

Abstract
Interferon γ (IFN-γ) induces rapid tyrosine phosphorylation of the latent cytoplasmic transcription factor, Stat1, which then forms homodimers, translocates to the nucleus and participates in IFN-γ-induced transcription. However, little is known of the interactions between Stat1 and the general transcription machinery during transcriptional activation. We show here that Stat1 can directly interact with the CREB-binding protein (CBP)/p300 family of transcriptional coactivators. Specifically, two interaction regions were identified: the amino-terminal region of Stat1 interacts with the CREB-binding domain of CBP/p300 and the carboxyl-terminal region of Stat1 interacts with the domain of CBP/p300 that binds adenovirus E1A protein. Transfection experiments suggest a role for these interactions in IFN-γ-induced transcription. Because CBP/p300-binding is required for the adenovirus E1A protein to regulate transcription of many genes during viral replication and cellular transformation, it is possible that the anti-viral effect of IFN-γ is based at least in part on direct competition by nuclear Stat1 with E1A for CBP/p300 binding.
Many site-specific eukaryotic DNA binding proteins, often termed “gene-specific” transcription factors (GS-TFs), raise the level of transcription of various genes by binding to DNA sites several hundred to many thousands of base pairs from polymerase II initiation sites (1). The mechanism by which these GS-TFs cooperate with the general transcription factors to alter polymerase II initiation rates is an area of intense research. Another group of proteins, most of which do not themselves bind DNA, termed coactivators or TBP-associated factors (TAFIIs), act through protein–protein interactions to integrate the activation potential of the GS-TFs with the general transcription factors (2–4). The GS-TFs include a variety of structural classes of proteins and a very large number of individual factors, highlighting the importance of attempts to determine which coactivators or TAFs recognize which GS-TFs.
Of special interest in this connection are GS-TFs that regulate gene expression in response to outside stimuli. One such group of proteins, the signal transducers and activators of transcription (STATs; seven are known at present), become phosphorylated on a single tyrosine residue in response to polypeptide ligands binding to their cell surface receptors (5, 6). The tyrosine phosphorylated STAT proteins dimerize and enter the nucleus to stimulate transcription. Stat1 and Stat3 can also be phosphorylated on a single serine residue (Ser-727) at the carboxyl end of the molecule (7). Interferon γ (IFN-γ) specifically induces phosphorylation of Stat1, which enters the nucleus as a homodimer and binds to DNA elements called GAS (IFN-γ-activated sites) (8–11). Due to alternative splicing, Stat1 exists in two forms: full-length Stat1α and Stat1β lacking 38 residues (including serine-727) at the carboxyl-terminus (12). Only Stat1α is able to activate transcription of IFN-γ-responsive genes or to confer the anti-viral state, indicating that the carboxyl terminus of Stat1 is the transactivation domain of the molecule (13, 14). How Stat1 contacts the general transcription machinery to induce transcription of otherwise silent genes is unknown.
The CREB-binding protein (CBP)/p300 family of transcriptional coactivators have been shown to potentiate the activity of several groups of transcription factors (15). There is an overall sequence similarity of CBP and p300, particularly in five separate domains, with which a number of transcription factors have been shown to interact (16, 17). CBP was first identified through its interaction with the cAMP response element-binding protein (CREB; ref. 18). This interaction was contingent on the phosphorylation of Ser-133 of the CREB protein (19, 20). The p300 protein was cloned through its interaction with E1A of adenovirus (21). E1A protein is highly phosphorylated on serines, although the possible role of serine phosphorylation in p300 binding has not been assessed. These early findings led us to investigate whether Stat1α, known to contain a serine phosphate at residue 727, might interact with CBP/p300 in IFN-γ-induced transcription activation.
Experiments described in this report have identified interaction between the carboxyl terminus of Stat1α and the E1A-binding domain of CBP/p300. In addition, there is another interaction between the CREB-binding domain of CBP/p300 and the amino terminus of Stat1α. Transfection experiments demonstrated that the interaction between the carboxyl terminus of Stat1α and the E1A-binding domain of CBP/p300 is required for transcriptional activation property of Stat1α homodimer during response to IFN-γ. The competition between the carboxyl-terminal region of Stat1 and the E1A protein for the same site on CBP/p300 could also in part explain the anti-viral effect of IFN-γ.
MATERIALS AND METHODS
Cell Culture and Antibodies.
