

Summary
DNA2, a helicase/nuclease family member, plays versatile roles in processing DNA intermediates during DNA replication and repair. Yeast Dna2 (yDna2) is essential in RNA primer removal during nuclear DNA replication and is important in repairing UV damage, base damage, and double-strand breaks. Our data demonstrate that, surprisingly, human DNA2 (hDNA2) does not localize to nuclei, as it lacks a nuclear localization signal equivalent to that present in yDna2. Instead, hDNA2 migrates to the mitochondria, interacts with mitochondrial DNA polymerase γ, and significantly stimulates polymerase activity. We further demonstrate that hDNA2 and flap endonuclease 1 synergistically process intermediate 5’ flap structures occurring in DNA replication and long-patch base excision repair (LP-BER) in mitochondria. Depletion of hDNA2 from a mitochondrial extract reduces its efficiency in RNA primer removal and LP-BER. Taken together, our studies illustrate an evolutionarily diversified role of hDNA2 in mitochondrial DNA replication and repair in a mammalian system.
Introduction
DNA replication and repair, which are central processes in all cells, require the involvement of nuclease and helicase activities to process different DNA intermediate structures and to maintain genomic stability. One such enzyme is DNA2, a member of the nuclease/helicase family (Budd and Campbell, 1995). DNA2 proteins from different organisms have distinct nuclease, ATPase, and helicase domains (Bae and Seo, 2000; Budd et al., 1995). Their nuclease activity is structure-specific in that they recognize and cleave 5’ flap DNA structures. However, unlike the typical flap endonuclease 1 (FEN1), which cleaves the single-stranded flap DNA at the junction between ssDNA and dsDNA (Harrington and Lieber, 1994), yeast Dna2 (yDna2) nuclease removes a portion of DNA in the middle of the ssDNA flap (Bae et al., 2001; Bae and Seo, 2000). The distinct cleavage pattern of yDna2 and FEN1 is consistent with a model suggesting that yDna2 and FEN1 sequentially remove RNA primers in vivo (Bae et al., 2001). In this model, yDna2 in complex with RPA cleaves a long flap tail of 25 nt or longer during Okazaki fragment maturation, generating a 5–8 nt short flap structure that is then removed by FEN1 to generate a ligatable DNA end. The sequential enzymatic reactions are controlled by RPA, which binds to the long flap strand, inhibiting FEN1 flap endonuclease activity. However, RPA can bind to the N-terminal region of yDna2 to stimulate Dna2 cleavage of flap substrates. The yDna2/RPA complex then falls off the short flap structure allowing FEN1 to interact and cut the substrate (Bae et al., 2001). yDna2 helicase activity can unwind the DNA duplex, generating a 5’ flap structure, which facilitates RNA primer cleavage by the nuclease activities of yDna2 and FEN1 (Bae et al., 2002). In addition, the yDna2 helicase may be important in the resolution of the secondary structures formed during DNA replication. yDna2 also plays a role in DNA repair. Yeast Dna2 deficiency causes hypersensitivity to MMS, UV and X-ray irradiation (Budd and Campbell, 2000).
Like its homologue in yeast, human DNA2 (hDNA2) also cleaves the single-stranded DNA flap and shows ATPase and helicase activity in vitro (Kim et al., 2006; Masuda-Sasa et al., 2006), suggesting that it may play similar roles in DNA replication and repair. However, our extensive sequence analysis revealed that the hDNA2 polypeptide was shorter than yDna2 and lacked the RPA binding domain and classic nuclear localization signals (NLS). In this study, we employed immunofluorescence staining and Western blotting analysis to demonstrate that hDNA2 did not localize to the nucleus. Instead, hDNA2 was detected in the mitochondria. Furthermore, hDNA2 formed a complex with mitochondrial DNA polymerase γ (Polγ), stimulating its polymerase activity. We also demonstrated that hDNA2 nuclease activity was important in processing RNA primers during DNA replication and in processing intermediates of LP-BER, in cooperation with FEN1.
