

Abstract
CDK6 promotes cell cycle progression and is over-expressed in human lymphoid malignancies. To determine the role of CDK6 in development and tumorigenesis, we generated and analyzed knockout mice. Cdk6-deficient mice show pronounced thymic atrophy due to reduced proliferative fractions and concomitant transitional blocks in the double negative (DN) stages. Using the OP9-DL1 system to deliver temporally controlled, Notch-receptor dependent signaling, we show that CDK6 is required for Notch-dependent survival, proliferation and differentiation. Furthermore, CDK6-deficient mice were resistant to lymphomagenesis induced by active AKT, a downstream target of Notch signaling. These results demonstrate a critical requirement for CDK6 in Notch/AKT-dependent T cell development and tumorigenesis and strongly support CDK6 as a specific therapeutic target in human lymphoid malignancies.
Keywords: CDK6, thymocytes, thymus, development, tumorigenesis
Introduction
Notch signaling is essential in early thymic T cell development (1). Multipotent T-cell precursors from the bone marrow commit to the T-cell lineage following stimulation of Notch upon entry to the thymus (2, 3). In vertebrates, Notch ligands consist of Jagged and Delta-like family members. Only Delta class ligands can efficiently support T-cell development (4). Interaction of Notch receptor with ligands results in proteolytic cleavage of Notch, releasing the intracellular domain (ICN), which then translocates to the nucleus and activates transcription of target genes for T cell development (2).
Notch signaling is also continuously required for specification, proliferation, differentiation and survival during the DN stages but must be attenuated in DP thymocytes to allow their further maturation and to prevent their oncogenic transformation (5). However, the mechanism underlying this requirement for T cell development is unknown. It has been shown that Notch-promoted survival and trophic effects in pre-T cells are mediated by the PI3K-AKT pathway (6). However, very little information exists connecting to Notch function to cell cycle control. Therefore, the identification of the cell cycle proteins involved in these responses is likely to provide important insights into development and tumorigenesis.
Cyclin-dependent kinase 6 (CDK6), a cyclin D-responsive regulator of the retinoblastoma protein pathway, appears to be of central importance in T cell development. CDK6 is a multifunctional protein that negatively regulates both retinoblastoma protein (pRB) and p27KIP1 (p27) activity (7) and modulates differentiation of certain cells (8). The activity of CDK6 is very tightly controlled by association with D-cyclins (7) and two families of cyclin-dependent kinase inhibitors (CKIs) including the CIP/KIP family and the inhibitors of cyclin-dependent kinase 4 (INK4) family proteins (9).
Of the three D-type cyclins, cyclins D2 and D3 are predominant in thymocytes (10). Cyclin D3 is strongly induced in the DN4 and ISP compartments following pre-TCR assembly. In contrast, cyclin D2 expression is high in the DN1-DN3 stages prior to pre-TCR assembly and barely detectable after pre-TCR assembly (10), suggesting that CDK6 may use different cyclin partners in the thymocyte developmental stages. Consistent with a key role for CDK6 as a D-cyclin partner in thymocytes, knockout of CDK4 or CDK2 was shown to have no significant effect on T cell development (11, 12), but significantly decreased thymic cellularity was observed in a CDK6 knockout animal (13). Moreover, its predominant expression in hematopoietic cell types (14, 15) and over-expression in human T-cell lymphoblastic lymphomãleukemia (T-LBL/ALL) (15-18), suggests a role for CDK6 in T cell malignancies. However, the specific role of CDK6 in thymocyte development is unclear and the effect of CDK6 loss on T cell tumorigenesis has not been ascertained.
In the present study, we tested the role of CDK6 in murine thymocyte development and tumorigenesis by generating Cdk6-deficient thymocytes using a knock-in approach. We report that CDK6 acts as a downstream target of Notch and AKT and is essential for T cell development and tumorigenesis.
Materials and Methods
Construction of Cdk6 knockout and knockin mice
To disrupt the mouse Cdk6 locus, we used a Lox-STOP-Lox (LSL) cassette as described in detail in Figure S1 and Supplemental Materials and Methods. Chimeric mice were generated by microinjecting two independent, karyotypically normal ES cell clones. Mating to nestin-cre mice allowed excision of the LSL cassette and re-expression of CDK6 (WT-Δ strains). Subsequent crosses to C57BL/6 wild-type mice ensured the removal of the cre transgene. Resulting heterozygotes were mated to homozygosity. Cdk6-LSL animals that were not mated to nestin-cre were defined as KO animals.
Western blotting, immunoprecipitation and in vitro kinase assays
Thymi were dissected and lysed. Western blotting, IP-Westerns, and in vitro kinase assays were performed as described (19). Antibodies used in this study included cyclin D1 (72-13G, Santa Cruz), cyclin D2 (M-20, Santa Cruz), cyclin D3 (C-16, Santa Cruz), p18 (M-20, Santa Cruz), p27 (Transduction Laboratories), CDK2 (BD Transduction Laboratories), CDK6 (C-21, Santa Cruz), CDK4 (C-22, Santa Cruz), Actin (Sigma), HA (Covance, mono HA.11), AKT (Cell signaling, phospho-Akt pathway sample kit), respectively.
Flow cytometry and FACS
For cell surface staining, 7AAD and Annexin V staining, four color flow cytometry was done as described (20, 21). Thymocytes were sorted by a MoFlo Cell Sorter. Mature cells were stained with antibodies against cell surface markers CD4, CD8 and TCR-β. For DN1-4 populations, cells were stained with the biotinylated ‘cocktail’ of lineage-specific antibodies described (20, 21), followed by depletion with Streptavidin-conjugated magnetic beads (Dynal, Oslo, Norway). Lineage-depleted cell suspensions were then surface-stained with anti-CD44, anti-CD25 and Streptavidin. For ISP-CD8+ population, cells were stained with the biotinylated ‘cocktail’ of lineage-specific antibodies. Enriched cell suspensions were then surface-stained with anti-CD4-FITC, anti-CD8-APC, anti-TCRβ-PE. The ISP-CD8+ population was defined as with CD8+/CD4- /TCR-β-. Sorted cells were processed immediately for Western blot analysis.
Purification of Bone Marrow cells and OP9/OP-9DL1 cocultures
Bone marrow (BM) cells obtained from WT and KO mice were depleted of CD3+, Gr-1+, Mac-1+, Ter119+, and B220+ cells through labeling with the corresponding biotinylated antibodies, followed by incubation with streptavidin-coated magnetic beads. Lineage-depleted BM cells were stained with c-Kit-APC and streptavidin-FITC, and c-Kit+Lin- cells were sorted.