U3A cells (provided by George Stark, Cleveland Clinic Foundation Research Institute; and Ian Kerr, Imperial Cancer Research Foundation, London) were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% cosmic calf serum (HyClone). U-2 OS human osteosarcoma cells (purchased from American Type Culture Collection) were maintained in DMEM supplemented with 10% fetal bovine serum (HyClone). Stable cell lines containing transfected expression plasmids were selected and maintained with G418 at 250 μg/ml (GIBCO/BRL). A monoclonal antibody against the SH2 domain of Stat1 was a gift from Robert Schreiber (Washington University School of Medicine, St. Louis). The anti-FLAG monoclonal antibody was purchased from Kodak/IBI. Recombinant human IFN-γ was a gift from Amgen.
Glutathione S-Transferase (GST)-Fusion Protein Interaction Assays and Western Blot Analysis.
Stat1 proteins were expressed in baculovirus and purified as previously described (22). GST-fusion proteins were purified from bacteria using glutathione-Sepharose beads as instructed by the manufacturer (Pharmacia). Nuclear extracts or whole cell extracts were prepared as previously described (7) from untreated or IFN-γ-treated (30 min at 5 ng/ml) U3A cells reconstituted with FLAG-tagged STAT1α or β. For binding assays with purified Stat1 proteins (see Fig. 1 B and C), 500 ng of purified Stat1 proteins was incubated with 10 μg of various GST-fusion proteins bound to glutathione-Sepharose beads (Pharmacia) at 4°C overnight in 1 ml of 10 mM Hepes (pH 7.4), 200 mM NaCl, 0.5 mM EDTA (pH 8.0), 0.1% Nonidet P-40, 5 mg of BSA per ml, and 1 mM DTT. For binding assays with cell extracts (see Fig. 2 A and B), ≈500 μg of nuclear or whole-cell extracts were incubated with 10–20 μg of GST-fusion proteins bound on beads at 4°C overnight in 1 ml of 150 mM NaCl, 20 mM Hepes (pH 7.9), 10 mM KCl, 13% glycerol, 1 mM EDTA (pH 8.0), 0.1 mM NaVO4, and 1 mM DTT. The resulting binding complexes were washed in the same binding buffer for five times, and the bound proteins were separated on 7% SDS/PAGE. Western blot analyses were done using a monoclonal antibody against Stat1 SH2 domain (see Fig. 1 B and C) or anti-FLAG monoclonal antibody (see Fig. 2 A and B). Western blot analyses were done using chemiluminescence as described by the manufacturer (DuPont/NEN).
Figure 1.
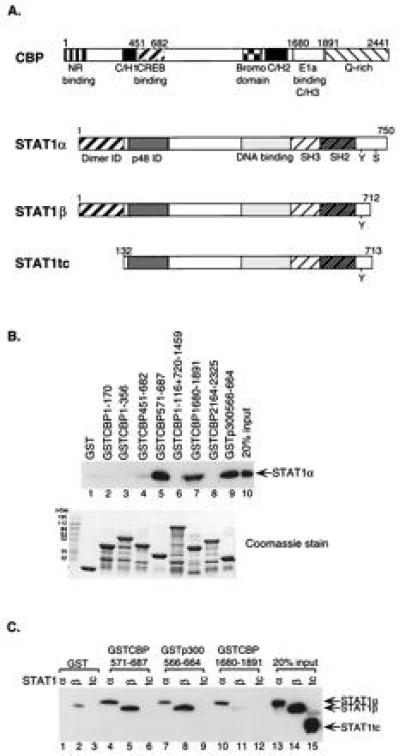
Stat1 interacts with CBP/p300 at two distinct regions. (A) A schematic diagram of CBP and variants of Stat1. The fragments of CBP used as GST-fusion proteins were residues: 1-170, 1-356, 451-682, 571-687, 1-116 plus 720-1459, 1680-1891, and 2164-2325. The fragment of p300 used was residues 566-664. NR, nuclear hormone receptor; C/H, cysteine/histidine-rich region; Stat1tc, a truncated version of Stat1 (residues 132-713); ID, interaction domain; Y, Tyr-701; S, Ser-727. (B) Stat1α interacts with two different regions of CBP/p300. Purified Stat1α protein was incubated with different regions of CBP/p300 as GST-fusion proteins bound to glutathione-Sepharose-beads. The bound proteins were analyzed by Western blotting using an anti-Stat1 antibody. Equivalent amounts of various GST-fusion proteins were used in the assay, as shown by Coomassie staining. (C) Two different interaction regions between Stat1 and CBP/p300. Purified Stat1α, Stat1β, or Stat1tc proteins were incubated with Sepharose beads-bound GST-fusion proteins containing fragments of CBP/p300 that interacted with Stat1α in B and analyzed by Western blotting.
