

Abstract
Combinations of new medications or existing therapies are gaining momentum over monotherapy to treat central nervous system (CNS) demyelinating diseases including multiple sclerosis (MS). Recent studies established that statins (HMG-CoA reductase inhibitors) are effective in experimental autoimmune encephalomyelitis (EAE), an MS model and are promising candidates for future MS medication. Another drug, rolipram (phosphodiesterase-4 inhibitor) ameliorates the clinical severity of EAE via induction of various anti-inflammatory and neuroprotective activities. In this study, we tested whether combining the suboptimal doses of these drugs can suppress the severity of EAE. Prophylactic studies revealed that combined treatment with suboptimal doses of statins perform better than their individually administered optimal doses in EAE as evidenced by delayed clinical scores, reduced disease severity, and rapid recovery. Importantly, combination therapy suppressed the progression of disease in an established EAE case via attenuation of inflammation, axonal loss and demyelination. Combination treatment attenuated inflammatory TH1 and TH17 immune responses and induced TH2-biased immunity in the peripheral and CNS as revealed by serological, quantitative, and immunosorbant assay-based analyses. Moreover, the expansion of T regulatory (CD25+/Foxp3+) cells and self-immune tolerance was apparent in the CNS. These effects of combined drugs were reduced or minimal with either drug alone in this setting. In conclusion, our findings demonstrate that the combination of these drugs suppresses EAE severity and provides neuroprotection thereby suggesting that this pharmacological approach could be a better future therapeutic strategy to treat MS patients.
Keywords: Combination therapy, EAE, Lovastatin, Rolipram, Inflammation, Demyelination, Neuroprotection
INTRODUCTION
Experimental autoimmune encephalomyelitis (EAE) is an experimentally induced inflammatory CNS demyelinating disease that mimics many aspects of multiple sclerosis (MS) (Lublin, 1985). Pathophysiology of the disease includes breaching of the blood-brain barrier (BBB), infiltration of inflammatory cells (i.e., myelin reactive CD4+ and CD8+ T cells, and monocytes) into the CNS which perpetuate inflammatory response via activation of resident glial cells and secretion of inflammatory mediators (Hafler, 2004). These events subsequently lead to axonal loss and demyelination through multiple effector mechanisms leading to neurological deficits in MS patients.
Various immunomodulatory agents attenuate EAE with different mechanisms of action are being tested for MS treatment because presently approved MS therapies are only partially effective and are associated with side effects and potential toxicities. For instance, interferon beta (IFN-β) and glatiramer acetate (GA) were promising in some patients, but many individuals experienced poor responses or adverse effects (Arbizu, et al., 2000, Dhib-Jalbut, et al., 2002). Because of the inherent complexity of MS and the involvement of multiple cell types such as brain, endothelial, and vascular immune cells, the challenge of monotherapy with either pre-existing or new MS drugs is limiting the chronic progressive disability observed in affected individuals. One approach to improve treatment is to develop more efficacious agents and another, more plausible, approach is to investigate possible combinations of existing or novel agents that together synergistically or additively attenuate the disease process in different cell types.
In addition to their cholesterol-lowering effects, statins (HMG-CoA reductase inhibitors) are reported to have immunomodulatory effects that can be exploited in the treatment of CNS demyelinating diseases including MS (Paintlia, et al., 2004, Youssef, et al., 2002). Promising results were obtained in an initial clinical trial of simvastatin in MS (Vollmer, et al., 2004) and rheumatoid arthritis (Leung, et al., 2003) patients. The immnomodulatory effect of statins include preservation of BBB integrity (Stanislaus, et al., 2001), inhibition of inflammatory cell infiltration into the CNS (Stanislaus, et al., 2001) and skewing of TH1 to TH2 immune responses (Paintlia, et al., 2004, Youssef, et al., 2002). Recent observations of statin-mediated protection of neuroprogenitor cells from inflammatory insult and resulting enhanced myelin repair in ameliorating EAE animals suggest that, in addition to immunomodulatory activity, statins mediate neuroprotection and possibly neuroregeneration (Paintlia, et al., 2005). Similarly, the selective inhibition of phosphodiesterase (PDE)-4 with rolipram, which increases intracellular cAMP, is reported to halt the induction of EAE (Genain, et al., 1995, Sommer, et al., 1995) via immunomodulation of the TH1 to TH2 immune responses (Bielekova, et al., 2000). Because the cellular targets of both statins and rolipram differ with respect to their immunomodulatory activities, we hypothesized that combination therapy with suboptimal doses of these drugs could be used to attenuate EAE severity. Thus, in this study, we documented the testing of the therapeutic efficacy of suboptimal doses of these drugs in combination in an EAE model.
MATERIALS AND METHODS
Reagents
Myelin basic protein (MBP) ~50% pure from guinea pig brain, complete Freund’s adjuvant (CFA), HRP-tagged anti-mouse IgG antibodies, pertussis toxin, and other chemicals were purchased from Sigma (St. Louis, MO). Lovastatin, rolipram (±; racemate), and PDE-4 inhibitor [PDI-4; 3, 5-Dimethyl-1-(3-nitrophenyl)-1H-pyrazole-4-carboxylic acid ethyl ester] were purchased from Calbiochem (San Diego, CA). ‘TRIZOL’ reagent was purchased from Invitrogen (Carlsbad, CA). Anti- Indole-amine-2, 3-dioxygenase (IDO), -CD25 and -IgG Texas Red antibodies were purchased from Abcam Inc. (Cambridge, MA). Anti-iNOS polyclonal antibodies were purchased from Upstate (Charlottesville, VA).
Animals
Female Lewis rats (Harlan Laboratory, Harlan, IN) weighing 225–250 g were housed in the animal care facility of the Medical University of South Carolina (MUSC), throughout the experiment and provided with food and water ad libitum. All experiments were performed according to the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals (NIH publication number 80-23, revised 1985) and were approved by the MUSC Animal Care and Use Committee.
EAE Induction and clinical evaluation
The procedure used for the induction of EAE has been described previously (Paintlia, et al., 2004). In brief, female rats received a subcutaneous injection in the hind limb of MBP (50 μg) in 0.1 ml of phosphate-buffered saline (PBS; pH 7.4) emulsified in an equal volume of CFA supplemented with 2 mg/ml of mycobacterium tuberculosis H37Ra (Difco, Detroit, MI) on days 0 and 7. Immediately thereafter and again 24 h later, rats received pertussis toxin (200 ng, intraperitoneally, ip) in 0.1 ml of PBS. Individual rats were observed daily and clinical scores were assessed by an experimentally blinded investigator using a 0 to 5 scale: 0, no clinical score; 1, piloerection; 2, loss in tail tonicity; 3, hind leg paralysis; 4, paraplegia, and 5, moribund or dead.
Lovastatin and rolipram treatments
Lovastatin (4 mg/ml) was dissolved in 0.8% of ethanol/0.6 N NaOH and adjusted to pH 7.4 and rolipram (2 mg/ml) was dissolved initially in 100 μl of DMSO and finally suspended in PBS (pH 7.4). Different doses of lovastatin (2 and 5 mg/kg, ip) and rolipram (2, 5, and 7.5 mg/kg, sc) were administered daily starting from the day of immunization in separate groups to determine their protective optimal doses to blunt EAE. For combination treatments, suboptimal doses of both drugs i.e., lovastatin (1 mg/kg) and rolipram (1 mg/kg) were administrated daily and every other day, respectively. Control EAE rats received an injection of vehicle (placebo, 0.8% ethanol in PBS, ip), daily. Drug treatment with suboptimal doses of lovastatin and rolipram in combination or separately was started either on the day of immunization or 10th days of postimmunization (dpi; animal with clinical score [CS] ≥2.0) and continued throughout the study. Corresponding control rats received an emulsion of CFA/PBS into their hind limb foot pads and a daily injection of vehicle (ip) or lovastatin (1 mg/kg) and rolipram (1 mg/kg) as described above as a drug control. Rats were sacrificed on the peak day of the disease (15th day post immunization, dpi) to collect sera and spinal cords (SC, lumbar region). Rats that developed severe EAE (CS; >4.5) in drug- or placebo-treated groups were euthanized on the same day as per the animal protocol approved by the animal care and use committee. Nine rats per treatment group were used in each experiment for analysis.
Neuropathological Evaluations
Because the most significant histopathological changes in animals with EAE are detected in the lumbar region of the SC (Eng, et al., 1989), this portion of the SC was removed for analysis. At the time of sacrifice, the rats were anesthetized with pentobarbital (65 mg/kg, ip) and perfused transcardially with 4% ice-cold paraformaldehyde in 0.1 M phosphate buffer (pH 7.2). The SC was removed from the column, cut to collect the lumbar region and then paraffin-embedded. Transverse sections (5 μm) were cut and four consecutive slices were placed on the same glass slide. A series of sections was stained with hematoxylin and eosin (H&E), Luxol Fast Blue (LFB) and Bielschowsky silver to detect inflammatory infiltrates, demyelination, and axonal loss, respectively. Neuropathological data were quantified on 6–8 sections per rat. Sections were mounted with Vectashield, a aqueous mounting media (Vector Labs, Burlingame, CA), examined under a light microscope (Olympus BX-60, Goleta, CA) and images were captured with an Olympus digital video camera (Optronics; Goleta, CA) using a dual band pass filter using Adobe Photoshop 7 software. The number of perivascular inflammatory infiltrates was calculated and expressed as the number of inflammatory infiltrates per section. Histological scores of the degree of demyelination and axonal loss of each rat were evaluated by two investigators blinded to the treatment, using a semi-quantitative system described in the literature (Giuliani, et al., 2005). Briefly, a score of zero indicates no disease; grade 1 refers to foci of demyelination/axonal loss, which is superficial and involves less than 25% of the lateral columns; grade 2 denotes deep foci that involve over 25% of the lateral columns; and grade 3 denotes diffuse and widespread demyelination/axonal loss.