OP9 and OP9-DL1 stromal cells were cultured in Minimum Essential Medium Alpha (Invitrogen), supplemented with 20% fetal bovine serum (Invitrogen), 100 U/mL penicillin, 100μg/mL streptomycin, and plated 1 or 2 days before use. Cocultures were initiated with ∼2 × 105 c-Kit+Lin- BM cells in the presence of 5 ng/mL Flt3-L and 5 ng/mL IL-7 (Peprotech). Cocultures were harvested by forceful pipetting at the indicated time points. Cells were counted and FACS analysis was performed as described above.
Statistics
Data from different pairs of mice are compared by a two-tailed Student’s t test.
Results
Generation of Cdk6 knockout mice
Recent analysis of mice lacking CDK6 suggested a role for this enzyme in thymocyte proliferation, but did not address the specific roles of CDK6 in thymocyte developmental stages. Further, these studies did not address the role of the kinase in leukemia/lymphoma (13). To study the role of CDK6 in T cell development and tumorigenesis, we generated KO mice using a Lox-STOP-Lox (LSL) cassette (Figure S1A). Animals with an intact LSL cassette in the Cdk6 locus are hereafter referred to as KO mice. To exclude the possibility that any defects observed in the KO strains resulted from non-specific effects, we also removed the LSL cassette through germline expression of Cre. The Cre-reactivated wild type allele (referred to as WT-Δ hereafter) expresses the wild-type CDK6 from the endogenous locus with regulatory controls intact. Southern blot and PCR analysis confirmed the expected allele structure (Figure S1, D-E), and sequence analysis of the entire coding region of Exon1 confirmed the presence of the expected LSL cassette and no other changes (data not shown).
Altered thymocyte numbers in Cdk6 KO mice
Consistent with a previous report (13), our CDK6 KO mice were found upon dissection to have defects in thymus development. KO mice display significant thymic hypoplasia (Figure 1A, left panel) at birth and this deficiency continues with increasing age. These thymuses contained about 4-fold fewer thymocytes than those from WT littermates (Figure 1A, middle panel) although body weights were comparable (Figure 1A, right panel). Therefore, our initial analysis of KO mice recapitulates a reported defect in thymocyte formation in a different strain, underscoring our success at achieving knockout of CDK6 with the LSL strategy.
Figure 1.

Defective thymocyte development in KO mice and analyses of cell cycle regulatory protein expression and function of kinases. A, left panel, Appearance of thymuses dissected from 2-month-old mice. A, middle panel, Total number of thymocytes from 1-3-month-old mice. The cell numbers are expressed as mean ± S.E (n= 15 for WT and KO, n=17 for WT-Δ). *, P value = 1.12508E-09 for WT and KO. Student T-test was used to confirm significant differences between pairs of animals. (A, right panel) Body weights of 1-3-month-old mice. The body weight is expressed as mean (g) ± S.E. B, left panel, Immunoblots were probed with different antibodies as indicated. Actin was used as internal control to ensure equal loading. B, middle and right panels, IP-Westerns. CDK6 or Cyclin D3 was immunoprecipitated from thymocyte extracts and the immunoblots were probed with the indicated antibodies, HC indicates IgG heavy chain. C, In vitro kinase assay. CDK6, CDK4, or CDK2 was immunoprecipitated from thymocyte extracts and an in vitro kinase assay was performed using the recombinant retinoblastoma protein (GST-RB) as a substrate. D, Immunoblot analysis of sorted thymocyte subsets (3 million cells of each subset) from wild-type mice. Upper, middle and bottom panels represent the same blot sequentially probed with antibodies against CDK6, CDK4 and CDK2.
Biochemical properties of cell cycle regulators in KO and WT-Δ thymocytes
Extracts of thymocytes derived from KO mice demonstrated a complete loss of detectable CDK6 protein (Figure 1B, left panel). Further, excision of the LSL cassette in WT-Δ animals restored expression of CDK6 to normal levels. Consistent with this, WT-Δ animals have normal appearing thymuses and thymocyte numbers. Next, we assessed the expression level of other cell cycle regulators in thymus extracts. Levels of CDK2 and CDK4 were comparable in all strains of mice. The expression levels of cyclins D3, D2, A, E, p18 and p27 were very similar in WT, KO and WT-Δ animals. No expression of cyclin D1, p15, p16, p21 or p57 was observed in the unstressed thymus (data not shown), consistent with other groups’ observations (14, 22).
In the absence of CDK6, profound differences in other CDK activities may be observed because D-type cyclins, p27 and INK4 family proteins no longer bind to CDK6 in KO mice and may bind in excess to CDK4 or CDK2 (13) instead. To examine potential cyclin/CDK/INK re-assortment, we analyzed the composition of CDK6/D3-containing complexes in thymus lysates (Figure 1B, middle, right). We found that the ability of CDK6 expressed in WT-Δ to bind cyclins D3, p18 and p27 is similar to that of WT. As expected, no cyclin D3, p18 or p27 co-immunoprecipitated with anti-CDK6 antibodies using extracts from KO thymuses. In the absence of CDK6 (KO lane), anti-cyclin D3 immunoprecipitation revealed that the level of cyclin D3-bound CDK4 increased, whereas the level of cyclin D3-bound CDK2 and p27 closely resembled that in extracts derived from WT littermate thymus (Figure 1B, right panel). Further, the level of p27- bound CDK4 or CDK2 (Figure S2A), and phosphorylation of pRB was very similar in WT, WT-Δ and KO thymocytes (Figure S2B), whereas the binding of p27 to cyclin E/CDK2 was reported to be increased and accompanied by impaired RB phosphorylation in CDK4-/- fibroblasts (23). This difference suggests that p27 may coordinate CDK4 and CDK2 activation, but not CDK6 and CDK2 activation. Thus, in the absence of CDK6, changes in p27 association with different cdk complexes are not a major determinant of the proliferative or differentiative effects observed.
To assess CDK activity in thymocyte lysates, we performed in vitro kinase assays (Figure 1C). CDK6 activity from WT-Δ mice was similar to that in WT animals. In contrast, KO extracts contain very little CDK6 kinase activity (Figure 1C, left panel). To determine whether CDK4 or CDK2 might compensate for the absence of CDK6 in thymocyte development, we performed in vitro kinase assays for these enzymes. Whole thymus lysates from KO mice have CDK4 and CDK2 kinase activity similar to that from WT or WT-Δ (Figure 1C, middle and right panels). Therefore, there is no obvious compensatory change in CDK4 or CDK2 kinase activity in KO mice in thymocyte populations analyzed together, nor did loss of CDK6 lead to inhibition of CDK2 activity.
