances between loci in centimorgans are shown to the left of the chromosome. The positions of loci cited in this study can be obtained from GDB (Genome Data Base, The Johns Hopkins University, Baltimore).
The central region of mouse chromosome 7 shares regions of homology with human chromosomes 16p and 10q (summarized in Fig. 3). In particular, Spn has been placed on human 16p11.2 and Fgfr2 has been placed on human 10q25.3–q26. The tight linkage between Spn, Fgfr2, and Sps2 in mouse suggests that Sps2 will reside on one of these chromosomes as well.
We have compared our interspecific map of chromosome 7 with a composite mouse linkage map that reports the map location of many uncloned mouse mutations (compiled by M. T. Davisson, T. H. Roderick, A. L. Hillyard, and D. P. Doolittle and provided from GBASE, a computerized data base maintained at The Jackson Laboratory, Bar Harbor, ME). Our lack of knowledge of the precise biological functions of this enzyme makes it difficult to predict the phenotype that might be expected for an alteration in its activity in the intact animal. Sps2 mapped in a region of the composite map that lacks mouse mutations with a phenotype that might be expected for an alteration in this locus (data not shown), but a careful screen of all these mutants, including minor mutations, must be investigated at the nucleotide sequence level.
Expression of the SPS2 Protein in COS-7 Cells.
To compare the expression levels of mouse Sps2 in the presence or absence of its 3′ UTR, we engineered constructs for expression in COS-7 cells which contained a FLAG peptide sequence to allow immunopurification and identification by Western blotting of the SPS2 protein. Anti-FLAG-immunopurified lysates from these cells (Fig. 4) show that the readthrough of the in-frame TGA codon of Sps2 can be achieved in this expression system in the absence of Sps2 3′ UTR elements. However, a construct containing the Sps2 3′ UTR shows that this segment increases by more than 20 times the production and the incorporation of 75Se into the SPS2 protein (no difference in mRNA stability was detected; data not shown), suggesting the existence of uncharacteristic SECIS elements, yet to be characterized, in the 3′ UTR of the Sps2 gene. In addition, this experiment shows that the protein product resulting from the expression of Sps2 migrates similarly regardless of the presence or absence of the region downstream of the second TGA codon, indicating that the codon located at nucleotide position 1494 is likely used for termination, as the readthrough of this codon would have resulted in the addition of 92 amino acid residues to SPS2, resulting in a predicted 66-kDa protein product. As shown in Fig. 4, the observed band is in agreement with the calculated Mr of 47,786 of mouse SPS2 protein up to the second TGA codon (the peptidesort software program from the GCG package was used to estimate the Mr, and a cysteine residue was used for approximation purposes in place of Sec). Accordingly, the similarity between the predicted protein sequence of mouse and human Sps2 drops from approximately 80% before the second TGA to an estimated 10% downstream of this codon (data not shown).
Figure 4.
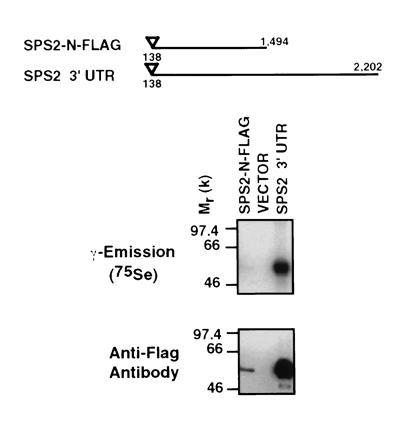
Readthrough of the in-frame TGA codon of mouse Sps2 in COS-7 cells and analysis of the contribution of the 3′ UTR to the degree of efficiency of translation. The inverted triangle represents the positioning of the FLAG sequence at the N terminus of the Sps2 cDNA constructs. Anti-FLAG-immunopurified lysates were analyzed by Western blotting using anti-FLAG M2 antibody. Mammalian cells were transfected with the pME18X expression vector without insert as control (VECTOR).
DISCUSSION
We have identified novel homologs of the SelD gene in humans, mice, H. influenzae, and M. jannaschii that exhibit a TGA codon within their ORFs. Using the mouse gene, we show that this codon is read through to form the nascent SPS2 protein; this has direct implications for the correct translation of the other homologs of SPS presented here.
Using a FLAG tag strategy, we were able to document the readthrough of the in-frame TGA codon of Sps2 and to detect 75Se incorporation into the immunopurified SPS2 protein. As Sec has been recognized as the 21st amino acid and is specifically incorporated in response to the TGA codon, these results are strongly suggestive that SPS2 is a selenoenzyme (1).