Results
Human DNA2 is not a nuclear protein
Previous studies reported that both hDNA2 and yDNA2 had conserved nuclease/helicase/ATPase domains; however, the human homologue lacked an RPA binding motif that was important for regulating the activity and action of yDna2 (Figure 1A) (Kim et al., 2006; Masuda-Sasa et al., 2006). Our sequence analysis revealed a striking aspect that yDna2 had classic Pat4, Pat7, and bipartite NLS sequences - none of which were found in hDNA2 (Figure 1A). Interestingly, NLS deficiency in hDNA2 has its evolutionary roots. We found that DNA2 proteins progressively lose NLS during the evolutionary process. DNA2 proteins in single-cell organisms such as S. cerevisiae, S. pombe, and A. fumigatus all have three types of classic NLS (Supplementary Table S1). DNA2 proteins in Arabidopsis, C. elegans, Drosophila, puffer fish, and frog lose one or two types of the NLS, whereas vertebrate DNA2 proteins (chicken, mice, rats, cattle, and humans) lack all three types of classic NLS (Supplementary Table S1).
Figure 1. Human DNA2 does not translocate into nuclei nor localize to replication foci.
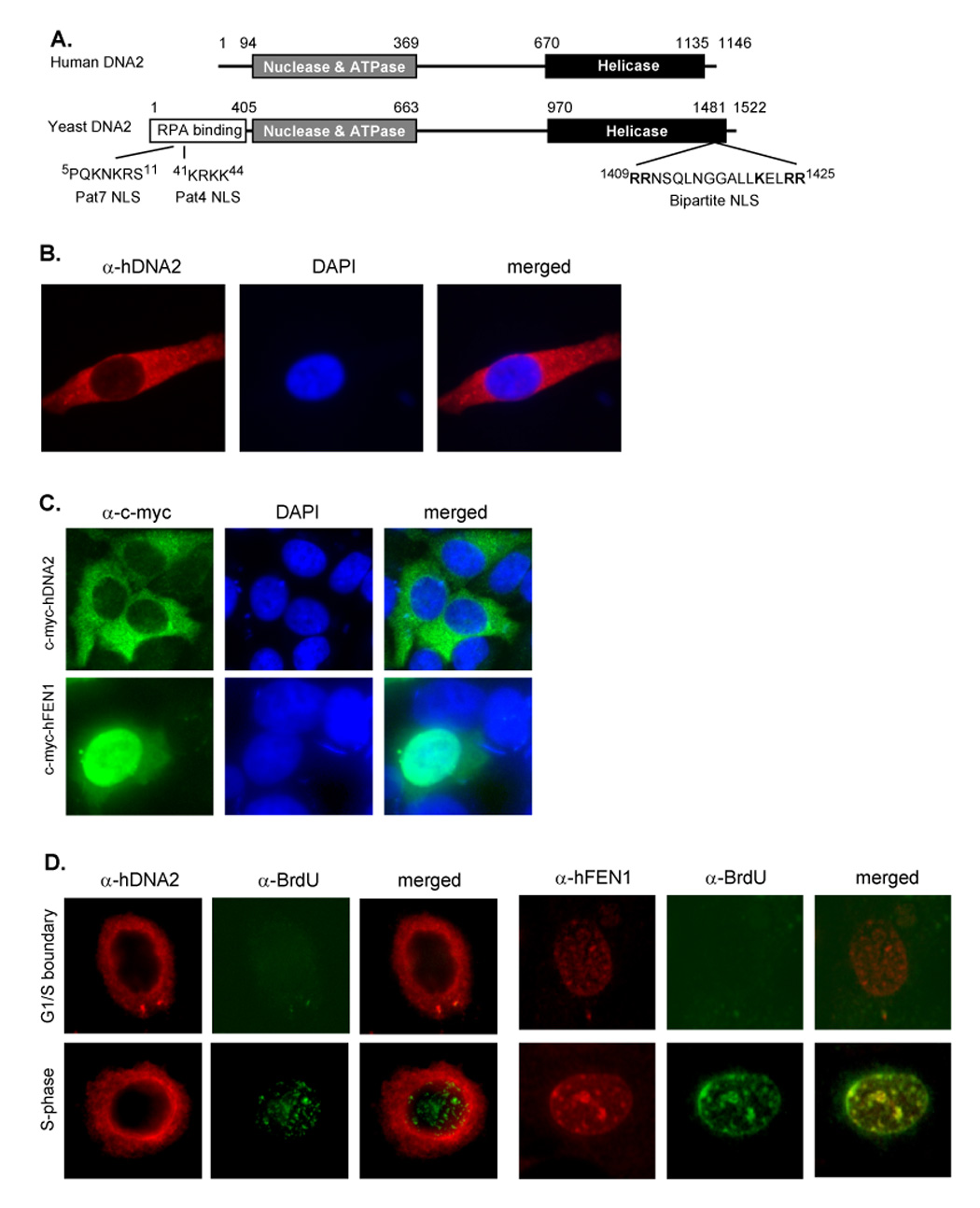
A. The domain structure of human and yeast DNA2. The RPA binding motif, nuclease, ATPase, and helicase domains. The classic Pat4, Pat7, and bipartite nuclear localization signals were identified with the pSORT II program (Nakai and Horton, 1999). B. Representative image of immunofluorescence staining of hDNA2 and nuclei of HeLa cells. hDNA2 (red) was stained with a polyclonal antibody to hDNA2; nucleus (blue) was stained with DAPI. C. hDNA2-c-myc and hFEN1-c-myc fusion proteins were transiently expressed in HeLa cells. The cells were co-stained with a monoclonal antibody against c-myc peptide and DAPI. D. Localization of hDNA2 and hFEN1 in the G1/S boundary and S phase. HeLa cells at G1/S boundary or in S phase