In summary, in unstressed thymocytes, CDK6 kinase activity is lost in CDK6 KO animals. Further, there is no apparent alteration in CDK4 or CDK2 kinase activity in the absence of CDK6. The data shown above clearly demonstrate that our KO and WT-Δ animals behave as expected at the biochemical level.
Altered thymocyte development in Cdk6 mutant mice
In effort to understand the observed deficiency in thymic development caused by CDK6 loss, we first analyzed expression of CDK6 during thymocyte development (Figure 1D). We found that expression of CDK6 is relatively constant in sorted subpopulations compared to that reported for cyclin D3, which is very dynamic (10). The level of CDK6 was high within DN2, DN3, DN4, ISP and CD8+ subsets. It remained relatively moderate upon transition to the DP and CD4+ single positive stage. The expression profile of CDK4 is very similar to that of CDK6 during thymocyte development. In contrast, the expression profile of CDK2 differed from those of CDK4 and CDK6. There was no detectable CDK2 within the DN2 stage. CDK2 was detectable in other subsets and peaked at the immature single-positive (ISP) stage. Thus, CDK4 and CDK6 protein levels in thymocytes are highest in pro-T cells, are lower after the DN to DP transition, and remain low in CD4+ SP and CD8+ SP cells.
To determine if Cdk6 loss impacted thymocyte development, we established the precise distribution of thymocytes in KO mice using flow cytometry (Figure 2A). The fraction of DP cells was decreased in KO mice, and SP populations increased. The DP compartment was reduced to ∼86±1.5% of that of WT (Figure 2A), whereas the CD4+ SP population increased by 210±16% and CD8+ SP population increased by 184±13%. In contrast, changes in the DN population did not reach statistical significance. The absolute number of thymocytes from each compartment was reduced to different degrees. DP thymocytes were reduced approximately 4-fold in KO mice, CD4+ SP 1.5-fold, CD8+SP 2-fold, and DN 3-fold (Figure 2B), compared to WT littermates. Hence, CDK6 loss causes reduction in thymocyte numbers in all the compartments, and immature compartments are more profoundly altered than the mature SP compartments. Importantly, CDK6 expression in WT-Δ restored the percentage of thymocyte subsets to a normal level (Figure 2A) and restored the normal fractions of DN subsets (Figure 2C), as discussed below. Coupled with the normal size and cellularity of the thymus in WT-Δ animals (Figure 1A), these data demonstrate that the thymic defects observed in our KO animals are the result of changes in CDK6 protein level resulting from the presence of the LSL cassette.
Figure 2.

Alteration of thymocyte subset fractions in thymuses of KO and WT-Δ mice. A, left panel, Examples of flow cytometric profiles of thymocytes from 2-month-old mice stained with anti-CD4, anti-CD8 and TCR-β antibodies. The percentage of cells in each quadrant is shown. A, right panel, The bar graph summarizes percentage of each subset from separate experiments (N=8). Data are expressed as mean ± S.E. *, p < 0.05, significantly different from the control levels which were arbitrarily defined as 1 unit (100%). B, Absolute cell numbers (×106) for the subsets were calculated and are shown as bar diagrams. The cell numbers are expressed as mean ± S.E. The statistical significance was calculated using the Student’s T-test; p-values are indicated above the bars. N=8 for WT and KO, *, p < 0.05, significantly different from the control levels which were arbitrarily defined as 1 unit (100%). C, Alteration in DN subsets. On the left, thymocytes from 2-month-old CDK6 KO or controls were stained with the ‘cocktail’ of lineage-specific antibodies and anti-CD44 and anti-CD25. Lineage-positive cells were electronically ‘gated out’ and CD44-versus-CD25 profiles of the lineage-negative compartments are presented. Numbers in quadrants indicate the percentage of cells in each subset. On the right, the bar graph summarizes percentage of each subset from separate (N=8) experiments. Data are expressed as mean ± S.E. *, p < 0.05, significantly different from the control levels which were arbitrarily defined as 1 unit. D, Examples of flow cytometric analysis of CD25 (left) or CD44 (middle) expression in DN thymocytes from WT and KO. On the right, examples of flow cytometric analysis of CD25 expression in CD4+, DP, and CD8+ subsets from WT and KO thymocytes.
To determine the impact of CDK6 loss on early thymocyte development more precisely, Lin-thymocytes were analyzed for the expression of CD44 and CD25, which define distinct immature T cell populations (24). Although the fraction of thymocytes was very similar in DN1 and DN2 compartments of WT and KO mice (Figure 2C), KO mice showed a ∼2±0.33-fold increase in DN3 cells, indicating a potential developmental block at this stage. In contrast, the fraction of DN4 cells was reduced 1.25±0.04-fold. The expression level of CD25 was elevated and that of CD44 reduced in DN cells from KO thymocytes (Figure 2D). CD25 was also elevated in CD8 SP cells. There was no difference in the expression level of CD25 and CD44 between WT and WT-Δ (data not shown). KO thymocytes expressed normal levels of TCR-β and of other pre-TCR components (data not shown). We found that DN3 stage cells from WT and KO animals were of similar cell size (data not shown).
Taken together, these data suggest that loss of CDK6 causes reduced thymocyte cell numbers in most developmental subsets. Further, loss of CDK6 resulted in increased CD25 expression and reduced CD44 expression. However, this alteration of CD25 and CD44 expression and partial block in DN3 is not sufficient to explain the profound reduction in thymic cellularity. Thus, additional effects of CDK6 loss on cellular proliferation or survival may also influence T cell development, particularly in the DN3 compartment.
Proliferation and apoptosis in thymocyte subsets
Reduced thymic cellularity in KO may reflect reduced proliferation and/or increased apoptosis. We assessed the proliferative and apoptotic fractions in thymocytes at different developmental stages using the radiometric DNA dye 7-amino-actinomycin D (7AAD) or Annexin V respectively, followed by flow cytometry. We found a significantly decreased fraction of cycling cells in KO thymocytes at the DN2, DN3, ISP-CD8+, and CD8+stages that averaged 29±2.9%, 43±7.1%, 59±5.6%, and 64±6.5% of WT, respectively (Figure S3A). Thus, the reduced proliferative fraction in KO mice may contribute to reduced thymic cellularity. However, Annexin V staining revealed a substantial reduction of apoptosis in DN3, DN4, CD4, and CD8 compartments in KO mice averaging 55±6.7%, 76±4.5%, 59±5.6%, and 56±6.0% of WT, respectively (Figure S3B). Therefore, in most compartments, the reduced proliferation rate is accompanied by a reduced apoptosis rate (Figure S3C), apparently limiting the impact of proliferative changes on thymocyte cell numbers. The exception to this is observed in the DN2 compartment in KO, where the severely reduced proliferative fraction occurs in the absence of changes in apoptosis (Figure S3C). A key implication of these data is that thymocyte cell number changes may not be fully explained by the changes observed in thymocyte development. Thus, alteration of thymic progenitors in the bone marrow, or their ability to respond properly to Notch or other signaling pathways upon populating the thymus, may be key to alterations in thymocyte cell numbers.