We showed that the presence of the 3′ UTR increases the expression of the SPS2 protein, indicating the existence of SECIS-like elements in the 3′ UTR. These sequence motifs may not fit the observed linear pattern of their previously described counterparts (6), as rigorous analyses of both mouse and human Sps2 mRNAs and their predicted secondary structure, using the mfold program (20), did not reveal a SECIS element in their 3′ UTRs.
On the basis of current knowledge of the biochemistry of selenium (1–5, 11) and especially the fact that the selD gene product appears to be the first component of the pathway that ultimately leads to the production of Sec, our results suggest that the synthesis of SPS2 is likely to be a key control point of an autoregulatory loop operating at the translational level to regulate the production of selenoproteins through the generation of MSP and Sec (Fig. 5 a and b). The up-regulation of SPS2 activity will likely lead to the synthesis of MSP that can be made available for production of Sec from seryl-tRNAs. Sec, a limiting factor for the synthesis of selenoproteins, could then be incorporated into a number of proteins, including SPS2 (Fig. 5). However, other factors, namely the levels of a specific elongation factor, yet to be characterized in eukaryotic organisms, and/or the level of tRNA(Ser)Sec are likely to be necessary, along with SPS1 and SPS2, to change significantly the profile of selenoproteins being synthesized at any given moment (1–5, 38, 39). An important point in this regulatory network is likely to be the basal level of Sec, as a certain amount of Sec has to be present in the cell for the production of SPS2 to occur. This may be manufactured by SPS1 in mammalian cells, since this SPS homolog does not require Sec for its own production and may be capable of maintaining basal levels of MSP and Sec (Fig. 5a); SPS2, which is likely to be more active (1), could then function as a privileged effector under stimulatory conditions (Fig. 5a). H. influenzae and M. jannaschii have only the Sec-containing form of the SPS enzyme, making these organisms particularly attractive systems for the study of the control of selenium metabolism (Fig. 5b). However, the existence of a nonspecific pathway of incorporation of Sec and selenomethionine into proteins (30), which appears to be of widespread distribution, may suggest the existence of alternative pathways for the production of Sec that do not involve SPS. The presence of TGA-containing selD homologs in organisms from all three different domains of life (Eukaryotes, Bacteria, and Archaea) suggests that this form of the SPS enzyme was present in early life forms (40). On the other hand, the apparent absence of a selD gene homolog in S. cerevisiae and Mycoplasma genitalium may indicate that this enzyme is not essential to all life forms and might have been lost through evolution in these organisms.
Figure 5.
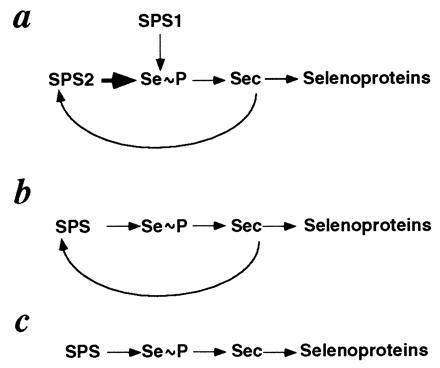
Simplified diagram to represent the three available scenarios in Sec metabolism: (a) Mus musculus and Homo sapiens (Eukaryotes). (b) H. influenzae (Bacteria) and M. jannaschii (Archaea). (c) E. coli (Bacteria). The wider arrow in SPS2 denotes the likely difference in activity, yet to be demonstrated (1), between this enzyme and SPS1.
We showed that Sps2 is expressed at very early stages of development in vivo, namely in sites of blood cell development. Together with the demonstration that Sps2 is a T-cell activation gene, this suggests that, in T cells, the up-regulation of Sps2 may have a role in directing the production of MSP to the synthesis of selenoproteins involved in the control of the immune response rather than in processes related with cell growth and division. This observation is especially intriguing if we consider the sparse information available concerning the potential role of selenium in the immune system (41). Furthermore, a recent report described the possible existence of novel genes in the human immunodeficiency virus type 1 (HIV-1) genome that potentially encode selenoproteins (42). Therefore, the up-regulation of Sps2 in CD4+ T lymphocytes upon stimulation and infection by HIV could influence the synthesis of HIV-encoded selenoproteins relevant for the life cycle of the virus inside T cells. Analogously, the dependence of H. influenzae SPS on Sec for its own synthesis may reveal a need for exogenous sources of this amino acid, and perhaps account for some of the biological properties of this important human pathogen.