Immunohistochemistry
For the immunohistochemistry analysis, sections were blocked with a serum-PBS solution and incubated with overnight at 4 °C with the primary antibody at the optimized working dilution prepared in PBS 0.1 M (pH 7.4) with triton (0.3%) and bovine serum albumin (BSA, 5 mg/ml). On the second day, the sections were incubated for 1 h with the secondary antibody (anti-IgG conjugated with Texas red) prepared in PBS 0.1 M plus BSA (1 mg/ml). Sections were also incubated with Texas red-conjugated IgM without primary antibody as negative controls and an appropriate mouse IgG and rabbit polyclonal IgG were used as isotype controls. After thorough washings, slides were mounted with aqueous mounting media and analyzed by immunofluorescence microscopy and images were captured by using Olympus digital camera as described above.
RNA extraction, cDNA synthesis and quantitative real time-PCR (QRT-PCR) analysis
Lumbar SC tissues were carefully processed for RNA isolation using ‘TRIZOL’ reagent according to the manufacturer’s protocol as described previously (Paintlia, et al., 2004). Single-stranded cDNA was synthesized from SC tissue RNA from each group of animals using a superscript pre-amplification system for first-strand cDNA synthesis (BIO-RAD Laboratories, Hercules CA). QRT-PCR was performed using the BIO-RAD Laboratories iCycler iQ Real-Time PCR Detection System. Primers for target genes (Table 1) were designed using Primer quest software available free at website www.idtdna.com and synthesized from integrated DNA technologies (IDT, Coralville, IA). For ORT-PCR reaction supermix (IQ™ SYBR Green) was purchased from BIO-RAD (BIO-RAD Laboratories, Hercules CA). Thermal cycling conditions were as follows: activation of iTaq™ DNA polymerase at 95 °C for 10 min, followed by 40 cycles of amplification at 95 °C for 30 s and 58–60 °C for 1 min. The detection threshold was set above the mean baseline fluorescence determined by the first 20 cycles. Amplification reactions in which the fluorescence increased above the threshold were defined as positive. A standard curve for each template was generated using a serial dilution of the template (cDNA). Specificity of assay was determined by melting-curve analysis in each experimental run of QRT-PCR. The quantities of target gene expression were normalized to the corresponding GAPDH or 18S rRNA expression in test samples and data is presented as arbitrary units (au) in Tables 2 and 3)
Table 1.
Genes and DNA sequences of primers i.e., forward primer (FP) and reverse primer (RP) used for QRT-PCR amplification
| Gene Name | Primers |
|---|---|
| GAPDH | FP: 5′-cctacccccaatgtatccgttgtg-3′; RP: 5′-ggaggaatgggagttgctgttgaa-3′ |
| 18S rRNA | FP: 5′-ccagagcgaaagcatttgccaaga-3′; RP: 5′-tcggcatcgtttatggtcggaact-3′ |
| IL-23 | FP: 5′-atcaccactgggagactcaacaga-3′; RP: 5′-tgcgaaggatcttggaacggagaa-3′ |
| IL-17 | FP: 5′-actcagctgaaaacgctgaggaaa-3′; RP: 5′-tgtgcacaccttactgagagacct-3′ |
| IFN-γ | FP: 5′-atttccctccccactccattag-3′; RP: 5′-ctggtgacagctggtgaatca-3′ |
| IL-1β | FP: 5′-gagagacaagcaacgacaaaatcc-3′; RP: 5′-ttcccatcttcttctttgggtattg-3′ |
| TNF-α | FP: 5′-cttctgtctactgaacttcggggt-3′; RP: 5′-tggaactgatgagagggagcc-3′ |
| iNOS | FP: 5′-ggaagaggaacaactactgctggt-3′; RP: 5′-gaactgagggtacatgctggagc-3′ |
| IL-4 | FP: 5′-ggtatccacggatgtaacgacagc-3′; RP: 5′-ccgtggtgttccttgttgccgtaa-3′ |
| IL-10 | FP: 5′-ctgtcatcgatttctccctgtgag-3′; RP: 5′-tgagtgtcgcgtaggcttctatgc-3′ |
| TGF-β1 | FP: 5′-tgatacgcctgagtggctgtcttt-3′; RP: 5′-aagcgaaagccctgtattccgtct-3′ |
| CD4 | FP: 5′-aggtctcccttcagtttgctggtt-3′; RP: 5′-tcaccaccaggttcacttcctgat-3′ |
| CD8 | FP: 5′-aaagcaagacctggaccaacgaga-3′; RP: 5′-taacgtgcctgaccattcacagga-3′ |
| CD25 | FP: 5′-ccaaacgcaagccaaaccaaacag-3′; RP: 5′-acacgaggctgacggtcaacataa-3′ |
| Foxp3 | FP: 5′-agagtttctcaagcactgccaagc-3′; RP: 5′-tgcatagctcccagcttctccttt-3′ |
| IDO | FP: 5′-aagcactggagaaggcactgtgta-3′; RP: 5′-atccacgaagtcacgcatcctctt-3′ |
| ICAM-1 | FP: 5′-gtccaattcacactgaatgccagc-3′; RP: 5′-ttaaacaggaactttcccgccacc-3′ |
| VCAM-1 | FP: 5′-gacaccgtcattatctcctgcact-3′; RP: 5′-gtgtacgagccatccacagacttt-3′ |
| MCP-1 | FP: 5′-gaccagaaccaagtgagatca-3′; RP: 5′-gcttcagatttatgggtcaagt-3′ |
| CCR-2 | FP: 5′-tctacttcttctggactccataca-3′; RP: 5′-ctaagtgcatgtcaaccacac-3′ |
Enzyme–linked immunosorbant assay (ELISA)
Anti-MBP specific IgG isotypes were detected in serum samples by solid phase ELISA. Briefly, plates were coated with MBP (2 μg/ml) diluted in PBS overnight in a humidified chamber followed by washing with PBS containing 0.05% Tween 20 and blocking for 1 h with 1% BSA in PBS before the addition of serum samples. Samples were diluted 1:100 in PBS following 2 h incubation at RT and then plates were washed with PBS containing 0.05% Tween 20 to remove any unbound primary antibody. Bound antibody was detected by incubation with alkaline phosphatase-labeled rat anti-mouse IgG1, anti-mouse IgG2a and anti-mouse IgG2b (1:2,000) from Serotec (Raleigh, NC) using p-nitrophenyl phosphate (Sigma-Aldrich, St. Louis, MO) in 0.1 M glycine buffer as a substrate. Absorbance was read at 405 nm in a microplate spectrophotometer (Bio-Tek Instruments, Winooski, VT). SC tissue homogenates were prepared in PBS with a tissue homogenizer, Ultra-Turbax (JK Lab, Germany) and centrifuged at 12,000 × g for 15 min at 4 °C to collect supernatants. Interleukin (IL)-4 and IL-10 OptEIA, ELISA kits (BD Biosciences, San Jose, CA) were used with a sandwich ELISA method (using the manufacturer’s protocol) to detect their levels in SC supernatants. Similarly, IL-17, interferon (IFN)-γ and tumor growth factor (TGF)-β1 were detected in the supernatant of SC homogenate using ELISA kits purchased from Rapidbio (Rapidbio sales, West Hills, CA). Data are computed as concentration of cytokine protein/mg of SC tissue protein or serum.
Immunoblotting
SC tissues were homogenized in ice-cold lysis buffer (50 mm Tris-HCl, pH 7.4, containing 50 mM NaCl, 1 mM EDTA, 0.5 mM EGTA, 10% glycerol, and protease inhibitor mixture) and sample protein concentration was determined with Bradford reagent (Bio-Rad, Hercules, CA). SDS-PAGE, Western blotting and immunoblotting were performed as described previously (Paintlia, et al., 2004). Autoradiographs of immunoblots were generated using enhanced chemiluminescence detection kits (Amersham Biosciences, Piscataway, NJ).
Statistical analysis
Using a student’s unpaired t-test for two data points and one-way ANOVA (student-Newman-Keuls to compare all pairs of columns) for multiple data points, p-values were determined for clinical scores, neuropathological scores, QRT-PCR analysis and ELISA in triplicate from three independent experiments using GraphPad software (GraphPad Software Inc. San Diego, CA). The criterion for statistical significance was p <0.05.