CDK6 is required for Notch-dependent proliferation, survival and differentiation
In the bone marrow (BM), progenitors with efficient T lineage potential are found within the Lin-Sca1+c-kit+ (LSK) fraction (25). We sought to investigate if CDK6 loss disrupts the proliferative capacity of these BM T cell precursors. The LSK populations from BM were similar between WT and KO mice (Figure 3A, left panel). When the absolute numbers of LSK in each strain were calculated, we found that the absolute number was also similar between WT and KO mice (Figure 3A, right panel), consistent with other groups’ observations (13). These data indicate that CDK6 loss does not have a profound effect prior to seeding of the thymus by progenitor cells, leaving open the possibility that CDK6 might affect the progenitor cells’ ability to respond properly to Notch or other signaling pathways upon entering the thymus.
Figure 3.

CDK6 is required for Notch-dependent proliferation. A, Bone marrow (BM) cells obtained from WT and KO mice (from humerus and femur) were counted and stained with PE-conjugated antibodies specific for lineage (Lin) markers (CD3, CD4, CD8, B220, Mac-1, Gr-1, TER119) and SCA-1-Fitc, c-KIT-APC. Labeled cells were subsequently analyzed by FACS. The percentage of Lin-Sca-1+c-Kit+ (LSK) population (on the left) was expressed as mean ± S.E (n=4 for WT and KO). The absolute cell numbers (×106) for the LSK (On the right) were calculated based on the total number of cells and the percentage of LSK in the total cell population and are shown as bar diagrams. The cell numbers are expressed as mean ± S.E. N=4 for WT and KO. B, c-Kit+Lin- bone marrow cells were cocultured on OP9-DL1 cells and counted at times indicated. The expansion fold change was calculated based on the ratio between the total number counted at the time indicated versus the total c-Kit+Lin- cell number plated at day 0, which were arbitrarily defined as 1 unit. Data are expressed as mean ± S.E. *, p < 0.05, (N=5), significantly different from the control levels. C, c-Kit+Lin- bone marrow cells were cocultured on OP9 or OP9-DL1 cells and counted at day 6 indicated. The bar graph summarizes the fold changes over day 0 (which were arbitrarily defined as 1 unit) from separate (N=6) experiments. Data are expressed as mean ± S.E. *, p < 0.05, significantly different from the control levels which were arbitrarily defined as 1 unit. D, Representative example of FACS analysis for surface expression of CD19 and c-Kit of pre-B cells sorted from bone marrow and cultured for 6 days on OP9 cells.
To investigate if CDK6 affects the ability of progenitor cells to respond properly to Notch signaling, we took advantage of a cell culture system (OP9-DL1) that has the capacity to induce the differentiation of hematopoietic progenitors into T-cells through expression of Delta-like-1 on BM stromal cells, thus mimicking the thymic stroma (4). c-Kit+Lin- BM cells were cocultured on OP-9DL1 cells in the presence of the cytokines Flt3-L and Il-7 and analyzed by cell counting. There was ∼8-fold increase in cellularity by day 6 in WT cells, with the cell yields continuing to increase by ∼12-fold by day 12. By day 18, the cell number reached a plateau (Figure 3B). In contrast, c-Kit+Lin- progenitors from KO BM failed to expand throughout the co-culture period. These findings suggested that CDK6 is required for either cytokine or Notch signaling in cell proliferation.
To further understand the origin of these defects, c-Kit+Lin- progenitors from WT and KO BM were cocultured on OP9 or OP9-DL1 stromal cells in parallel and analyzed by cell counting at day 6. Notably, both systems employ the cytokines Flt3-L and Il-7 in the growth medium; the difference between these two culture systems is the presence of Delta-like-1 on OP9-DL1 (4). In the absence of this Notch ligand, OP9 cells support proliferation and differentiation of B cell precursors. Consistent with published results, WT progenitors showed the expected expansion on OP9-control cells, resulting in a 28-fold population expansion by day 6 of culture (Figure 3C) and differentiation of about 40% of the cells to mature B cells as evidenced by surface expression of CD19+c-Kit- (Figure 3D). We observed a similar increase in the number of KO cells during the first 6 days of culture on OP9 cells, with the development of over 60% mature B cells (Figure 3D). Therefore, there was no significant difference in cell expansion between WT and KO progenitor cells when plated on OP9 cells, suggesting the profound reduction in response to plating on OP9-DL1 cells reflected a lack of response to Delta-like 1 mediated Notch signaling.
We next examined the levels of proliferation and apoptosis of cells cultured on OP9-DL1 cells. As shown in Figure S4A, we observed an approximately 3-fold reduction in the proliferative fractions in KO cells as compared to WT cells at 12 days after plating. Moreover, the apoptotic fraction of thymocyte subsets at day 12 was increased by 1.4 to 6.5-fold in each developmental stage of KO thymocyte cultures (Figure S4B and C). Thus, the reduced proliferation and increased apoptosis of KO progenitors cultured on OP9-DL1 cells supports a role for CDK6 in mediating proliferation and survival in response to Notch signals and may explain the significant reduction in thymic cellularity in vivo. Because OP9-DL1 cocultures can recapitulate most stages of thymocyte development, we next compared differentiation of KO and WT progenitors plated on OP9-DL1. Notably, the cells from KO mice showed a moderate decrease in the percentage of mature single positive CD8+ cells and a dramatic decrease in DP cells (24-fold, 5-fold, and 7 fold reduction at day 6, 12 and 18, respectively) (Figure 4A). Re-expression of CDK6 in WT-Δ mice restored the differentiation into mature thymocytes to a normal level, comparable to WT (Figure S5).
Figure 4.