Figure 2.
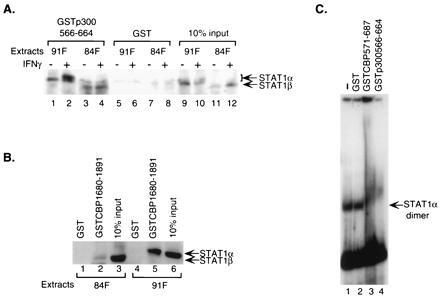
Endogenous Stat1 homodimers interact with CBP/p300. (A) Both unphosphorylated and phosphorylated cellular Stat1α and β interact with the CREB-binding domain of CBP/p300. GST-fusion proteins of CBP/p300 bound on glutathione-Sepharose beads were incubated with nuclear extracts from untreated or IFN-γ-treated U3A cells deficient for endogenous Stat1 and stably expressing FLAG-tagged Stat1α or β. The specifically bound Stat1 proteins were analyzed by Western blotting using an anti-FLAG antibody. 84F, extracts from cells expressing FLAG-tagged Stat1β; 91F, extracts from cells expressing FLAG-tagged Stat1α. (B) Interaction between cellular Stat1α and the E1A-binding domain of CBP requires the carboxyl terminus of Stat1α. Whole-cell extracts from treated cells stably expressing FLAG-tagged Stat1α or β as in A were incubated with GST-fusion proteins and the resulting bound Stat1 analyzed as above. (C) The CREB-binding domain of CBP/p300 interacts with Stat1α dimer bound to DNA. Nuclear extracts from IFN-γ-treated U3A cells reconstituted with FLAG-tagged Stat1α were incubated with a 32P-labeled Stat1-DNA-binding site before being exposed to various purified GST-fusion proteins, and the resulting DNA–protein complexes were analyzed by EMSA.
Plasmid Constructions.
GST-fusion constructs GSTCBP1-170 and GSTCBP1-356 were made by PCR using primers containing 5′ BamHI site and 3′ EcoRI site to amplify CBP fragments encompassing residues 1-170 and 1-356. Amplified products were digested with appropriate enzymes and cloned into pGEX2TK (Pharmacia). Plasmids containing CBP1-116+720-1459 and CBP2164-2325 were gifts from Marc Montminy (Harvard Medical School, Boston). The appropriately digested DNA fragments were further subcloned into pGEX2T (Pharmacia). GSTCBP451-682, GSTCBP571-687, GSTCBP1680-1891, and GSTp300/566-664 were gifts from Richard Goodman (Vollum Institute, Oregon Health Sciences University, Portland, OR). Construction of expression vectors Rc/CMV (Invitrogen) containing Stat1α or β was as previously described (7). The FLAG-epitope (23) was inserted at the 3′ end of the Stat1α or β coding sequence to generate FLAG-tagged Stat1α or β expression vectors. The 3xLy6E GAS–LUC reporter was constructed as previously described (7). pCMVβ construct was purchased from Invitrogen. RSVCBP was a gift from Richard Goodman (Vollum Institute, Oregon Health Sciences University). Rc/CMVE1A12S and Rc/CMVE1A(Δ64-68) were gifts from Tony Kouzarides (University of Cambridge, Cambridge, United Kingdom).
Electrophoretic Mobility-Shift Assay (EMSA).
Nuclear extracts (≈5–10 μg) from IFN-γ-treated (30 min at 5 ng/ml) U3A cells reconstituted with FLAG-tagged Stat1α were incubated with 1 ng of 32P-labeled M67 probe (24) for 5 min at room temperature. Eluted GST-fusion proteins (6 μg) were then added into the mixture and incubated further on ice for 30 min. The protein–DNA complexes were analyzed by EMSA as previously described (25).
Transfection Experiments.
Transient transfections were done in triplicates on six-well plates with 5 × 104 cells per well using the calcium phosphate method as instructed by the manufacturer (GIBCO/BRL). Total amount of DNA transfected was brought up to 6 μg using sonicated salmon sperm DNA. Twenty-four hours after transfection, cells were treated with IFN-γ for 6 hr or left untreated. Luciferase assays were performed according to the manufacturer (Promega) and β-galactosidase (β-gal) assays were done as previously described (26). Except for Fig. 3B, all results shown are luciferase activities normalized against the internal control β-gal activity. Because the internal control β-gal activity increased consistently with overexpression of exogenous CBP, raw data of luciferase activities from four individual experiments were used to calculate the fold of induction in Fig. 3B.