RESULTS
Combination therapy with suboptimal doses of lovastatin and rolipram are complementary in EAE
We first evaluated the therapeutic efficacy of both drugs i.e., lovastatin and rolipram to determine their optimal doses for blunting the progression of EAE. Clinical signs of EAE were evident in MBP-immunized and vehicle-treated rats from the 8–9th dpi onwards, followed by acute disease resulting in 80–90% mortality (clinical score (CS) ≥4.5) by the 13–15th dpi (Fig. 1A). Remaining rats in a vehicle-treated group had CS ≥4.0 till 20th dpi (Fig. 1A) and euthanized per animal protocol guidelines. Consistent with previous reports, individually administered optimal doses of lovastatin and rolipram were ≥ 2 mg/kg (Paintlia, et al., 2004) and ≥5 mg/kg (Genain, et al., 1995), respectively, to attenuate the progression of EAE (Fig. 1A–B). Of note, the 2 mg/kg dose of rolipram, however, attenuated the progression of EAE (Fig. 1B), but a complete return to normal was delayed until the 30th dpi. Results of suboptimal doses of lovastatin (1 mg/kg) and rolipram (1 mg/kg) in combination were impressive when compared with their individual performances at optimal doses (Fig. 1A–C). Interestingly, combination treatment delayed the onset and limited the severity of EAE (CS; ≤3.0) when started from the day of immunization (0th dpi, Fig. 1C), and disease improvement was sooner (17th dpi) than with individually administered optimal doses (19th dpi) (Fig. 1A–C). Of note, the suboptimal dose of lovastatin was more effective than rolipram (mortality, 70%), when administered individually from 0th dpi (Fig. 1C). Similar to the combination of lovastatin with rolipram, another inhibitor of PDE-4 (PDI-4, 1 mg/kg) also behaved similarly when used in combination with lovastatin (1 mg/kg) in this setting (Fig. 1C), thus substantiating the significance of combining of these drugs for CNS demyelinating diseases. Similar to rolipram, PDI-4 is the potent inhibitor phosphodiesterase IV and metabolites of this compound are not toxic as mentioned in product safety data sheet. Rolipram reported to have shorter half life 1–2 hrs in vivo (Krause, et al., 1989), but no such information for PDI-4 is currently available. Corresponding with clinical symptoms, disease-mediated weight loss in EAE rats was inhibited by the combination of lovastatin and rolipram (data not shown). We observed no antagonism between rolipram and lovastatin, although reports in literature suggest that certain immunomodulatory agents act antagonistically (Brod, et al., 2000).
Figure 1. Combination therapy with suboptimal doses of lovastatin and rolipram are complementary in EAE.

EAE was induced in female Lewis rats using guinea pig MBP antigen emulsified in CFA and treatment with lovastatin (LOV) and rolipram (RLP) in combination or individually was performed as described under Materials and Methods. Drug treatments were started from the day of immunization of rats with MBP and continued until the end of the study. (A) Plot depicts clinical score associated with EAE in treated/untreated rats with different doses of LOV. (B) Plot depicts clinical score associated with EAE in treated/untreated rats with different doses of RLP. (C) Plot depicts clinical score associated with EAE in treated/untreated rats with suboptimal doses of LOV and RLP in combination and individually including PDEI-4 (another inhibitor of phosphodiesterase-4). Vehicle (VEH)- and LOV plus RLP-treated rats immunized with CFA emulsion without MBP were referred to as CON (VEH) and CON (LOV + RLP), respectively. Data in plots are presented as Mean ± SD of 9 rats treated similarly in a group. Statistical significance indicated as ** p<0.01 and *** p<0.001 versus EAE (VEH).
Therapeutic treatment with combination of lovastatin and rolipram improves histological outcomes and impedes neurodegeneration
Generally, MS treatment is initiated after patients have developed clinical signs or brain lesions. Therefore, it is imperative to test a therapeutic regimen which can prevent EAE induction and effectively attenuate an established case of EAE. Next, we evaluated whether combination therapy of these drugs could inhibit established EAE. Drug treatment was initiated after the onset of EAE on 10th dpi when individual rat developed a CS ≥2.0. Combination therapy drastically inhibited EAE development, whereas no such inhibition of EAE was observed in rats treated with lovastatin or rolipram separately with the same dose (Fig. 2A).
Figure 2. Combined treatment of lovastatin and rolipram attenuates cellular infiltration and demyelination in the SC of EAE rats.
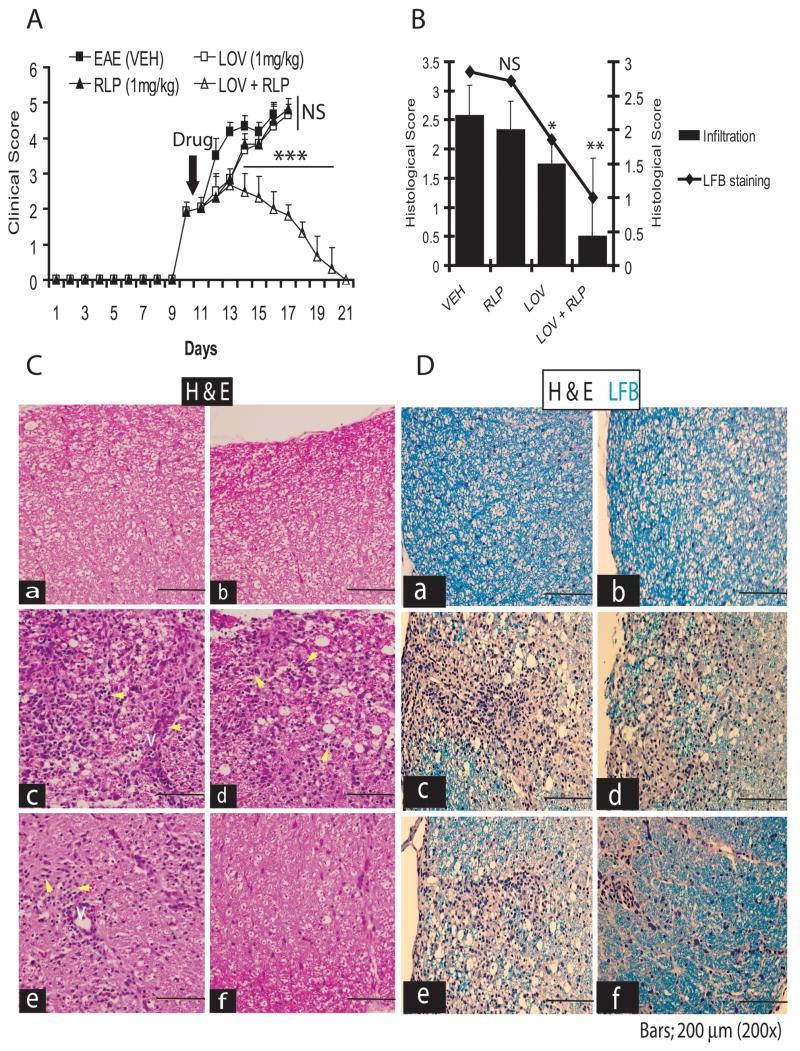
Treatment with lovastatin (LOV; 1 mg/kg) and rolipram (RLP; 1 mg/kg) in combination or individually was commenced in rats after the establishment of EAE (clinical score ≥2.0) as described under methods. Plot depicts clinical score in EAE animals treated with LOV and RLP in combination or individually (A). Plot depicts cellular infiltration and demyelination in the SC of EAE rats treated with LOV and RLP in combination or individually (B). Representative photograph of the lateral funiculus of SC stained with H&E (C) and both H&E and LFB (D) from each group of rats i.e., a) CON (VEH), b) CON (LOV + RLP), c) EAE (VEH), d) EAE (RLP), e) EAE (LOV) and f) EAE (LOV + RLP) demonstrates existing cellular infiltration and demyelination. Yellow arrowhead depicts cellular infiltration in the white mater and V indicates ‘vessel’ (C). Data in plots are expressed as Mean ± SD from three independent experiments (A) and histological images of SC (n = 9)/group (B). Statistical significance * p<0.05, ** p<0.01 and *** p<0.001 versus EAE (VEH).
Blinded analysis revealed that the number of infiltrates per section in rats on peak clinical day (15th dpi) was significantly reduced in rats treated with lovastatin and rolipram combination, compared to the vehicle-treated EAE rats (Fig. 2B). This reduction in the number of infiltrating inflammatory cells was also significant with lovastatin, but not with rolipram alone, compared to vehicle-treated EAE rats (Fig. 2B). The infiltration of inflammatory cells was specifically apparent in the white matter of the lateral and dorsal funiculi of the SC of EAE rats (Fig. 2C). Next, the presence of neuroinflammation and neurodegeneration was evaluated by LFB staining. Corresponding with cellular infiltration, combination therapy with lovastatin and rolipram significantly inhibited demyelination in the white matter of the SC of EAE rats, compared with vehicle-treated ones (Fig. 2B). Corresponding with cellular infiltration, there was a significant decrease in demyelination in the SC of EAE rats treated with lovastatin, but not with rolipram alone (Fig. 2B), compared to vehicle, but these effects were not as profound as those resulting from their combination. Representative sections depicting the reduction of cellular infiltration and demyelination in the lateral funiculi of the SC by combination therapy of lovastatin and rolipram in EAE rats compared with vehicle are displayed in Fig. 2D. Demyelination observed in the SC of EAE rats was further supported by immunoblotting for myelin protein i.e., MBP. Combination treatment with lovastatin and rolipram attenuated the inflammation-mediated breakdown of the myelin sheath in the SC of EAE rats (Fig. 3A). But this reduction of myelin breakdown was partial in the SC of EAE rats treated with lovastatin or rolipram alone (Fig. 3A). Likewise, axonal loss was markedly reduced by combination treatment with lovastatin and rolipram in the SC of EAE rats, compared with vehicle (Fig. 3B). The axonal loss was however, significantly attenuated by lovastatin and rolipram alone in EAE rats, compared with vehicle, but this reduction was not as impressive as that was resulting from their combination (Fig. 3B). In addition to the quantification of axonal loss, silver impregnation of axons also revealed the improved integrity of white matter in the affected lateral funiculi of SC sections of EAE rats (Fig. 3C). Taken together these data provide evidence that combination therapy with lovastatin and rolipram attenuates CNS invasion and subsequently impedes neurodegeneration in the EAE rats.