CDK6 is required for Notch-dependent differentiation. A, Flow cytometry of WT and KO cells stained with CD4 and CD8. c-Kit+Lin- bone marrow cells were cocultured on OP9-DL1 stromal cells and analyzed by FACS at various time points as indicated. B, Flow cytometry of WT and KO cells stained with the ‘cocktail’ of lineage-specific antibodies and anti-CD44 and anti-CD25. Lineage-positive cells were electronically ‘gated out’ and CD44-versus-CD25 profiles of the lineage-negative compartments are presented. Numbers in quadrants indicate the percentage of cells in each subset stained with CD44 and CD25. C, Comparisons of flow cytometric analysis of CD25 expression in DN thymocytes from WT and KO cells (Figure 4B) collected from OP9-DL1 cocultures.
Upon analysis of CD25 and CD44 expression, we found no apparent difference in DN subsets between WT and KO (Figure 4B, top panel) littermates at day 6, except CD25 was up-regulated in KO cells (Figure 4C, top panel). At day 12, ∼94% of KO cells had developed to the DN2 and DN3 stages and CD44- CD25- (DN4) cells were virtually undetectable (Figure 4B, middle panel). At day 18, surprisingly, the majority of KO cells in the DN compartment were found in the DN1 stage, suggesting that a fraction of the BM cells may respond poorly to OP9-DL1 culture. Thus, these data largely reproduce the defect seen in KO thymocytes in vivo. Surviving cells are delayed or blocked at the DN3 stage, while a fraction of thymocytes that progressed to the DP stage efficiently developed into CD4+ or CD8+ single positives.
In summary, both in vivo and cell culture results strongly suggest that CDK6 loss interferes with the proliferation and survival signals instigated by Notch signaling. Further, those thymocytes that survive CDK6 loss display defects in maturation consistent with a role for CDK6 in potentiating early stages and inhibiting late stages of Notch-regulated differentiation.
CDK6-deficient mice are resistant to thymic lymphoma formation induced by active AKT
Notch-promoted survival and trophic effects in pre-T cells are mediated by the PI3K-AKT pathway (6), and activation of Notch is critical to proliferation in both normal and tumor cells. We thus sought to examine if CDK6 loss impairs AKT1 activation in T cell progenitors or if AKT1 might depend on CDK6, for example, as a partner for D cyclins stabilized by activated AKT (26, 27). To explore this notion, we employed a transgenic mouse strain expressing constitutively active, myristylated Akt1 (MyrAkt1) from the proximal Lck gene promoter. The transgenic mice develop thymic lymphomas at about 5 months of age (28). Because recent evidence indicates that Notch and PKB/AKT are frequently hyper-active in many types of human and murine T cell tumors (28-30), this model may well reflect the critical role of AKT1 in T cell tumors. The Cdk6-/- allele was crossed onto the Lck-MyrAkt background and the resulting Akt1;Cdk6-/-, Akt1;Cdk6+/-, and Akt1;Cdk6+/+ compound mutant mice were observed for tumor occurrence. As expected, Akt1;Cdk6+/+ mice began to develop thymic lymphomas and became moribund beginning at approximately 5 weeks of age (Figure 5A), and virtually all had developed tumors by 26 weeks of age, consistent with previous reports (28). Similarly, most Akt1;Cdk6+/- mice had developed tumors by 22 weeks of age. In contrast, none of the Akt1;Cdk6-/- mice developed tumors by the age of 26 weeks (Figure 5A). Indeed, we continued to follow these mice out to 52 weeks of age, and never observed thymic lymphomas. Based on these results, we conclude that mice lacking CDK6 are resistant to T cell malignancies triggered by constitutively active AKT1.
Figure 5.

CDK6 operates downstream of Akt kinase. A, Comparison of tumor susceptibility between WT; Akt (n=21), Het;Akt (n=17), and KO;Akt (n=15) mice. B, Western blot analysis of thymocyte lysates derived from 4-week old WT or KO mice (left panel) or WT (lane 1) or WT;Akt (lane 2, pretumor) or 4-month old KO;Akt (lane 3) mice or WT;Akt (tumor cells, lane 4, right panel). Immunoblots were probed with different antibodies as indicated. Actin was used as an internal control to ensure equal loading. Anti-HA was used to detect transgene MyrAkt and distinguish it from endogenous Akt. C, On the left, the MyrAkt transgene failed to promote β-selection in the absence of CDK6. DN thymocytes from 4-week old WT;Akt and KO;Akt mice stained with the ‘cocktail’ of lineage-specific antibodies and anti-CD44 and anti-CD25. Lineage-positive cells were electronically ‘gated out’ and CD44-versus-CD25 profiles of the lineage-negative compartments are presented. Numbers in quadrants indicate the percentage of cells in each subset stained with CD44 and CD25. The bar graph (right panel) summarizes percentage of each subset from separate (n=3 for WT;Akt, n=4 for KO;Akt) experiments. Data are expressed as mean ± S.E. *, p < 0.05, significantly different from the control levels. D, Comparisons of flow cytometric analysis of CD44 (left panel) or CD25 (right panel) expression in some compartments from WT;Akt and KO;Akt.
To determine how CDK6 loss impaired tumor formation, we examined the phosphorylation of AKT on Ser473 and Thr308, since activation of AKT depends on these modifications (31). No difference between WT and KO animals was observed (Figure 5B, left panel). This is consistent with a role for CDK6 downstream of AKT1 activation. Next, we analyzed the expression of the MyrAkt1 transgene and the level of phosphorylation on S473 and T308 in transgene-positive animals. To achieve this, thymocytes were prepared from young healthy (Figure 5B, right panel, lane 2) or tumor bearing (lane 4) WT;Akt1 mice. Thymocytes were also prepared from young WT (lane 1) and KO;Akt1 (lane 3) thymi. Thymocyte extracts were blotted with anti-HA antibody, which specifically recognizes the HA-tagged, transgene-encoded MyrAkt1. This revealed that HA-MyrAkt1 was readily detected in WT;Akt1 thymocyte extracts, but not in those lacking the transgene (lane 1). Surprisingly, KO;Akt1 (lane 3) thymocyte extracts contained much higher levels of transgene-encoded Akt1 despite the complete lack of tumor formation noted above. This may indicate that CDK6 can negatively impact the expression from the LCK promoter, thus increasing transgene expression in KO cells. More likely, however, is the possibility that CDK6 KO cells accumulate in a stage that allows high levels of Lck-Myr-Akt1 expression, but are blocked from further progression to tumorigenesis.
To examine this possibility, we next analyzed the expression of cyclin D2 and D3. As had been previously observed, cyclin D3 was readily detectable in WT;Akt1 thymocyte extracts, but at a slightly lower level than is found in total thymocytes lacking transgene Akt1 (Figure 5B, right panel). Consistent with a defect in progression to tumorigenesis, we observed that the cyclin D2 level was much higher in WT;Akt1 tumors than is found in KO;Akt1 thymocytes at the same age.