Figure 3.
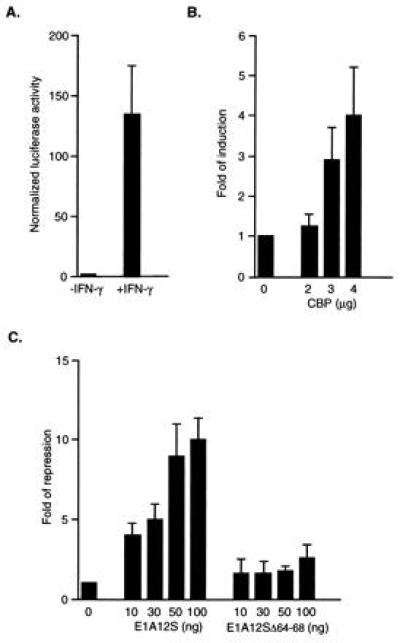
IFN-γ-induced transcriptional activation by Stat1α is enhanced by CBP and repressed by E1A. (A) IFN-γ induces reporter activity in cells expressing endogenous Stat1α. (B) Overexpression of CBP enhances IFN-γ-induced transcription. (C) E1A inhibits IFN-γ-induced Stat1α transactivation by competing for CBP/p300-binding. U2-OS cells were transfected with 0.5 μg of luciferase reporter, 0.3 μg of CMVβgal, and various amounts of RSVCBP, CMVE1A12S, and CMVE1A(Δ64-68) as indicated. Twenty-four hours after transfection, cells were treated with 5 ng of IFN-γ per ml for 6 hr and harvested for luciferase assay and β-gal assay. All transfections were done in triplicates. Results shown are the mean ± SD of two to four experiments. Results from only the IFN-γ-treated samples are presented in B and C as reporter activities from untreated cells did not change significantly.
RESULTS
A diagram of CBP/p300 and variants of Stat1 protein used in the present experiments is presented in Fig. 1A. The previously described seven regions of CBP (and one analogous region of p300) were expressed as GST-fusion proteins. Various forms of Stat1 were purified: Stat1α (full-length 750 aa), Stat1β (lacking the carboxyl-terminal 38 aa due to differential splicing of the Stat1 mRNA), and Stat1tc, containing residues 132-713. The boundaries of Stat1tc were chosen because this segment was resistant to proteolytic digestion (22).
We first examined interactions between the various CBP/p300 fragments and full-length unphosphorylated Stat1α (Fig. 1B). The GST-fusion proteins bound on glutathione-Sepharose beads were incubated with purified unphosphorylated Stat1α. The final bound proteins were eluted and subjected to SDS/PAGE and Western blot analyses with an anti-Stat1 antibody. The results showed that two regions of CBP bound Stat1α. One was a segment including residues from 571 to 687 of CBP (Fig. 1B, lane 5), which has been shown to bind to CREB (18). An equivalent region of p300 (residues 566-664) also bound strongly to Stat1α (Fig. 1B, lane 9). A longer fragment including residues from 451 to 682 of CBP did not interact with Stat1α (Fig. 1B, lane 4). Assuming this protein fragment to be correctly folded, this result hints at a complicated structure in the full-length CBP that affects the choice of binding partners in vivo. A second Stat1α-binding region in CBP was located in the region containing residues from 1680 to 1891 (Fig. 1B, lane 7), which is known to bind E1A (21). Equivalent amounts of each GST fusion protein were used in the binding assays as shown by Coomassie staining (Fig. 1B Lower).
To determine the regions in the Stat1α molecule that interact with the two different CBP domains, purified Stat1α, Stat1β, or Stat1tc was incubated with each of the three interacting fusion domains, CBP/571-687, p300/566-664, and CBP/1680-1891, and, as a control, GST alone. The bound proteins were analyzed as before using a Stat1 antibody against a segment of the molecule (the SH2 domain) that is present in all of three variants of Stat1. The CREB-binding region of CBP/p300 bound both Stat1α and Stat1β (Fig. 1C, lanes 4, 5, 7, and 8) but did not bind Stat1tc, which lacks both the amino and carboxyl termini of Stat1α (Fig. 1C, lanes 6 and 9). On the other hand, the E1A-binding region of CBP bound only the full-length Stat1α (Fig. 1C, lane 10), but not Stat1β or Stat1tc (Fig. 1C, lanes 11 and 12). Thus the amino terminus of Stat1, lacking in Stat1tc but present in Stat1β, was required for interacting with the CREB-binding domain, while the E1A-binding domain must interact with the carboxyl terminus of Stat1α, since this domain is missing in both Stat1β and Stat1tc.