Acknowledgments
This work was serially conducted in the Zlotnik and Bazan laboratories. M.J.G. thanks Drs. J. M. Pina Cabral for guidance; T. C. Stadtman, M. J. Berry, R. F. Burk, K. E. Hill, and E. Ching for fruitful discussions; M. J. Berry for comunicating unpublished results; J. Chiller, G. Hardiman, A. Kornberg, M. de Sousa, and M. Teixeira da Silva for encouragement; G. Zurawski for reading the manuscript; H. Lepper, D. Liggett, D. B. Householder, and L. Routhier for assistance; and S. McCarthy and M. McMahon for providing the 3T3 ΔB-Raf:ER fibroblast-derived total RNA. M.J.G. is supported by the Junta Nacional de Investigação Científica e Technológica, Portugal (CIÊNCIA/BD/2685/93). This research was supported, in part, by the National Cancer Institute, under contract with ABL. DNAX is supported by Schering–Plough Corp.
Footnotes
Abbreviations: SPS, selenophosphate synthetase; Sec, selenocysteine; MSP, monoselenophosphate; ORF, open reading frame; UTR, untranslated region.
Data deposition: The sequences reported in this paper have been deposited in in the GenBank data base (accession nos. U43285U43285 and U43286U43286 for mouse and human Sps2 cDNAs, respectively).
References
- 1.Stadtman T C. Annu Rev Biochem. 1996;65:83–100. doi: 10.1146/annurev.bi.65.070196.000503. [DOI] [PubMed] [Google Scholar]
- 2.Stadtman T C. J Biol Chem. 1991;266:16257–16260. [PubMed] [Google Scholar]
- 3.Burk R F, Hill K E. Annu Rev Nutr. 1993;13:65–81. doi: 10.1146/annurev.nu.13.070193.000433. [DOI] [PubMed] [Google Scholar]
- 4.Bock A, Forchhammer K, Heider J, Leinfelder W, Sawers G, Veprek B, Zinoni F. Mol Microbiol. 1991;5:515–520. doi: 10.1111/j.1365-2958.1991.tb00722.x. [DOI] [PubMed] [Google Scholar]
- 5.Bock A, Forchhammer K, Heider J, Baron C. Trends Biochem Sci. 1991;16:463–467. doi: 10.1016/0968-0004(91)90180-4. [DOI] [PubMed] [Google Scholar]
- 6.Berry M J, Banu L, Chen Y, Mandel S J, Kieffer J D, Harney J W, Larsen P R. Nature (London) 1991;353:273–276. doi: 10.1038/353273a0. [DOI] [PubMed] [Google Scholar]
- 7.Baron C, Heider J, Bock A. Proc Natl Acad Sci USA. 1993;90:4181–4185. doi: 10.1073/pnas.90.9.4181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Leinfelder W, Zehelein E, Mandrad-Berthelot M-A, Bock A. Nature (London) 1988;331:723–725. doi: 10.1038/331723a0. [DOI] [PubMed] [Google Scholar]
- 9.Veres Z, Tsai L, Scholz T D, Politino M, Balaban R S, Stadtman T C. Proc Natl Acad Sci USA. 1992;89:2975–2979. doi: 10.1073/pnas.89.7.2975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Glass R S, Singh A P, Jung W, Veres Z, Scholz T D, Stadtman T C. Biochemistry. 1993;32:12555–12559. doi: 10.1021/bi00210a001. [DOI] [PubMed] [Google Scholar]
- 11.Stadtman T C. Annu Rev Biochem. 1990;59:111–127. doi: 10.1146/annurev.bi.59.070190.000551. [DOI] [PubMed] [Google Scholar]
- 12.Leinfelder W, Forchhammer K, Veprek B, Zehelein E, Bock A. Proc Natl Acad Sci USA. 1990;87:543–547. doi: 10.1073/pnas.87.2.543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Low S C, Harney J W, Berry M J. J Biol Chem. 1995;270:21659–21664. doi: 10.1074/jbc.270.37.21659. [DOI] [PubMed] [Google Scholar]
- 14.Guimarães M J, Lee F, Zlotnik A, McClanahan T. Nucleic Acids Res. 1995;23:1832–1833. doi: 10.1093/nar/23.10.1832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guimarães M J, Bazan J F, Zlotnik A, Wiles M V, Grimaldi J C, Lee F, McClanahan T. Development (Cambridge, UK) 1995;121:3335–3346. doi: 10.1242/dev.121.10.3335. [DOI] [PubMed] [Google Scholar]
- 16.Cocks B G, Malefyt R W, Galizzi J-P, de Vries J E, Aversa G. Int Immun. 1993;5:657–663. doi: 10.1093/intimm/5.6.657. [DOI] [PubMed] [Google Scholar]