We next determined the impact of CDK6 loss on early lymphomagenesis by analyzing Lin-thymocytes, since constitutively active Lck-MyrAkt1 transgenes promote the transition of DN3 cells to the DN4 stage (32). Consistent with previous observations, staining DN thymocytes from WT;Akt1 with anti-CD44 and anti-CD25 revealed an unexpected dramatic increase of DN1 cells, with a parallel decrease of both DN3 and DN4 cells (Figure 5C). The increased DN1 population results from the Akt1-mediated up-regulation of CD44 in DN4 thymocytes, making them appear as DN1 cells (32). In contrast, the majority of DN thymocytes from KO;Akt mice were found in the DN3 stage, similar to the observation in KO mice (Figure 2C). As expected, the expression level of CD44 was increased in all the compartments examined in WT;Akt1 thymocytes but not in those from KO;Akt1 mice (Figure 5D, left panel), whereas the expression level of CD25 in KO;Akt1 mice was increased in DN, ISP and CD8+ stages, compared with that of WT;Akt1 (Figure 5D, right panel). There was no change in the expression level of intracellular TCR-β in WT;Akt1 and KO;Akt1 mice (data not shown). Therefore, we conclude that the MyrAkt1 transgene cannot promote the transition of DN3 cells to the DN4 stage in the absence of CDK6, nor can the transgene drive high levels of CD44 expression in KO;Akt1 mice. Thus, constitutive Akt1 signaling requires CDK6 to promote tumorigenesis in the thymus.
Discussion
The G1 cell cycle control protein CDK6 has been linked to thymocyte proliferation (13), and CDK6 gene amplification or over-expression is commonly observed in T-LBL/ALL (15, 17). Using CDK6 knockout and reconstituted control mice, we have found that immature thymocytes from CDK6 knockout mice fail to undergo normal expansion and show complete resistance to lymphomagenesis triggered by constitutively active MyrAkt1. Using the OP9-DL1 thymic stroma mimetic system (4) to deliver temporally controlled Notch-receptor dependent signaling, we show that CDK6 is required for Notch-dependent T-lineage survival, proliferation and differentiation. The failed expansion of BM progenitors in the absence of CDK6 stems in part from reduced proliferative fractions, increased apoptosis and concomitant transitional blocks in the DN stages. Furthermore, loss of CDK6 resulted in increased CD25 expression and reduced CD44 expression, indicating that CDK6 acts in part to regulate these key T cell markers. Taken together, these data demonstrate a critical requirement for CDK6 in Notch-dependent thymocyte development and AKT1-dependent T cell tumorigenesis and strongly support CDK6 as a specific therapeutic target in human lymphoid malignancies.
Because CDK6 is an obligate catalytic partner for cyclin D3, the developmental defects in thymocytes from CDK6-/- mice would be predicted to resemble the phenotype of cyclin D3-/- mice. However, a distinct difference in phenotype is also observed, perhaps due to differences in the expression pattern of CDK6 and D cyclins. For instance, the proliferative fractions of CDK6 KO thymocytes were dramatically reduced in DN2 (∼30% of WT) and DN3 (∼50% of WT) stages (Figure S3A) prior to pre-TCR assembly, whereas the proliferative fractions were dramatically reduced in cyclin D3-/- mice only in the DN4 and ISP stages (10) after pre-TCR assembly. Therefore, cyclin D3 acts primarily downstream of the pre-TCR but CDK6 can act prior to pre-TCR formation. Nevertheless, cyclin D3 is required for Notch-driven lymphomagenesis. It is intriguing that CDK6 loss has such profound consequences, given the readily detectable expression of CDK4 in all T cell developmental stages. This in turn implies that CDK6 plays specific roles in T cell development (and cancer) that cannot be fulfilled by CDK4.
This notion is further supported by the observation that CDK6 knockout progenitors failed to undergo normal proliferation and differentiation in vitro by using the OP9-DL1 system (Figure 3, 4 and S4). These data strongly support the hypothesis that CDK6 is required for Notch-dependent initiation of T cell commitment by BM progenitors, likely acting downstream of the serine-threonine kinase AKT. This key component of the signal transduction cascade regulating pre-T cell growth and survival downstream of Notch (6) can potentially phosphorylate over 9000 proteins in mammalian cells (33). However, it is not known which downstream effectors of AKT are most critical for the genesis of cancer. Our data clearly indicate that CDK6 is a crucial downstream target of AKT, but the nature of the relationship between AKT and CDK6 remains to be determined. The activation of CDK6 may be due to upregulation of cyclin D2 (26, 27). Alternatively, or in addition, CDK6 gene expression may be regulated by Notch activation, because Notch signaling collaborates with the pre-TCR to suppress the activity of E2A by inducing E2A ubiquitination and degradation (34-36), which in turn relieves suppression of CDK6 gene expression (18). The aberrant activation of CDK6 in E47-deficient T-lineage cells contributes to the development of lymphoid malignancy (18).
Based on our data and published evidence, we propose a working model of the role of CDK6 in transmitting signals from Notch/AKT to regulate T cell development and tumorigenesis (Figure 6). Activation of Notch leads to or is accompanied by (1) activation of AKT; (2) inhibition of E2A; and (3) induction of pre-Tα transcript (one component of the pre-TCR) (37). AKT has also been shown to work downstream of the pre-TCR at the β-selection checkpoint (32). CDK6 can then be activated by (1) AKT; (2) elevated D-cyclins; (3) increased gene expression because of reduced E2A proteins. The activated CDK6 in turn suppresses the expression of CD25, ensuring DN3 stage cells transit to DN4. This model leads to the hypothesis that cdk6 loss would also block Notch-driven lymphomagenesis; this speculative connection between Notch, AKT and CDK6 in tumor formation and the molecular basis for these unique functions of CDK6 are currently under investigation.
Figure 6.