The above experiments were all done with purified proteins, which were not phosphorylated. To further demonstrate that the E1A-binding domain and CREB-binding domain of CBP/p300 could interact with phosphorylated and unphosphorylated Stat1 molecules in the presence of other cellular proteins, nuclear extracts of IFN-γ treated and untreated cells were prepared and tested for binding to the GST-fusion proteins as above. The cells used were U3A cells deficient for endogenous Stat1 (13) and reconstituted with FLAG-tagged (23) Stat1α or Stat1β. Western blot analyses of bound proteins were done with an anti-FLAG antibody. The results showed that the CREB-binding domain of p300 could interact with both Stat1α and β (Fig. 2A, lanes 1–4), confirming the results obtained with purified proteins (Fig. 1C). For Stat1α, two bands could be observed with IFN-γ-treated extracts (Fig. 2A, lane 2): the slower migrating band has been shown in earlier experiments to be the tyrosine-phosphorylated Stat1α molecule (10), indicating that phosphorylated homodimer of Stat1α as well as unphosphorylated monomer can interact with the CREB-binding domain of CBP/p300.
The requirement of the carboxyl terminus of cellular Stat1α for interaction with the E1A-binding domain of CBP is shown in Fig. 1B. Whole-cell extracts from IFN-γ-treated cells used above were incubated with GST-fusion proteins containing residues 1680-1891 of CBP and analyzed by Western blotting. The E1A-binding domain of CBP only interacted with Stat1α (Fig. 2B, lane 5), not Stat1β (Fig. 2B, lane 2), further confirming that the 38 residues at the carboxyl end is required for Stat1α to interact with the E1A-binding domain.
To show that DNA-bound Stat1α could interact with the CREB-binding domain of CBP/p300, GST-fusion proteins containing the CREB-binding domain were used to test their effects on Stat1α–DNA complexes by EMSA. Nuclear extracts from IFN-γ-treated U3A cells reconstituted with FLAG-tagged Stat1α were first incubated with an oligonucleotide probe, M67, which represents a high-affinity Stat1-binding site (11, 24). Various purified GST-fusion proteins were then added to the Stat1α–DNA complex. As shown in Fig. 2C, the Stat1α–DNA complex was completely “super-shifted” by GST-fusion proteins containing the CREB-binding domain of CBP/p300 (Fig. 2C, lanes 3 and 4), but not by GST alone (Fig. 2C, lane 2), indicating that the CREB-binding domain could interact with Stat1 homodimers bound to DNA. Due to background bands migrating at the same position as Stat1α dimers, it was not possible to see the effects of E1A-binding domain on the Stat1α–DNA complex by this assay.
These results demonstrate that both phosphorylated homodimeric and unphosphorylated monomeric Stat1α can interact with CBP/p300, and their interactions occur at two different regions of both Stat1α and CBP/p300.
In the final set of experiments, we explored the possible participation of CBP in Stat1 driven gene expression. A luciferase reporter construct containing three binding sites for Stat1 was transfected into human U2-OS cells (27) alone or with the following expression constructs: CBP, E1A12S, and E1A12SΔ64-68, which has been shown to be defective in p300 binding (28). Transfected cells were then either treated with IFN-γ or left untreated. The results showed that the expression of the reporter gene was greatly induced by IFN-γ (Fig. 3A), indicating the endogenous Stat1α is phosphorylated as confirmed by EMSA and activates transcription by binding to the GAS sites in the reporter construct (data not shown). Overexpression of exogenous CBP produced a dose-dependent increase in reporter activity (Fig. 3B). E1A has been shown previously by transfection experiments to inhibit transcriptional activation mediated by CBP/p300, and this inhibition is dependent on its binding to CBP/p300 (16, 17). Fig. 3C shows that the wild-type E1A12S completely inhibited IFN-γ-induced reporter activity, while the mutant E1A12SΔ64-68 defective in CBP/p300-binding, had only a marginal inhibitory effect at high doses, suggesting a possible competition between Stat1α and E1A for the same site on CBP/p300. Reporter activities from untreated cells did not change significantly (data not shown). These results demonstrate that CBP can potentiate the IFN-γ-induced transcriptional activation by Stat1α, and the interaction between Stat1α and the E1A-binding domain is important for this process.