Figure 3. Attenuation of axonal loss and demyelination in the SC of EAE rats by combination therapy of lovastatin and rolipram.

Treatment with lovastatin (LOV; 1 mg/kg)) and rolipram (RLP; 1 mg/kg) in combination or individually was commenced in rats after the establishment of EAE (clinical score ≥2.0) as described under Methods. Autoradiograph of immunoblotting depicts level of MBP isoforms in the SC of treated/untreated control and EAE animals (A). Plot depicts histological score of axonal loss in the white matter of lateral funiculi of SC of EAE rats treated with drugs in combination or individually or with vehicle (VEH) only (B). Representative photograph of the lateral funiculus of SC demonstrate impregnation of axons by Beilschowsky’s silver staining (C). Labeling of micrographs indicates a) CON (VEH), b) CON (LOV + RLP), c) EAE (VEH), d) EAE (RLP), e) EAE (LOV) and f) EAE (LOV + RLP) in both B and D. Data in plot is presented as Mean ± SD from three independent experiments with 9 rats/group and histological images of SC (n = 9)/group. Statistical significance * p<0.05, ** p<0.01 and *** p<0.001 versus EAE (VEH).
Combination treatment of lovastatin and rolipram attenuates infiltration of mononuclear cells and improves endothelial functions
Because the observed histological changes and neurodegeneration in the MS brain are associated with the invasion of the CNS by invading cells including myelin-reactive T cells (CD4+ and CD8+) and monocytes (CD11b+) (Hafler, 2004), we next examined SC tissues for the presence of these cells using QRT-PCR. As expected, the transcripts for CD4, CD8, and CD11b proteins were elevated in the SC of vehicle-treated EAE rats, suggestive of CNS invasion by autoreactive T cells and monocytes (Fig. 4A–B). Conversely, combination therapy with lovastatin and rolipram significantly attenuated this invasion of CNS by autoreactive T cells and monocytes as compared to vehicle (Fig. 4A–B). Of note, the observed effect of combination treatment with suboptimal dose of lovastatin and rolipram to attenuate the CNS invasion was better than their same doses when used separately (Fig. 4A–B).
Figure 4. Expression of transcripts for inflammatory infiltrates, adhesion molecules and chemokines including receptors in the SC of EAE rats treated with lovastatin and rolipram in combination or individually.

Plots depict the level of transcripts A; CD4 and CD8 T cells, B; CD11b monocytes, C; ICAM-1 and VCAM-1, and D; MCP-1 and CCR-2 in the SC of EAE (n = 9) and control (n = 6) rats treated with lovastatin (LOV) and rolipram (RLP) in combination or individually as determined by QRT-PCR as described under methods. Data in plots are expressed as Mean ± SD from three independent experiments. Statistical significance * p<0.05, ** p<0.01, *** p<0.001 and ‘NS’ (not-significant) versus EAE (VEH).
CNS invasion is considered to be facilitated via interaction between adhesion molecules (ICAM-1 and VCAM-1) on endothelial cells and LFA-1 on leukocytes including the release of chemokines (MCP-1) and expression of their receptors (CCR2) on invading leukocytes (Dopp, et al., 1994). Therefore, we next examined the expression of these mediators in the SC involved in the CNS invasion and endothelial dysfunction. Corresponding with cellular infiltration, the increased level of transcripts for ICAM-1, VCAM-1, MCP-1, and CCR2 were significantly attenuated by combination treatment with lovastatin and rolipram in the SC of EAE rats compared to vehicle (Fig. 4C–D). Of note, an increased level of MCP-1 and CCR2 transcripts in vehicle-treated EAE rats is comparable to their previously documented mRNA levels and immunohistochemistry staining data in the SC of EAE rats as reported earlier (Paintlia, et al., 2004). It is noteworthy that the reduction of these mediators in the SC of EAE rats was reduced to 50% and 20% by individually administered lovastatin and rolipram, respectively, compared with vehicle, but it was not as profound as that observed with their combination (Fig. 4A–D). Together, these data suggest that the combination therapy of lovastatin and rolipram reverses EAE pathogenesis via attenuation of cellular infiltration and improvement of endothelial function in the CNS.
The combination of lovastatin and rolipram promotes a bias towards TH2 immunity
Previously, we had documented that transcripts of immune cells associated with TH1 immune responses were elevated in the SC of EAE rats (Paintlia, et al., 2004). Recent studies revealed that an IL-23-induced subset of T cells producing IL-17 (TH17) is involved in EAE progression (Kleinschek, et al., 2007). Interestingly, combination treatment with lovastatin and rolipram significantly reduced transcripts for pro-inflammatory cytokines associated with EAE development i.e., IL-23, IL-17, IFN-γ, TNF-α, and IL-1β in the SC of EAE animals compared with vehicle (Fig. 5A–B). In addition, the level of transcription factor, retinoic acid orphan receptor gamma t (RORγt), involved in the expression of IL-17 by TH17 cells was also reduced significantly by combination therapy compared to vehicle (Fig. 5B). ELISA-based assays further supported these observations and showed the significant reduction of IFN-γ (Fig. 6A) and IL-17 (Fig. 6B) proteins in the SC of EAE rats by combination therapy compared with vehicle. Likewise, iNOS transcripts (Fig. 5A) and protein (Fig. 6C–D) were reduced significantly in the SC of EAE rats by drug combination therapy compared to vehicle. Of note, no significant reduction of these mediators was observed in the SC of EAE rats treated with rolipram alone when compared with vehicle (Fig. 5A–B and Fig. 6A–D). Although expression of these mediators in the SC of EAE rats treated with lovastatin alone was significantly reduced when compared with vehicle, this reduction was not as profound as that observed when lovastatin was combined with rolipram (Fig. 5A–B and Fig. 6A–D).
Figure 5. Level of transcripts for pro-inflammatory and anti-inflammatory mediators in the SC and anti-MBP immunoglobulin isotypes in the serum of EAE rats treated with lovastatin and rolipram in combination or individually.

Plots depict the level of transcripts A; IFN-γ, TNF-α, IL-1β and iNOS, B; IL-23, IL-17, RORγt and CD25, C; IL-4, IL-10 and TGF-β1, and D; Foxp3 and IDO in the SC of EAE (n = 9) and control (n = 6) rats treated with lovastatin (LOV) and rolipram (RLP) in combination or individually as determined by QRT-PCR as described under methods. Plot depicts the level of ant-MBP immunoglobulin isotypes in the serum of EAE (n = 9) and control (n = 6) rats treated with LOV and RLP in combination or individually as determined by ELISA-based assay described under methods (E). Data in plots are expressed as Mean ± SD from three independent experiments. Statistical significance * p<0.05, ** p<0.01, *** p<0.001 and ‘NS’ (not-significant) versus EAE (VEH).
Figure 6. Protein level of pro-inflammatory and anti-inflammatory mediators in the SC of EAE rats following treatment with lovastatin and rolipram in combination or individually.

Plots depict the level of IFN-γ (A), IL-17 (B), IL-4 (E) and IL-10 (F) in the SC homogenate of EAE (n = 9) and control (n = 6) rats treated with lovastatin (LOV) and rolipram (RLP) in combination or individually as determined by ELISA-based assay described under methods. Autoradiograph (C) and plot (D) depict immunoblotting of iNOS protein in the SC of EAE rats in each group. Data in plots are expressed as Mean ± SD from three independent experiments. Statistical significance * p<0.05, ** p<0.01 and ‘NS’ (not-significant) versus EAE (VEH).
Because TH2 immunity play an important role in the suppression of autoimmune disease i.e., EAE (O’Garra, et al., 1997), we next measured TH2 cytokines i.e., IL-4 and IL-10 in the SC. Combination treatment with lovastatin and rolipram significantly increased IL-4 and IL-10 transcripts (Fig. 5C) and protein (Fig. 6E–F) in the SC of EAE rats compared to vehicle. These cytokines, however, were elevated significantly in the SC of lovastatin-treated EAE rats, especially IL-10 when compared with vehicle, but this was not as profound as that observed with its combination with rolipram (Fig. 5C and Fig. 6E–F). No significant change in these cytokines was observed in the SC of rolipram-treated EAE rats compared to vehicle (Fig. 5C and Fig. 6E–F).
The above data imply that combination therapy may additively modulate T-cell function in vivo. The reduction in SC inflammation is suggestive of its potential for selective reduction in the TH1 cell reactivity that primarily mediates the immune response in the periphery generally associated with EAE progression. If this is the case, a consequence of combination therapy may be a bias toward TH2 immunity. Because the TH1 immunity predominantly elicits IgG2a, whereas TH2 immunity produces higher levels of IgG1 in mice (Hooper, et al., 1998), we next assessed whether combination therapy influences humoral immune response to MBP in the peripheral immune system in EAE rats. Anti-MBP antibodies of the IgG2a subclass that were elevated in the sera of vehicle-treated EAE rats were significantly reduced by combination therapy of these drugs (Fig. 5E). Conversely, anti-MBP antibodies of IgG2b and IgG1 subclasses were significantly elevated in the sera of EAE rats treated with these drugs in combination, compared with vehicle (Fig. 5E). No such change was observed in the level of IgG2a in the sera of EAE rats treated with lovastatin or rolipram alone when compared with vehicle (Fig. 5E). IgG1 and IgG2b were significantly elevated in EAE rats treated with lovastatin but not in those treated with rolipram alone, compared with vehicle (Fig. 5E). Altogether, these data suggest that combination therapy of lovastatin and rolipram attenuates inflammatory TH1 and TH17 immune responses and causes a bias towards TH2 immunity in the peripheral and CNS.