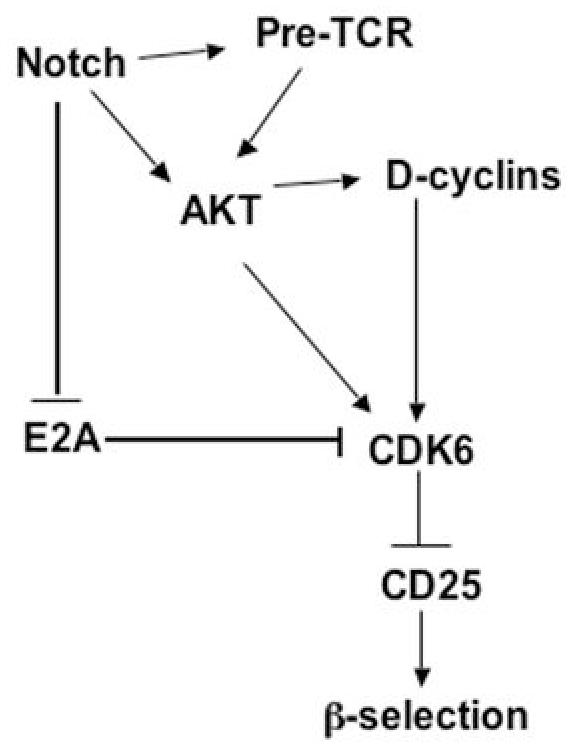
Working model for CDK6 regulation of T cell development and tumorigenesis. Activation of Notch leads to (1) activation of AKT; (2) inhibition of E2A; and (3) induction of pre-Tα transcript (one component of pre-TCR). Akt has also been shown to work downstream of pre-TCR at the β-selection checkpoint. CDK6 as a downstream mediator of Akt, can be activated by (1) Activated Akt; (2) elevated D-cyclins; (3) Increased gene expression because of reduced E2A proteins. The activated CDK6 in turn suppresses the expression of CD25, ensuring DN3 stage cells transit to DN4. Work presented here demonstrates a need for CDK6 in Notch-dependent development of thymocytes and in AKT-driven lymphomagenesis; the model postulates that CDK6 also has a role in Notch-dependent tumor formation and in mediating AKT-dependent development. Future verification of this model would provide a clear indication of links between important developmental and cell cycle pathways in thymocytes and their exploitation by tumorigenic processes.
It is interesting to note that the apparently absolute requirement for CDK6 in AKT-driven thymic lymphoma joins an increasing list of tumors highly dependent on G1 cyclin/CDK functions. This contrasts with the development of thymocytes in vivo, which show a significant but more limited regulation of thymocyte reduction in the CDK6 KO animals. Finally, we note that CDK6 is expressed in ES cells and is a transcriptional target of nanog, a pluripotency factor, yet development of most tissues in mice is not profoundly altered by CDK6 loss (38). Together, these observations support the notion that normal stem and progenitor cells have considerable plasticity in the event of loss of individual cell cycle control genes, but tumor initiating cells may be much more sensitive to such disruption. This in turn suggests that enzymes such as CDK6 need serious consideration as therapeutic targets in cancer and other developmental diseases where the initiating cell may differ significantly from its normal counterpart.
Supplementary Material
Acknowledgments
The authors gratefully acknowledge the dedication and assistance of Allen Parmelee at the Flow Cytometry Core in Pathology at Tufts University School of Medicine. This work was supported by NIH grants CA096527 and DE015302 to PWH.
References
- 1.Wilson A, MacDonald HR, Radtke F. Notch 1-deficient common lymphoid precursors adopt a B cell fate in the thymus. J Exp Med. 2001;194:1003–12. doi: 10.1084/jem.194.7.1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Artavanis-Tsakonas S, Rand MD, Lake RJ. Notch signaling: cell fate control and signal integration in development. Science. 1999;284:770–6. doi: 10.1126/science.284.5415.770. [DOI] [PubMed] [Google Scholar]
- 3.Radtke F, Wilson A, Stark G, et al. Deficient T cell fate specification in mice with an induced inactivation of Notch1. Immunity. 1999;10:547–58. doi: 10.1016/s1074-7613(00)80054-0. [DOI] [PubMed] [Google Scholar]
- 4.Schmitt TM, Zuniga-Pflucker JC. Induction of T cell development from hematopoietic progenitor cells by delta-like-1 in vitro. Immunity. 2002;17:749–56. doi: 10.1016/s1074-7613(02)00474-0. [DOI] [PubMed] [Google Scholar]
- 5.Aster JC, Pear WS, Blacklow SC. Notch Signaling in Leukemia. Annu Rev Pathol. 2008;3:587–613. doi: 10.1146/annurev.pathmechdis.3.121806.154300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ciofani M, Zuniga-Pflucker JC. Notch promotes survival of pre-T cells at the beta-selection checkpoint by regulating cellular metabolism. Nat Immunol. 2005;6:881–8. doi: 10.1038/ni1234. [DOI] [PubMed] [Google Scholar]
- 7.Sherr CJ, Roberts JM. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev. 1999;13:1501–12. doi: 10.1101/gad.13.12.1501. [DOI] [PubMed] [Google Scholar]
- 8.Grossel MJ, Hinds PW. From cell cycle to differentiation: an expanding role for cdk6. Cell Cycle. 2006;5:266–70. doi: 10.4161/cc.5.3.2385. [DOI] [PubMed] [Google Scholar]
- 9.Pavletich NP. Mechanisms of cyclin-dependent kinase regulation: structures of Cdks, their cyclin activators, and Cip and INK4 inhibitors. J Mol Biol. 1999;287:821–8. doi: 10.1006/jmbi.1999.2640. [DOI] [PubMed] [Google Scholar]
- 10.Sicinska E, Aifantis I, Le Cam L, et al. Requirement for cyclin D3 in lymphocyte development and T cell leukemias. Cancer Cell. 2003;4:451–61. doi: 10.1016/s1535-6108(03)00301-5. [DOI] [PubMed] [Google Scholar]
- 11.Rane SG, Dubus P, Mettus RV, et al. Loss of Cdk4 expression causes insulin-deficient diabetes and Cdk4 activation results in beta-islet cell hyperplasia. Nat Genet. 1999;22:44–52. doi: 10.1038/8751. [DOI] [PubMed] [Google Scholar]
- 12.Martin A, Odajima J, Hunt SL, et al. Cdk2 is dispensable for cell cycle inhibition and tumor suppression mediated by p27(Kip1) and p21(Cip1) Cancer Cell. 2005;7:591–8. doi: 10.1016/j.ccr.2005.05.006. [DOI] [PubMed] [Google Scholar]
- 13.Malumbres M, Sotillo R, Santamaria D, et al. Mammalian cells cycle without the D-type cyclin-dependent kinases Cdk4 and Cdk6. Cell. 2004;118:493–504. doi: 10.1016/j.cell.2004.08.002. [DOI] [PubMed] [Google Scholar]
- 14.Ragione F Della, Borriello A, Mastropietro S, et al. Expression of G1-phase cell cycle genes during hematopoietic lineage. Biochem Biophys Res Commun. 1997;231:73–6. doi: 10.1006/bbrc.1997.5938. [DOI] [PubMed] [Google Scholar]