DISCUSSION
The experiments described in this paper show that the STATs join a growing list of transcription factors in using CBP/p300 family of transcriptional coactivators to bring about their effects on transcription (15). One new aspect of our findings compared with the previous findings on interactions between transcription factor and CBP/p300 is the participation of two widely separated regions (both ends) of a single Stat1 molecule.
The carboxyl end of Stat1α has been shown to be required for transcriptional activation by Stat1α (7, 13, 14). Our findings demonstrate that this region of Stat1α directly interacts with CBP/p300 and this interaction is important for the IFN-γ-induced transcriptional activation by Stat1α. The interaction between the amino terminus of Stat1 and the CREB-binding domain of CBP/p300 is of particular interest, because recently the amino terminus has been implicated in stabilizing dimer–dimer interactions of the STATs bound to neighboring weak DNA binding sites (22, 29). Also, the amino-terminal portion of Stat1 has been implicated in prolongation of the STAT activation signal, which may be due to interaction of this region with phosphotyrosine phosphatases (30). Exactly how this interaction between the amino terminus of Stat1α and the CREB-binding domain of CBP/p300 affects transcription activation by Stat1 remains unknown and how this interaction is integrated with its other functions presents a very interesting problem. Another point of difference with the CREB and c-Jun interactions with CBP is that Stat1α, like the RXR and TR fragments of nuclear hormone receptors, bound to CBP without being serine-phosphorylated (19, 20, 31, 32). Since it has been shown that serine phosphorylation on Ser-727 in STATs is required for maximal transcriptional activity (7), it is possible that an even tighter interaction would occur with the STATs when Ser-727 is phosphorylated.
As this manuscript was being prepared, Livingston and collaborators reported that the carboxyl-terminal region of Stat2 interacted with the C/H1 region of CBP/p300 during IFN-α response (33). While like Stat1, the carboxyl-terminal region of Stat2 is also the transcription activation domain, it has a distinctly different character from the Stat1 carboxyl terminus. The Stat2 carboxyl terminus is a highly acidic region and does not undergo serine phosphorylation (34, 35). IFN-γ is different from IFN-α in many aspects, including its gene structure, receptor, and anti-viral effect (5, 6). In response to IFN-α, either Stat1α or β can form a heterotrimeric complex with Stat2 and p48 (34, 36–38). This complex binds to a DNA element called ISRE (interferon stimulation response element) to activate IFN-α-responsive genes (39). However, in response to IFN-γ, only Stat1 dimers are formed and bind to dyad symmetrical DNA elements termed GAS (6, 8, 9). Moreover, only the full-length Stat1α can activate transcription in response to IFN-γ, even though Stat1β dimer can bind to DNA efficiently (13, 40). Therefore, it seems likely that the structurally different carboxyl termini of Stat1 and Stat2 contact different regions of CBP/p300 during the response to different cytokines—i.e., IFN-γ and IFN-α, respectively. Thus these earlier and present findings suggest that IFN-γ and Stat1 use a different mechanism of gene activation from IFN-α and Stat2. In addition, because CBP/p300-binding is required for E1A to regulate positively the transcription of many genes during viral replication and cellular transformation by adenovirus (41, 42), our results imply that IFN-γ could exert its anti-viral effect, in part, by drastically increasing the nuclear level of Stat1 that competes directly with E1A for the binding site in CBP/p300.
Acknowledgments
We gratefully acknowledge Dr. Goodman for the GST-fusion constructs and CBP expression vectors, Dr. M. Montminy for recombinant CBP fragments, Drs. George Stark and Ian Kerr for the U3A cell line, Dr. Robert Schreiber for anti-Stat1 monoclonal antibody, Dr. Kouzarides for the E1A expression vectors, and Lois Cousseau for preparing the manuscript. This work was supported by National Institute Grants AI34420 and AI32489 to J.E.D. J.J.Z. was supported by a Postdoctoral Fellowship from the Aaron Diamond Foundation. D.C. was supported by a postdoctoral fellowship from the Jane Coffin Childs Memorial Funds for Medical Research.