The combination of lovastatin and rolipram promotes expansion of T regulatory (Treg) cells and immune tolerance in the CNS
Since T regulatory (Treg) cells play an important role in the suppression and reversal of EAE (Yu, et al., 2005), we next measured the level of transcripts for CD25 and its transcription factor, Forkhead box P3 (Foxp3) proteins as a marker of Treg (CD25+/Foxp3+) cells in the CNS. Combination treatment with lovastatin and rolipram markedly increased CD25 transcript (Fig. 5B) and protein (Fig. 7A) including Foxp3 transcript (Fig. 5D) in the SC of EAE rats compared to vehicle. Furthermore, TGF-β1 transcripts (Fig. 5C) and protein (Fig. 7B) were also increased significantly in the SC of EAE rats by combination therapy compared to vehicle. No change in the expression of these mediators was observed upon treatment with rolipram in the SC of EAE rats compared to vehicle except CD25 (Fig. 5B–D and Fig. 7A–B). Although, the expression of these mediators in the SC of EAE rats treated with lovastatin was significantly elevated when compared with vehicle, this change was not as dramatic as that observed when lovastatin was combined with rolipram (Fig. 5B–D and Fig. 7A–B).
Figure 7. Level of proteins associated with Treg cells and immunotolerance in the SC of EAE rats.

Immunoblot depicts CD25, IDO, and β-actin in the SC of EAE animals treated/untreated with LOV and RLP in combination or individually (A). Plot depicts TGF-β1 level in the SC homogenate of EAE (n = 9) and control (n = 6) animals treated with lovastatin (LOV) and rolipram (RLP) in combination or individually as determined by ELISA-based assay described under methods (B). Representative photograph of the lateral funiculus of SC of EAE animals treated with LOV and RLP i.e., a) CON (VEH), b) CON (LOV + RLP), c) EAE (VEH), d) EAE (RLP), e) EAE (LOV) and f) EAE (LOV + RLP) demonstrating the expression of IDO in the SC (C). Data in plot is expressed as Mean ± SD from three independent experiments. Statistical significance * p<0.05, ** p<0.01 and ‘NS’ (not-significant) versus EAE (VEH).
The increased expression of indole-amine-2, 3-dioxygenase (IDO) in dendritic cells is important for the suppression of EAE (Kwidzinski, et al., 2005) via suppression of T-cell responses and induction of immune tolerance (Mellor and Munn, 2004); thus, we next determined the status of IDO (transcript and protein) in the CNS. Combination treatment with lovastatin and rolipram significantly increased IDO transcripts in the SC of EAE rats compared with vehicle (Fig. 5D). These results were further supported by immunoblotting of IDO protein (Fig. 7A) and immunohistochemistry of transverse SC sections (Fig. 7C). IDO immunostaining was evident in the dorsal funiculi of the SC of EAE rats treated with lovastatin and rolipram in combination (Fig. 7C). The level of IDO transcripts (Fig. 5D) and protein (Fig. 7A and C) in the SC of EAE rats treated with lovastatin was significantly elevated when compared with vehicle, but it was not as impressive as that observed with the drug combination. No significant change in IDO transcripts (Fig. 5D) and protein (Fig. 7A and C) was observed in the SC of EAE rats treated with rolipram alone when compared with vehicle. Together, these data imply that combined therapy of lovastatin and rolipram also promotes Treg expansion and immune tolerance in the CNS as mechanism of protection to suppress EAE.
DISCUSSION
Several reports suggest that combination therapy with existing or novel MS therapeutics in the treatment of MS have better clinical outcomes than with monotherapy. Combination therapy is considered to be advantageous if both drugs (a) have different mechanisms of action, (b) have excellent safety profiles, and (c) have no additional toxicities when used in combination for additive or synergistic effects (Stuve, et al., 2006). In this regard, statins (Paintlia, et al., 2004, Youssef, et al., 2002) and rolipram (Bielekova, et al., 2000, Sommer, et al., 1995) meet these criteria and both characteristically prevent/ameliorate EAE. Also, both statins (Johnson-Anuna, et al., 2005) and rolipram cross the BBB (Krause and Kuhne, 1988) to interfere in the neurodegenerative process in the CNS. In this study, we show that combined treatment with lovastatin and rolipram, compared to individual drug treatment, was associated with significant suppression of EAE severity via inhibition of cellular infiltration and improved endothelial function in the CNS. The attenuation of inflammatory TH1 and TH17 immune responses and induction of biased TH2 immunity was apparent in the peripheral and CNS including the expansion of Treg cells and immune tolerance. Subsequently, it was associated with profound reduction of demyelination and axonal loss in the CNS.
The EAE model is useful for initial testing of potential combination therapies for MS. Combination therapies are tested in animals (Brod, et al., 2000, Soos, et al., 2002) and humans (Dhib-Jalbut, et al., 2002, Ytterberg, et al., 2007), including phase-I clinical trials (Jeffery, et al., 2005, Rudick, et al., 2006) with major FDA-approved immunomodulators including IFN-β and GA in the MS regimen. Moreover, other combination therapies are also being investigated in MS and EAE with FDA-approved therapies and other novel therapeutics for MS including statins and minocycline (Giuliani, et al., 2005, Luccarini, et al., 2008, Stuve, et al., 2006). Results of a recently conducted phase 1 trial of IFN-β and atorvastatin combined therapy in MS patients are quite impressive and provide a rationale for conducting phase II/III trials (Paul, et al., 2008). In vitro studies revealed that immunomodulatory actions of statins are comparable to IFN-β (Neuhaus, et al., 2005). Considering statins to be future MS therapeutics, we previously demonstrated an immunomodulatory synergy with lovastatin and another novel therapeutic drug, 5-aminoimidazole-4-carboxamide-1-beta-D-ribofuranoside in EAE (Paintlia, et al., 2006). Overall, these studies support the notion that immunomodulatory agents with different mechanisms of action can be combined for the treatment of MS and provide a rationale for testing the combination of lovastatin plus rolipram. The present study establishes that the combining of suboptimal doses of lovastatin and rolipram provides better outcomes by impeding EAE severity and neurodegeneration than their individually administered doses. In particular, despite significant reduction of the some of the inflammatory mediators by an individually administered dose of these drugs especially lovastatin, the disease severity was not attenuated thereby suggestive of the potential benefits of combination therapy over monotherapy for treating CNS demyelinating diseases. Of note, the combined therapy with suboptimal doses of these drugs was better than their optimal individually administered doses. The observed effects of these drugs in combination are complementary to their immunomodulatory and neuroprotective activities as discussed below.
In organ-specific autoimmunity, the balance of cytokines is a key determinant of resistance or susceptibility. In EAE, disease susceptibility is thought to correlate with the expression of proinflammatory cytokines such as IL-23, IFN-γ, TNF-α, IL-6, and IL-1β including iNOS. Conversely, TH2 cytokines such as IL-4, IL-10 and IL-13 are important for preventing or ameliorating EAE (O’Garra, et al., 1997). IFN-γ producing cells can induce EAE, and their presence within the CNS likely contributes to acute inflammation, whereas the IL-17 immune pathway is suggested to play an important role in the disease process (Kleinschek, et al., 2007). Inflammation is considered to be the cause of tissue damage as a result of CNS invasion by inflammatory infiltrates. Combined treatment with lovastatin and rolipram significantly attenuated the infiltration of inflammatory cells and induced a biased anti-inflammatory TH2 immunity which attenuates peripheral and central inflammation through bystander suppression. Consistent with these findings, statins were previously shown to attenuate EAE development by promoting a TH1 to TH2-biased immune responses (Paintlia, et al., 2004, Youssef, et al., 2002). Moreover, a recent study demonstrated that statins can inhibit IL-17 transcription and secretion by human CD4+ T cells (Zhang, et al., 2008). Similar to statins, rolipram has been reported to attenuate the induction of chronic EAE (Bielekova, et al., 2000, Genain, et al., 1995) via immunomodulation of TH1 to TH2 immune responses (Abbas, et al., 2000, Bielekova, et al., 2000). It is worth mentioning that statins act on multiple targets in EAE modulation. Statins, by inhibiting HMG-CoA reductase, play a critical role in the mevalonate pathway that regulates the synthesis of cholesterol, and they interact with isoprenoids which mediate the membrane association of many GTPases (Zhang and Casey, 1996). Depletion of isoprenoid intermediates in vivo by statins, promotes a TH2 bias by reducing the membrane association of Ras and RhoA, which have an important role in extracellular signal-regulated kinases and p38 kinases (Dunn, et al., 2006).
An invasion of the CNS by autoreactive T cells and monocytes is facilitated by the breaching of the BBB and the expression of adhesion molecules in endothelial cells during EAE progression (Eralinna, et al., 1996). We and others previously documented that statins inhibit the infiltration of inflammatory cells into the CNS (Stanislaus, et al., 2001, Walters, et al., 2002, Youssef, et al., 2002). Detailed studies revealed that the inhibitory effects of statins on the preservation of the BBB are mediated by attenuation of isoprenylation of endothelial cell proteins such as Rho GTPAses (Walters, et al., 2002). In agreement with these findings, combination therapy significantly reduced transcripts for T cells, monocytes, and adhesion molecules in the SC, suggesting improved endothelial functions and attenuation of CNS invasion (please see Fig. 4). The additional mechanism by which statins inhibit inflammatory cell transmigration through the BBB is by reducing the activity of matrix metalloproteinase (MMP) such as MMP-9. Statins are reported to attenuate the expression MMP-9 in endothelial cells by the RhoA/ROCK pathway through an isoprenoid-dependent mechanism (Turner, et al., 2005). Likewise, rolipram treatment has been shown to stabilize the BBB in EAE animals (Folcik, et al., 1999). In addition, rolipram has been shown to inhibit NF-κB and MMP-9 activities in activated T cells (Sanchez, et al., 2005).