were pulse labeled with BrdU as previously described (Zheng et al., 2007a). The cells were co-stained with antibodies to hDNA2 and BrdU or with antibodies to FEN1 and BrdU. BrdU signals (green) indicate the replication foci. Images were viewed and recorded with a 40× objective and a 10× ocular lens.
These data suggested that hDNA2 might have different subcellular localization and functions compared to yDna2. To experimentally determine whether hDNA2 localizes to the nucleus, we first conducted immunostaining of HeLa cells with a polyclonal antibody raised against hDNA2. The result revealed that hDNA2 was predominantly localized outside the nucleus (Figure 1B). Similar results were observed with immunostaining of primary human fibroblast and melanoma H294T cells (data not shown). To validate this observation, we expressed c-myc tagged hDNA2 proteins in HeLa cells. While no c-myc-tagged hDNA2 was noted in the nucleus, c-myc-tagged hFEN1 predominantly localized to the nucleus (Figure 1C). Furthermore, hDNA2 did not migrate to the nucleus, neither did it co-localize with the replication foci in S phase (Figure 1D). In contrast, hFEN1 co-localized with nuclear replication foci (Figure 1D). Taken together, these data suggested that hDNA2 was not a nuclear protein, consistent with its apparent lack of NLS.
Human DNA2 localizes to mitochondria
We next tested if hDNA2 could localize to the mitochondria. Co-immunostaining of HeLa cells for hDNA2 and mitochondrial heat shock protein 70 (mtHSP70) revealed co-localization of hDNA2 and mtHSP70 (Figure 2A, arrows). hDNA2 signals were also observed in cytosolic regions other than mitochondria. In addition, signals of transiently expressed hDNA2-EGFP fusion protein co-localized with mito-tracker red signals, confirming that hDNA2 localized to mitochondria (Figure 2B).
Figure 2. Human DNA2 localizes to the mitochondria.