Footnotes
Abbreviations: GS-TF, “gene-specific” transcription factor; CREB, cAMP response element-binding protein; CBP, CREB-binding protein; GST, glutathione S-transferase; IFN-γ, interferon γ; EMSA, electrophoretic mobility-shift assay; STAT, signal transducer and activator of transcription; β-gal, β-galactosidase.
References
- 1.Roeder R G. Trends Biochem Sci. 1996;21:327–335. [PubMed] [Google Scholar]
- 2.Bjorklund S, Kim Y-J. Trends Biochem Sci. 1996;21:335–337. doi: 10.1016/s0968-0004(96)10051-7. [DOI] [PubMed] [Google Scholar]
- 3.Verrijzer C P, Tjian R. Trends Biochem Sci. 1996;21:338–342. [PubMed] [Google Scholar]
- 4.Kaiser K, Meisterernst M. Trends Biochem Sci. 1996;21:342–345. [PubMed] [Google Scholar]
- 5.Darnell J E, Jr, Kerr I M, Stark G M. Science. 1994;264:1415–1421. doi: 10.1126/science.8197455. [DOI] [PubMed] [Google Scholar]
- 6.Schindler C, Darnell J E., Jr Annu Rev Biochem. 1995;64:621–651. doi: 10.1146/annurev.bi.64.070195.003201. [DOI] [PubMed] [Google Scholar]
- 7.Wen Z, Zhong Z, Darnell J E., Jr Cell. 1995;82:241–250. doi: 10.1016/0092-8674(95)90311-9. [DOI] [PubMed] [Google Scholar]
- 8.Lew D, Decker T, Strehlow I, Darnell J E., Jr Mol Cell Biol. 1991;11:182–191. doi: 10.1128/mcb.11.1.182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Decker T, Lew D J, Mirkovitch J, Darnell J E., Jr EMBO J. 1991;10:927–932. doi: 10.1002/j.1460-2075.1991.tb08026.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shuai K, Schindler C, Prezioso V R, Darnell J E., Jr Science. 1992;259:1808–1812. doi: 10.1126/science.1281555. [DOI] [PubMed] [Google Scholar]
- 11.Shuai K, Horvath C M, Tsai-Huang L H, Qureshi S, Cowburn D, Darnell J E., Jr Cell. 1994;76:821–828. doi: 10.1016/0092-8674(94)90357-3. [DOI] [PubMed] [Google Scholar]
- 12.Schindler C, Fu X-Y, Improta T, Aebersold R, Darnell J E., Jr Proc Natl Acad Sci USA. 1992;89:7836–7839. doi: 10.1073/pnas.89.16.7836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Muller M, Laxton C, Briscoe J, Schindler C, Improta T, Darnell J E, Jr, Stark G R, Kerr I M. EMBO J. 1993;12:4221–4228. doi: 10.1002/j.1460-2075.1993.tb06106.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Horvath C M, Darnell J E., Jr J Virol. 1996;70:647–650. doi: 10.1128/jvi.70.1.647-650.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Janknecht R, Hunter T. Nature (London) 1996;383:22–23. doi: 10.1038/383022a0. [DOI] [PubMed] [Google Scholar]
- 16.Lundblad J R, Kwok R P S, Laurance M E, Harter M L, Goodman R H. Nature (London) 1995;374:85–88. doi: 10.1038/374085a0. [DOI] [PubMed] [Google Scholar]
- 17.Arany Z, Newsome D, Oldread E, Livingston D M, Eckner R. Nature (London) 1995;374:81–84. doi: 10.1038/374081a0. [DOI] [PubMed] [Google Scholar]
- 18.Chrivia J C, Kwok R P S, Lamb N, Hagiwara M, Montminy M R, Goodman R H. Nature (London) 1993;365:855–859. doi: 10.1038/365855a0. [DOI] [PubMed] [Google Scholar]
- 19.Kwok R P S, Lundblad J R, Chrivia J C, Richards J P, Bachlinger H P, Brennan R G, Roberts S G E, Green M R, Goodman R H. Nature (London) 1994;370:223–226. doi: 10.1038/370223a0. [DOI] [PubMed] [Google Scholar]
- 20.Arias J, Alberts A S, Brindle P, Claret F X, Smeal T, Karin M, Feramisco J, Montminy M. Nature (London) 1994;370:226–229. doi: 10.1038/370226a0. [DOI] [PubMed] [Google Scholar]
- 21.Eckner R, Ewen M E, Newsome D, Gerdes M, DeCaprio J A, Lawrence J B, Livingston D M. Genes Dev. 1994;8:869–884. doi: 10.1101/gad.8.8.869. [DOI] [PubMed] [Google Scholar]