- 17.Sambrook J, Fritsch E F, Maniatis T. Molecular Cloning: A Laboratory Manual. 2nd Ed. Plainview, NY: Cold Spring Harbor Lab. Press; 1989. [Google Scholar]
- 18.Thompson J D, Higgins D G, Gibson T J. Nucleic Acids Res. 1995;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rost B, Sander C. J Mol Biol. 1993;232:584–599. doi: 10.1006/jmbi.1993.1413. [DOI] [PubMed] [Google Scholar]
- 20.Jaeger J A, Turner D H, Zucker M. Methods Enzymol. 1990;183:281–306. doi: 10.1016/0076-6879(90)83019-6. [DOI] [PubMed] [Google Scholar]
- 21.Pritchard C A, Samuels M, Bosch E, McMahon M. Mol Cell Biol. 1995;15:6430–6442. doi: 10.1128/mcb.15.11.6430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ransom J, Wu R, Fischer M, Zlotnik A. Cell Immunol. 1991;134:180–190. doi: 10.1016/0008-8749(91)90341-8. [DOI] [PubMed] [Google Scholar]
- 23.Copeland N G, Jenkins N A. Trends Genet. 1991;7:113–118. doi: 10.1016/0168-9525(91)90455-y. [DOI] [PubMed] [Google Scholar]
- 24.Avraham K B, Givol D, Avivi A, Yayon A, Copeland N G, Jenkins N A. Genomics. 1994;21:656–658. doi: 10.1006/geno.1994.1330. [DOI] [PubMed] [Google Scholar]
- 25.Green E L. Genetics and Probability in Animal Breeding Experiments. New York: Oxford Univ. Press; 1981. pp. 77–113. [Google Scholar]
- 26.Hopp T P, Prickett K S, Price U, Libby R T, March C J, Cerretti P, Urdal D L, Conlon P J. Bio/Technology. 1988;6:1205–1210. [Google Scholar]
- 27.Kim I Y, Veres Z, Stadtman T C. J Biol Chem. 1992;267:19650–19654. [PubMed] [Google Scholar]
- 28.Fleischmann R D, Adams M D, White O, Clayton R A, Kirkness E F, et al. Science. 1995;269:496–512. doi: 10.1126/science.7542800. [DOI] [PubMed] [Google Scholar]
- 29.Bult C J, White O, Olsen G J, Zhou L, Fleischmann R D, et al. Science. 1996;273:1058–1073. doi: 10.1126/science.273.5278.1058. [DOI] [PubMed] [Google Scholar]
- 30.Stadtman T C, Davis J N, Zehelein A, Bock A. Biofactors. 1989;2:35–44. [PubMed] [Google Scholar]
- 31.Casari G, Daruvar A, Sander C, Schneider R. Trends Genet. 1996;12:244–245. doi: 10.1016/0168-9525(96)30057-7. [DOI] [PubMed] [Google Scholar]
- 32.Fraser C M, Gocayne J D, White O, Adams M D, Clayton R A, et al. Science. 1995;270:397–403. doi: 10.1126/science.270.5235.397. [DOI] [PubMed] [Google Scholar]
- 33.Saraste M, Sibbald P R, Whittinghorter A. Trends Biochem Sci. 1990;15:430–434. doi: 10.1016/0968-0004(90)90281-f. [DOI] [PubMed] [Google Scholar]
- 34.Zvelebil M J, Sternberg M J. Protein Eng. 1988;2:127–138. doi: 10.1093/protein/2.2.127. [DOI] [PubMed] [Google Scholar]
- 35.Berry M J, Banu L, Larsen P R. Nature (London) 1991;349:438–440. doi: 10.1038/349438a0. [DOI] [PubMed] [Google Scholar]
- 36.Karimpour I, Cutler M, Shih D, Smith J, Kleene K. DNA Cell Biol. 1992;11:693–699. doi: 10.1089/dna.1992.11.693. [DOI] [PubMed] [Google Scholar]
- 37.Hill K E, Lloyd R S, Burk R F. Proc Natl Acad Sci USA. 1993;90:537–541. doi: 10.1073/pnas.90.2.537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Berry M J, Harney J W, Ohama T, Hatfield D L. Nucleic Acids Res. 1994;22:3753–3759. doi: 10.1093/nar/22.18.3753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hatfield D, Lee B J, Hampton L, Diamond A M. Nucleic Acids Res. 1992;19:939–943. doi: 10.1093/nar/19.4.939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Green P, Lipman D, Hillier L, Waterston R, States D, Claverie J M. Science. 1993;259:1711–1716. doi: 10.1126/science.8456298. [DOI] [PubMed] [Google Scholar]
- 41.Beck M A, Shi Q, Morris V C, Levander O A. Nat Med. 1995;5:433–436. doi: 10.1038/nm0595-433. [DOI] [PubMed] [Google Scholar]
- 42.Taylor E W, Ramanathan C S, Jalluri R K, Nadimpalli R G. J Med Chem. 1994;37:2637–2654. doi: 10.1021/jm00043a004. [DOI] [PubMed] [Google Scholar]