Recent studies revealed that Tregs (CD25+Foxp3+ T regulatory) are important in maintaining self tolerance to inhibit the function of effector T cells especially during TH17 immune responses (Yu, et al., 2005). TGF-β1 is key cytokine in the generation of Treg cells and a recent study revealed that its secretion by neurons is important for enhancing Treg cell expansion in the CNS to suppress EAE progression (Liu, et al., 2006). Consistent with our previous documentation, combination therapy of lovastatin and rolipram showed increased expression of TGF-β1 in the CNS (Paintlia, et al., 2004). A very recent study demonstrated that statin treatment promotes the generation of Treg cells in treated peripheral naïve CD4+ T cells (Mausner-Fainberg, et al., 2008). Phosphodiesterase inhibitors are also reported to upregulate Treg cells in experimental autoimmune myasthenia gravis (Aricha, et al., 2006). Another important candidate, IDO (tryptophan metabolizing enzyme), in the dendritic cells is also reported to involved in the suppression of EAE progression (Kwidzinski, et al., 2005) via suppression of the proliferation of effector T cells and an induction of immune tolerance (Mellor and Munn, 2004). Combined treatment with lovastatin and rolipram significantly increased these mediators (transcripts and proteins) in the CNS of an established EAE case (Table 3, and Fig. 5). These observed effects of these drugs in combination whether resulting from the reduced inflammation by an effect on immune cells in the periphery or due to the modulation of immune cells in the CNS is needed to be ascertained. Overall, these findings suggest that the observed immunomodulatory activities of these drugs in combination are attributed in part to the induction of TH2 biased immunity and generation of Treg cells and immune tolerance.
Local antigen presentation is a critical requirement for the initiation and perpetuation of the chronic inflammatory response within the CNS. While the CNS is devoid of professional antigen presenting cells, MHC class II antigens and costimulatory CD80 and CD86 molecules are upregulated in microglia and macrophages in response to local cytokine production (Youssef, et al., 2002). CNS resident astrocytes can also upregulate MHC class II after activation by IFN-γ (Cornet, et al., 2000). Current studies revealed that peripherally-derived myeloid dendritic cells (mDCs) accumulate in the CNS during relapsing EAE (Bailey, et al., 2007), and mDCs localize in the central parts of the active lesions and induce IL-17 production by CD4 cells. This effect of T-cell differentiation mediated by mDCs in the CNS plays important role in the chronic inflammatory response. Lovastatin has been reported to inhibit the expression of TNF-α and IL-1β in microglia and astrocytes (Pahan, et al., 1997). Treatment of microglia with a statin attenuated IFN-γ inducible transcription of MHC class II expression (Kwak, et al., 2001). Likewise, rolipram has been reported to inhibit the secretion of TNF-α by macrophages and monocytes via blocking the degradation of cAMP (Torphy and Undem, 1991). In addition, rolipram has been shown to downregulate the expression of pro-inflammatory cytokine but upregulate the expression of anti-inflammatory cytokines in the CNS (Yoshikawa, et al., 1999). These effects of statins and rolipram on modulation of the inflammatory response in brain cells are critical for their neuroprotective activities in the CNS. In addition, statins and rolipram are reported to inhibit NMDA-induced neuronal cell death (Zacco, et al., 2003, Zou and Crews, 2006). Recent studies provide evidence that statins protect neuroprogenitor cells from inflammatory insult and enhance myelin repair in ameliorating EAE animals thereby suggesting that lovastatin may promote neurorepair in EAE (Lee, et al., 2004, Paintlia, et al., 2005, Paintlia, et al., 2008, Sim, et al., 2008). Likewise, PDE-4 inhibitor-mediated preservation of cAMP has been shown to protect neurons against β-amyloid-induced neurotoxicity (Echeverria, et al., 2005). In addition, cAMP has been implicated in the survival of hippocampal neurons in vitro under conditions of reduced energy availability during glucose deprivation and glutamate excitotoxicity (Culmsee, et al., 2001).
In conclusion, our study demonstrated for the first time that combination of lovastatin and rolipram provide better protection in EAE than either drug when used individually at suboptimal doses. The partial reduction of myelin breakdown and axonal degeneration in EAE animals treated with these drugs separately may be attributed to their partial inhibition of inflammatory response in the CNS. In addition, the consistently observed weaker effects of rolipram as a single therapy than lovastatin are in agreement with a previous report demonstrating rolipram induced strong preventive effects with limited therapeutic efficacy in EAE Lewis rats (Jung, et al., 1996). The major drawback of rolipram administration in humans could be its tendency to induce vomiting as observed in ferrets (Robichaud, et al., 2001). Importantly, it could be subjugated by an intramuscular injection of ondansetron hydrochloride dehydrate (0.3 mg/kg) 20 min prior to rolipram administration as reported earlier (Genain, et al., 1995). It is noteworthy that administration of ondansteron hydrochloride in EAE rats neither aggravates nor attenuates EAE disease associated symptoms (data not shown). Although other mechanisms may contribute to the additive/synergistic immunomodulatory activities of combined therapy of lovastatin and rolipram, as mentioned above, the observed effects of these drugs in combination are complementary. At this time, the precise mechanism and contribution of these drugs in combination in the attenuation of EAE pathogenesis is not fully understood. Overall, the combined therapy of lovastatin and rolipram is an excellent therapeutic approach for the treatment of MS. Future clinical trials are warranted to investigate the effect of these drugs in combination therapy in MS.
Acknowledgments
This study was supported by grants from the NIH (NS-22576, NS-34741, NS-37766, NS-40144, NS-038236, C06-RR015455, and C06-RR018823 and support from Merck & Company.
Abbreviations
- EAE
Experimental autoimmune encephalomyelitis
- MS
Multiple sclerosis
- SC
spinal cord
- CNS
central nervous system
- dpi
Days of post-immunization
- QRT-PCR
Quantitative real-time-polymerase chain reaction
- LOV
Lovastatin
- RLP
Rolipram
- PDE-4
Phosphodiesterase-4
- CS
Clinical score
- LFB
Luxol fast blue
- H & E
Hematoxlin and Eosin
- IFN
Interferon
- IL
Interleukin, TGF, Tumor growth factor
- ICAM
Intracellular adhesion molecule
- VCAM
Vascular adhesion molecule
- Foxp3
Forkhead box P3
- RORγt
retinoic acid orphan receptor gamma t
- IDO
Indole-amine-2,3-dioxygenase
- BBB
Blood brain barrier
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Abbas N, Zou LP, Pelidou SH, Winblad B, Zhu J. Protective effect of Rolipram in experimental autoimmune neuritis: protection is associated with down-regulation of IFN-gamma and inflammatory chemokines as well as up-regulation of IL-4 in peripheral nervous system. Autoimmunity. 2000;32:93–99. doi: 10.3109/08916930008994078. [DOI] [PubMed] [Google Scholar]
- 2.Arbizu T, Alvarez-Cermeno JC, Decap G, Fernandez O, Uria DF, Garcia Merino A, Izquierdo G, Montalban X. Interferon beta-1b treatment in patients with relapsing--remitting multiple sclerosis under a standardized protocol in Spain. Acta Neurol Scand. 2000;102:209–217. doi: 10.1034/j.1600-0404.2000.102004209.x. [DOI] [PubMed] [Google Scholar]
- 3.Aricha R, Feferman T, Souroujon MC, Fuchs S. Overexpression of phosphodiesterases in experimental autoimmune myasthenia gravis: suppression of disease by a phosphodiesterase inhibitor. Faseb J. 2006;20:374–376. doi: 10.1096/fj.05-4909fje. [DOI] [PubMed] [Google Scholar]
- 4.Bailey SL, Schreiner B, McMahon EJ, Miller SD. CNS myeloid DCs presenting endogenous myelin peptides ‘preferentially’ polarize CD4+ T(H)-17 cells in relapsing EAE. Nat Immunol. 2007;8:172–180. doi: 10.1038/ni1430. [DOI] [PubMed] [Google Scholar]