- 15.Chilosi M, Doglioni C, Yan Z, et al. Differential expression of cyclin-dependent kinase 6 in cortical thymocytes and T-cell lymphoblastic lymphoma/leukemia. Am J Pathol. 1998;152:209–17. [PMC free article] [PubMed] [Google Scholar]
- 16.Lien HC, Lin CW, Huang PH, Chang ML, Hsu SM. Expression of cyclin-dependent kinase 6 (cdk6) and frequent loss of CD44 in nasal-nasopharyngeal NK/T-cell lymphomas: comparison with CD56-negative peripheral T-cell lymphomas. Lab Invest. 2000;80:893–900. doi: 10.1038/labinvest.3780093. [DOI] [PubMed] [Google Scholar]
- 17.Nagel S, Leich E, Quentmeier H, et al. Amplification at 7q22 targets cyclin-dependent kinase 6 in T-cell lymphoma. Leukemia. 2008;22:387–92. doi: 10.1038/sj.leu.2405028. [DOI] [PubMed] [Google Scholar]
- 18.Schwartz R, Engel I, Fallahi-Sichani M, Petrie HT, Murre C. Gene expression patterns define novel roles for E47 in cell cycle progression, cytokine-mediated signaling, and T lineage development. Proc Natl Acad Sci U S A. 2006;103:9976–81. doi: 10.1073/pnas.0603728103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hu MG, Hu GF, Kim Y, et al. Role of p12(CDK2-AP1) in transforming growth factor-beta1-mediated growth suppression. Cancer Res. 2004;64:490–9. doi: 10.1158/0008-5472.can-03-2284. [DOI] [PubMed] [Google Scholar]
- 20.Gounari F, Chang R, Cowan J, et al. Loss of adenomatous polyposis coli gene function disrupts thymic development. Nat Immunol. 2005;6:800–9. doi: 10.1038/ni1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gounari F, Aifantis I, Martin C, et al. Tracing lymphopoiesis with the aid of a pTalpha-controlled reporter gene. Nat Immunol. 2002;3(5):489–96. doi: 10.1038/ni778. [DOI] [PubMed] [Google Scholar]
- 22.Veiga-Fernandes H, Rocha B. High expression of active CDK6 in the cytoplasm of CD8 memory cells favors rapid division. Nat Immunol. 2004;5:31–7. doi: 10.1038/ni1015. [DOI] [PubMed] [Google Scholar]
- 23.Tsutsui T, Hesabi B, Moons DS, et al. Targeted disruption of CDK4 delays cell cycle entry with enhanced p27(Kip1) activity. Mol Cell Biol. 1999;19:7011–9. doi: 10.1128/mcb.19.10.7011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Godfrey DI, Zlotnik A. Control points in early T-cell development. Immunol Today. 1993;14:547–53. doi: 10.1016/0167-5699(93)90186-O. [DOI] [PubMed] [Google Scholar]
- 25.Schwarz BA, Bhandoola A. Circulating hematopoietic progenitors with T lineage potential. Nat Immunol. 2004;5:953–60. doi: 10.1038/ni1101. [DOI] [PubMed] [Google Scholar]
- 26.Diehl JA, Cheng M, Roussel MF, Sherr CJ. Glycogen synthase kinase-3beta regulates cyclin D1 proteolysis and subcellular localization. Genes Dev. 1998;12:3499–511. doi: 10.1101/gad.12.22.3499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Muise-Helmericks RC, Grimes HL, Bellacosa A, Malstrom SE, Tsichlis PN, Rosen N. Cyclin D expression is controlled post-transcriptionally via a phosphatidylinositol 3-kinase/Akt-dependent pathway. J Biol Chem. 1998;273:29864–72. doi: 10.1074/jbc.273.45.29864. [DOI] [PubMed] [Google Scholar]
- 28.Malstrom S, Tili E, Kappes D, Ceci JD, Tsichlis PN. Tumor induction by an Lck-MyrAkt transgene is delayed by mechanisms controlling the size of the thymus. Proc Natl Acad Sci U S A. 2001;98:14967–72. doi: 10.1073/pnas.231467698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nicholson KM, Anderson NG. The protein kinase B/Akt signalling pathway in human malignancy. Cell Signal. 2002;14:381–95. doi: 10.1016/s0898-6568(01)00271-6. [DOI] [PubMed] [Google Scholar]
- 30.Zweidler-McKay PA, Pear WS. Notch and T cell malignancy. Semin Cancer Biol. 2004;14:329–40. doi: 10.1016/j.semcancer.2004.04.012. [DOI] [PubMed] [Google Scholar]
- 31.Alessi DR, Andjelkovic M, Caudwell B, et al. Mechanism of activation of protein kinase B by insulin and IGF-1. Embo J. 1996;15:6541–51. [PMC free article] [PubMed] [Google Scholar]
- 32.Mao C, Tili EG, Dose M, et al. Unequal contribution of Akt isoforms in the double-negative to double-positive thymocyte transition. J Immunol. 2007;178:5443–53. doi: 10.4049/jimmunol.178.9.5443. [DOI] [PubMed] [Google Scholar]
- 33.Lawlor MA, Alessi DR. PKB/Akt: a key mediator of cell proliferation, survival and insulin responses? J Cell Sci. 2001;114:2903–10. doi: 10.1242/jcs.114.16.2903. [DOI] [PubMed] [Google Scholar]
- 34.Nie L, Xu M, Vladimirova A, Sun XH. Notch-induced E2A ubiquitination and degradation are controlled by MAP kinase activities. Embo J. 2003;22:5780–92. doi: 10.1093/emboj/cdg567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Talora C, Campese AF, Bellavia D, et al. Pre-TCR-triggered ERK signalling-dependent downregulation of E2A activity in Notch3-induced T-cell lymphoma. EMBO Rep. 2003;4:1067–72. doi: 10.1038/sj.embor.7400013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Engel I, Murre C. Disruption of pre-TCR expression accelerates lymphomagenesis in E2A-deficient mice. Proc Natl Acad Sci U S A. 2002;99:11322–7. doi: 10.1073/pnas.162373999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Reizis B, Leder P. Direct induction of T lymphocyte-specific gene expression by the mammalian Notch signaling pathway. Genes Dev. 2002;16:295–300. doi: 10.1101/gad.960702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Boyer LA, Lee TI, Cole MF, et al. Core transcriptional regulatory circuitry in human embryonic stem cells. Cell. 2005;122:947–56. doi: 10.1016/j.cell.2005.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.