- 22.Vinkemeier U, Cohen S L, Moarefi I, Chait B T, Kuriyan J, Darnell J E., Jr EMBO J. 1996;15:5616–5626. [PMC free article] [PubMed] [Google Scholar]
- 23.Chiang C-M, Roeder R G. Pept Res. 1993;6:62–64. [PubMed] [Google Scholar]
- 24.Wagner B J, Hayes T E, Hoban C J, Cochran B H. EMBO J. 1990;9:4477–4484. doi: 10.1002/j.1460-2075.1990.tb07898.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fried M, Crothers D M. Nucleic Acids Res. 1981;9:6505–6525. doi: 10.1093/nar/9.23.6505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ausubel F M, Brent R, Kingston R E, Moore D D, Seidman J G, Smith J A, Struhl K. Current Protocols in Molecular Biology. New York: Wiley; 1994. [Google Scholar]
- 27.Huang H-J S, Yee J-K, Shew J-Y, Chen P-L, Bookstein R, Friedmann T, Lee E Y-H P, Lee W-H. Science. 1988;242:1563–1566. doi: 10.1126/science.3201247. [DOI] [PubMed] [Google Scholar]
- 28.Bannister A J, Kouzarides T. EMBO J. 1995;14:4758–4762. doi: 10.1002/j.1460-2075.1995.tb00157.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xu X, Sun Y-L, Hoey T. Science. 1996;273:794–797. doi: 10.1126/science.273.5276.794. [DOI] [PubMed] [Google Scholar]
- 30.Shuai K, Liao J, Song M M. Mol Cell Biol. 1996;16:4932–4941. doi: 10.1128/mcb.16.9.4932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kamei Y, Xu L, Heinzel T, Torchia J, Kurokawa R, Gloss B, Lin S-C, Heyman R A, Rose D W, Glass C K, Rosenfeld M G. Cell. 1996;85:403–414. doi: 10.1016/s0092-8674(00)81118-6. [DOI] [PubMed] [Google Scholar]
- 32.Chakravarti D, LaMorte V J, Nelson M C, Nakajima T, Schulman I G, Juguilon H, Montminy M, Evans R M. Nature (London) 1996;383:99–103. doi: 10.1038/383099a0. [DOI] [PubMed] [Google Scholar]
- 33.Bhattacharya S, Eckner R, Grossman S, Oldread E, Arany Z, D’Andrea A, Livingston D M. Nature (London) 1996;383:344–347. doi: 10.1038/383344a0. [DOI] [PubMed] [Google Scholar]
- 34.Schindler C, Shuai K, Prezioso V R, Darnell J E., Jr Science. 1992;257:809–815. [PubMed] [Google Scholar]
- 35.Qureshi S A, Leung S, Kerr I M, Stark G R, Darnell J E., Jr Mol Cell Biol. 1996;16:288–293. doi: 10.1128/mcb.16.1.288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fu X-Y, Kessler D S, Veals S A, Levy D E, Darnell J E., Jr Proc Natl Acad Sci USA. 1990;87:8555–8559. doi: 10.1073/pnas.87.21.8555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Veals S A, Schindler C, Leonard D, Fu X-Y, Aebersold R, Darnell J E, Jr, Levy D E. Mol Cell Biol. 1992;12:3315–3324. doi: 10.1128/mcb.12.8.3315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Qureshi S A, Salditt-Georgieff M, Darnell J E., Jr Proc Natl Acad Sci USA. 1995;92:3929–3833. doi: 10.1073/pnas.92.9.3829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Levy D E, Kessler D S, Pine R, Reich N, Darnell J E., Jr Genes Dev. 1988;2:383–393. doi: 10.1101/gad.2.4.383. [DOI] [PubMed] [Google Scholar]
- 40.Shuai K, Stark G R, Kerr I M, Darnell J E., Jr Science. 1993;261:1744–1746. doi: 10.1126/science.7690989. [DOI] [PubMed] [Google Scholar]
- 41.Nevins J R. Science. 1992;258:424–429. doi: 10.1126/science.1411535. [DOI] [PubMed] [Google Scholar]
- 42.Wang H-G H, Rikitake Y, Carter M C, Yaciuk P, Abraham S E, Zerler B, Moran E. J Virol. 1993;67:476–488. doi: 10.1128/jvi.67.1.476-488.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]