- 5.Bielekova B, Lincoln A, McFarland H, Martin R. Therapeutic potential of phosphodiesterase-4 and -3 inhibitors in Th1-mediated autoimmune diseases. J Immunol. 2000;164:1117–1124. doi: 10.4049/jimmunol.164.2.1117. [DOI] [PubMed] [Google Scholar]
- 6.Brod SA, Lindsey JW, Wolinsky JS. Combination therapy with glatiramer acetate (copolymer-1) and a type I interferon (IFN-alpha) does not improve experimental autoimmune encephalomyelitis. Ann Neurol. 2000;47:127–131. [PubMed] [Google Scholar]
- 7.Cornet A, Bettelli E, Oukka M, Cambouris C, Avellana-Adalid V, Kosmatopoulos K, Liblau RS. Role of astrocytes in antigen presentation and naive T-cell activation. J Neuroimmunol. 2000;106:69–77. doi: 10.1016/s0165-5728(99)00215-5. [DOI] [PubMed] [Google Scholar]
- 8.Culmsee C, Monnig J, Kemp BE, Mattson MP. AMP-activated protein kinase is highly expressed in neurons in the developing rat brain and promotes neuronal survival following glucose deprivation. J Mol Neurosci. 2001;17:45–58. doi: 10.1385/JMN:17:1:45. [DOI] [PubMed] [Google Scholar]
- 9.Dhib-Jalbut S, Chen M, Henschel K, Ford D, Costello K, Panitch H. Effect of combined IFNbeta-1a and glatiramer acetate therapy on GA-specific T-cell responses in multiple sclerosis. Mult Scler. 2002;8:485–491. doi: 10.1191/1352458502ms862oa. [DOI] [PubMed] [Google Scholar]
- 10.Dopp JM, Breneman SM, Olschowka JA. Expression of ICAM-1, VCAM-1, L-selectin, and leukosialin in the mouse central nervous system during the induction and remission stages of experimental allergic encephalomyelitis. J Neuroimmunol. 1994;54:129–144. doi: 10.1016/0165-5728(94)90239-9. [DOI] [PubMed] [Google Scholar]
- 11.Dunn SE, Youssef S, Goldstein MJ, Prod’homme T, Weber MS, Zamvil SS, Steinman L. Isoprenoids determine Th1/Th2 fate in pathogenic T cells, providing a mechanism of modulation of autoimmunity by atorvastatin. J Exp Med. 2006;203:401–412. doi: 10.1084/jem.20051129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Echeverria V, Clerman A, Dore S. Stimulation of PGE receptors EP2 and EP4 protects cultured neurons against oxidative stress and cell death following beta-amyloid exposure. Eur J Neurosci. 2005;22:2199–2206. doi: 10.1111/j.1460-9568.2005.04427.x. [DOI] [PubMed] [Google Scholar]
- 13.Eng LF, D’Amelio FE, Smith ME. Dissociation of GFAP intermediate filaments in EAE: observations in the lumbar spinal cord. Glia. 1989;2:308–317. doi: 10.1002/glia.440020504. [DOI] [PubMed] [Google Scholar]
- 14.Eralinna JP, Soilu-Hanninen M, Roytta M, Hukkanen V, Salmi AA, Salonen R. Blood-brain barrier breakdown and increased intercellular adhesion molecule (ICAM-1/CD54) expression after Semliki Forest (A7) virus infection facilitates the development of experimental allergic encephalomyelitis. J Neuroimmunol. 1996;66:103–114. doi: 10.1016/0165-5728(96)00031-8. [DOI] [PubMed] [Google Scholar]
- 15.Folcik VA, Smith T, O’Bryant S, Kawczak JA, Zhu B, Sakurai H, Kajiwara A, Staddon JM, Glabinski A, Chernosky AL, Tani M, Johnson JM, Tuohy VK, Rubin LL, Ransohoff RM. Treatment with BBB022A or rolipram stabilizes the blood-brain barrier in experimental autoimmune encephalomyelitis: an additional mechanism for the therapeutic effect of type IV phosphodiesterase inhibitors. J Neuroimmunol. 1999;97:119–128. doi: 10.1016/s0165-5728(99)00063-6. [DOI] [PubMed] [Google Scholar]
- 16.Genain CP, Roberts T, Davis RL, Nguyen MH, Uccelli A, Faulds D, Li Y, Hedgpeth J, Hauser SL. Prevention of autoimmune demyelination in non-human primates by a cAMP-specific phosphodiesterase inhibitor. Proc Natl Acad Sci U S A. 1995;92:3601–3605. doi: 10.1073/pnas.92.8.3601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Giuliani F, Fu SA, Metz LM, Yong VW. Effective combination of minocycline and interferon-beta in a model of multiple sclerosis. J Neuroimmunol. 2005;165:83–91. doi: 10.1016/j.jneuroim.2005.04.020. [DOI] [PubMed] [Google Scholar]
- 18.Hafler DA. Multiple sclerosis. J Clin Invest. 2004;113:788–794. doi: 10.1172/JCI21357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hooper DC, Morimoto K, Bette M, Weihe E, Koprowski H, Dietzschold B. Collaboration of antibody and inflammation in clearance of rabies virus from the central nervous system. J Virol. 1998;72:3711–3719. doi: 10.1128/jvi.72.5.3711-3719.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jeffery DR, Chepuri N, Durden D, Burdette J. A pilot trial of combination therapy with mitoxantrone and interferon beta-1b using monthly gadolinium-enhanced magnetic resonance imaging. Mult Scler. 2005;11:296–301. doi: 10.1191/1352458505ms1154oa. [DOI] [PubMed] [Google Scholar]
- 21.Johnson-Anuna LN, Eckert GP, Keller JH, Igbavboa U, Franke C, Fechner T, Schubert-Zsilavecz M, Karas M, Muller WE, Wood WG. Chronic administration of statins alters multiple gene expression patterns in mouse cerebral cortex. J Pharmacol Exp Ther. 2005;312:786–793. doi: 10.1124/jpet.104.075028. [DOI] [PubMed] [Google Scholar]
- 22.Jung S, Zielasek J, Kollner G, Donhauser T, Toyka K, Hartung HP. Preventive but not therapeutic application of Rolipram ameliorates experimental autoimmune encephalomyelitis in Lewis rats. J Neuroimmunol. 1996;68:1–11. doi: 10.1016/0165-5728(96)00051-3. [DOI] [PubMed] [Google Scholar]
- 23.Kleinschek MA, Owyang AM, Joyce-Shaikh B, Langrish CL, Chen Y, Gorman DM, Blumenschein WM, McClanahan T, Brombacher F, Hurst SD, Kastelein RA, Cua DJ. IL-25 regulates Th17 function in autoimmune inflammation. J Exp Med. 2007;204:161–170. doi: 10.1084/jem.20061738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Krause W, Kuhne G. Pharmacokinetics of rolipram in the rhesus and cynomolgus monkeys, the rat and the rabbit. Studies on species differences. Xenobiotica. 1988;18:561–571. doi: 10.3109/00498258809041693. [DOI] [PubMed] [Google Scholar]
- 25.Krause W, Kuhne G, Matthes H. Pharmacokinetics of the antidepressant rolipram in healthy volunteers. Xenobiotica. 1989;19:683–692. doi: 10.3109/00498258909042306. [DOI] [PubMed] [Google Scholar]
- 26.Kwak B, Mulhaupt F, Veillard N, Pelli G, Mach F. The HMG-CoA reductase inhibitor simvastatin inhibits IFN-gamma induced MHC class II expression in human vascular endothelial cells. Swiss Med Wkly. 2001;131:41–46. doi: 10.4414/smw.2001.06144. [DOI] [PubMed] [Google Scholar]
- 27.Kwidzinski E, Bunse J, Aktas O, Richter D, Mutlu L, Zipp F, Nitsch R, Bechmann I. Indolamine 2,3-dioxygenase is expressed in the CNS and down-regulates autoimmune inflammation. Faseb J. 2005;19:1347–1349. doi: 10.1096/fj.04-3228fje. [DOI] [PubMed] [Google Scholar]
- 28.Lee OK, Ko YC, Kuo TK, Chou SH, Li HJ, Chen WM, Chen TH, Su Y. Fluvastatin and lovastatin but not pravastatin induce neuroglial differentiation in human mesenchymal stem cells. J Cell Biochem. 2004;93:917–928. doi: 10.1002/jcb.20241. [DOI] [PubMed] [Google Scholar]
- 29.Leung BP, Sattar N, Crilly A, Prach M, McCarey DW, Payne H, Madhok R, Campbell C, Gracie JA, Liew FY, McInnes IB. A novel anti-inflammatory role for simvastatin in inflammatory arthritis. J Immunol. 2003;170:1524–1530. doi: 10.4049/jimmunol.170.3.1524. [DOI] [PubMed] [Google Scholar]
- 30.Liu Y, Teige I, Birnir B, Issazadeh-Navikas S. Neuron-mediated generation of regulatory T cells from encephalitogenic T cells suppresses EAE. Nat Med. 2006;12:518–525. doi: 10.1038/nm1402. [DOI] [PubMed] [Google Scholar]
- 31.Lublin FD. Relapsing experimental allergic encephalomyelitis. An autoimmune model of multiple sclerosis. Springer Semin Immunopathol. 1985;8:197–208. doi: 10.1007/BF00197296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Luccarini I, Ballerini C, Biagioli T, Biamonte F, Bellucci A, Rosi MC, Grossi C, Massacesi L, Casamenti F. Combined treatment with atorvastatin and minocycline suppresses severity of EAE. Exp Neurol. 2008;211:214–226. doi: 10.1016/j.expneurol.2008.01.022. [DOI] [PubMed] [Google Scholar]
- 33.Mausner-Fainberg K, Luboshits G, Mor A, Maysel-Auslender S, Rubinstein A, Keren G, George J. The effect of HMG-CoA reductase inhibitors on naturally occurring CD4+CD25+ T cells. Atherosclerosis. 2008;197:829–839. doi: 10.1016/j.atherosclerosis.2007.07.031. [DOI] [PubMed] [Google Scholar]
- 34.Mellor AL, Munn DH. IDO expression by dendritic cells: tolerance and tryptophan catabolism. Nat Rev Immunol. 2004;4:762–774. doi: 10.1038/nri1457. [DOI] [PubMed] [Google Scholar]
- 35.Neuhaus O, Stuve O, Archelos JJ, Hartung HP. Putative mechanisms of action of statins in multiple sclerosis--comparison to interferon-beta and glatiramer acetate. J Neurol Sci. 2005;233:173–177. doi: 10.1016/j.jns.2005.03.030. [DOI] [PubMed] [Google Scholar]
- 36.O’Garra A, Steinman L, Gijbels K. CD4+ T-cell subsets in autoimmunity. Curr Opin Immunol. 1997;9:872–883. doi: 10.1016/s0952-7915(97)80192-6. [DOI] [PubMed] [Google Scholar]
- 37.Pahan K, Sheikh FG, Namboodiri AM, Singh I. Lovastatin and phenylacetate inhibit the induction of nitric oxide synthase and cytokines in rat primary astrocytes, microglia, and macrophages. J Clin Invest. 1997;100:2671–2679. doi: 10.1172/JCI119812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Paintlia AS, Paintlia MK, Khan M, Vollmer T, Singh AK, Singh I. HMG-CoA reductase inhibitor augments survival and differentiation of oligodendrocyte progenitors in animal model of multiple sclerosis. Faseb J. 2005;19:1407–1421. doi: 10.1096/fj.05-3861com. [DOI] [PubMed] [Google Scholar]
- 39.Paintlia AS, Paintlia MK, Singh AK, Singh I. Inhibition of rho family functions by lovastatin promotes myelin repair in ameliorating experimental autoimmune encephalomyelitis. Mol Pharmacol. 2008;73:1381–1393. doi: 10.1124/mol.107.044230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Paintlia AS, Paintlia MK, Singh AK, Stanislaus R, Gilg AG, Barbosa E, Singh I. Regulation of gene expression associated with acute experimental autoimmune encephalomyelitis by Lovastatin. J Neurosci Res. 2004;77:63–81. doi: 10.1002/jnr.20130. [DOI] [PubMed] [Google Scholar]
- 41.Paintlia AS, Paintlia MK, Singh I, Singh AK. Immunomodulatory effect of combination therapy with lovastatin and 5-aminoimidazole-4-carboxamide-1-beta-D-ribofuranoside alleviates neurodegeneration in experimental autoimmune encephalomyelitis. Am J Pathol. 2006;169:1012–1025. doi: 10.2353/ajpath.2006.051309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Paul F, Waiczies S, Wuerfel J, Bellmann-Strobl J, Dorr J, Waiczies H, Haertle M, Wernecke KD, Volk HD, Aktas O, Zipp F. Oral high-dose atorvastatin treatment in relapsing-remitting multiple sclerosis. PLoS ONE. 2008;3:e1928. doi: 10.1371/journal.pone.0001928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Robichaud A, Savoie C, Stamatiou PB, Tattersall FD, Chan CC. PDE4 inhibitors induce emesis in ferrets via a noradrenergic pathway. Neuropharmacology. 2001;40:262–269. doi: 10.1016/s0028-3908(00)00142-8. [DOI] [PubMed] [Google Scholar]
- 44.Rudick RA, Stuart WH, Calabresi PA, Confavreux C, Galetta SL, Radue EW, Lublin FD, Weinstock-Guttman B, Wynn DR, Lynn F, Panzara MA, Sandrock AW. Natalizumab plus interferon beta-1a for relapsing multiple sclerosis. N Engl J Med. 2006;354:911–923. doi: 10.1056/NEJMoa044396. [DOI] [PubMed] [Google Scholar]
- 45.Sanchez AJ, Puerta C, Ballester S, Gonzalez P, Arriaga A, Garcia-Merino A. Rolipram impairs NF-kappaB activity and MMP-9 expression in experimental autoimmune encephalomyelitis. J Neuroimmunol. 2005;168:13–20. doi: 10.1016/j.jneuroim.2005.03.024. [DOI] [PubMed] [Google Scholar]
- 46.Sim FJ, Lang JK, Ali TA, Roy NS, Vates GE, Pilcher WH, Goldman SA. Statin treatment of adult human glial progenitors induces PPARgamma-mediated oligodendrocytic differentiation. Glia. 2008;56:954–962. doi: 10.1002/glia.20669. [DOI] [PubMed] [Google Scholar]
- 47.Sommer N, Loschmann PA, Northoff GH, Weller M, Steinbrecher A, Steinbach JP, Lichtenfels R, Meyermann R, Riethmuller A, Fontana A, et al. The antidepressant rolipram suppresses cytokine production and prevents autoimmune encephalomyelitis. Nat Med. 1995;1:244–248. doi: 10.1038/nm0395-244. [DOI] [PubMed] [Google Scholar]
- 48.Soos JM, Stuve O, Youssef S, Bravo M, Johnson HM, Weiner HL, Zamvil SS. Cutting edge: oral type I IFN-tau promotes a Th2 bias and enhances suppression of autoimmune encephalomyelitis by oral glatiramer acetate. J Immunol. 2002;169:2231–2235. doi: 10.4049/jimmunol.169.5.2231. [DOI] [PubMed] [Google Scholar]
- 49.Stanislaus R, Singh AK, Singh I. Lovastatin treatment decreases mononuclear cell infiltration into the CNS of Lewis rats with experimental allergic encephalomyelitis. J Neurosci Res. 2001;66:155–162. doi: 10.1002/jnr.1207. [DOI] [PubMed] [Google Scholar]
- 50.Stuve O, Youssef S, Weber MS, Nessler S, von Budingen HC, Hemmer B, Prod’homme T, Sobel RA, Steinman L, Zamvil SS. Immunomodulatory synergy by combination of atorvastatin and glatiramer acetate in treatment of CNS autoimmunity. J Clin Invest. 2006;116:1037–1044. doi: 10.1172/JCI25805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Torphy TJ, Undem BJ. Phosphodiesterase inhibitors: new opportunities for the treatment of asthma. Thorax. 1991;46:512–523. doi: 10.1136/thx.46.7.512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Turner NA, O’Regan DJ, Ball SG, Porter KE. Simvastatin inhibits MMP-9 secretion from human saphenous vein smooth muscle cells by inhibiting the RhoA/ROCK pathway and reducing MMP-9 mRNA levels. Faseb J. 2005;19:804–806. doi: 10.1096/fj.04-2852fje. [DOI] [PubMed] [Google Scholar]
- 53.Vollmer T, Key L, Durkalski V, Tyor W, Corboy J, Markovic-Plese S, Preiningerova J, Rizzo M, Singh I. Oral simvastatin treatment in relapsing-remitting multiple sclerosis. Lancet. 2004;363:1607–1608. doi: 10.1016/S0140-6736(04)16205-3. [DOI] [PubMed] [Google Scholar]
- 54.Walters CE, Pryce G, Hankey DJ, Sebti SM, Hamilton AD, Baker D, Greenwood J, Adamson P. Inhibition of Rho GTPases with protein prenyltransferase inhibitors prevents leukocyte recruitment to the central nervous system and attenuates clinical signs of disease in an animal model of multiple sclerosis. J Immunol. 2002;168:4087–4094. doi: 10.4049/jimmunol.168.8.4087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yoshikawa M, Suzumura A, Tamaru T, Takayanagi T, Sawada M. Effects of phosphodiesterase inhibitors on cytokine production by microglia. Mult Scler. 1999;5:126–133. doi: 10.1177/135245859900500210. [DOI] [PubMed] [Google Scholar]
- 56.Youssef S, Stuve O, Patarroyo JC, Ruiz PJ, Radosevich JL, Hur EM, Bravo M, Mitchell DJ, Sobel RA, Steinman L, Zamvil SS. The HMG-CoA reductase inhibitor, atorvastatin, promotes a Th2 bias and reverses paralysis in central nervous system autoimmune disease. Nature. 2002;420:78–84. doi: 10.1038/nature01158. [DOI] [PubMed] [Google Scholar]
- 57.Ytterberg C, Johansson S, Andersson M, Olsson D, Link H, Holmqvist LW, von Koch L. Combination therapy with interferon-beta and glatiramer acetate in multiple sclerosis. Acta Neurol Scand. 2007;116:96–99. doi: 10.1111/j.1600-0404.2007.00801.x. [DOI] [PubMed] [Google Scholar]
- 58.Yu P, Gregg RK, Bell JJ, Ellis JS, Divekar R, Lee HH, Jain R, Waldner H, Hardaway JC, Collins M, Kuchroo VK, Zaghouani H. Specific T regulatory cells display broad suppressive functions against experimental allergic encephalomyelitis upon activation with cognate antigen. J Immunol. 2005;174:6772–6780. doi: 10.4049/jimmunol.174.11.6772. [DOI] [PubMed] [Google Scholar]
- 59.Zacco A, Togo J, Spence K, Ellis A, Lloyd D, Furlong S, Piser T. 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitors protect cortical neurons from excitotoxicity. J Neurosci. 2003;23:11104–11111. doi: 10.1523/JNEUROSCI.23-35-11104.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhang FL, Casey PJ. Protein prenylation: molecular mechanisms and functional consequences. Annu Rev Biochem. 1996;65:241–269. doi: 10.1146/annurev.bi.65.070196.001325. [DOI] [PubMed] [Google Scholar]
- 61.Zhang X, Jin J, Peng X, Ramgolam VS, Markovic-Plese S. Simvastatin inhibits IL-17 secretion by targeting multiple IL-17-regulatory cytokines and by inhibiting the expression of IL-17 transcription factor RORC in CD4+ lymphocytes. J Immunol. 2008;180:6988–6996. doi: 10.4049/jimmunol.180.10.6988. [DOI] [PubMed] [Google Scholar]
- 62.Zou J, Crews F. CREB and NF-kappaB transcription factors regulate sensitivity to excitotoxic and oxidative stress induced neuronal cell death. Cell Mol Neurobiol. 2006;26:385–405. doi: 10.1007/s10571-006-9045-9. [DOI] [PMC free article] [PubMed] [Google Scholar]