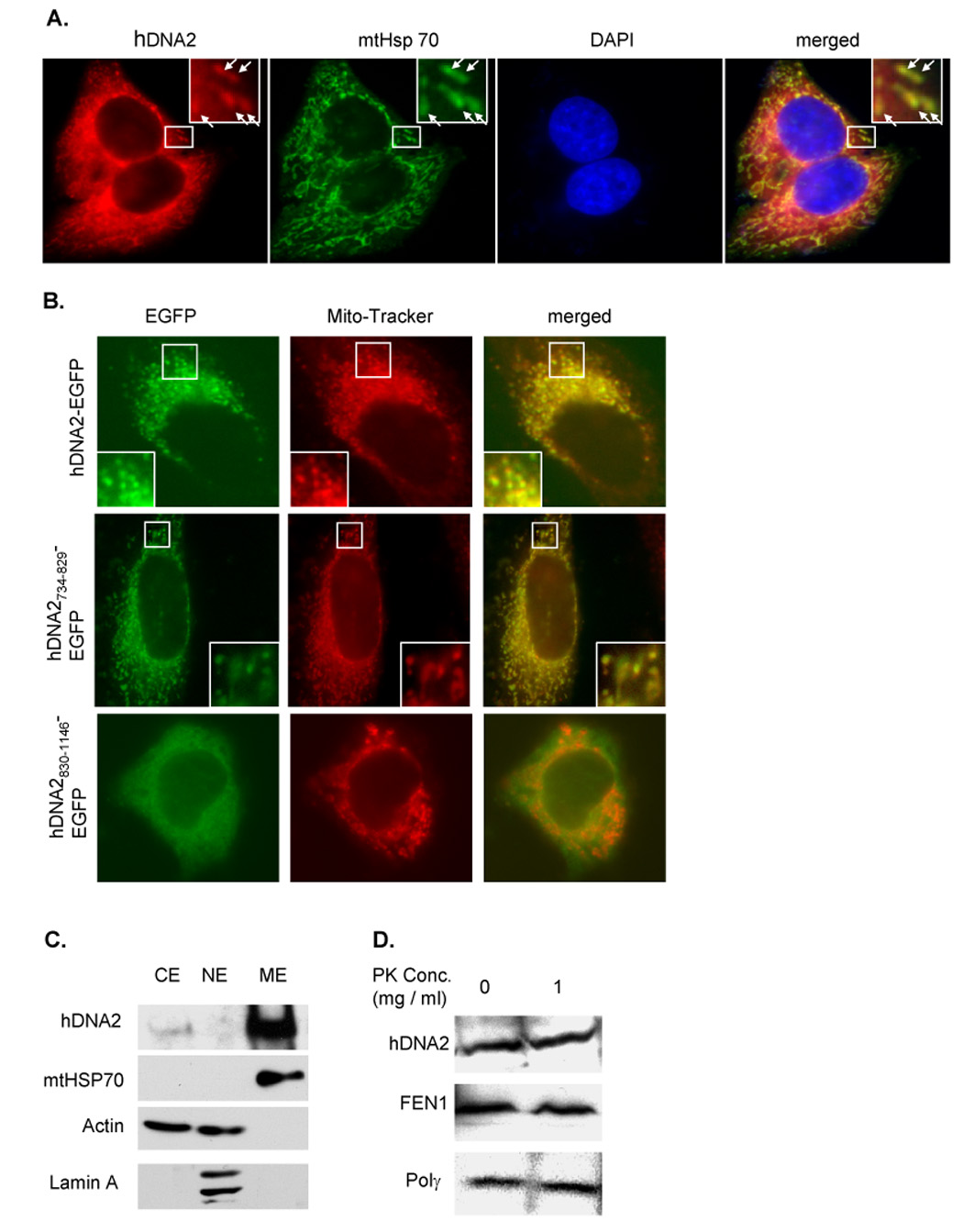
A. Localization of hDNA2 and mitochondrial specific heat shock protein 70 (mtHSP70) in HeLa cells. hDNA2 (red) and mtHSP70 (green) were stained with antibodies to hDNA2 and mtHSP70. The nucleus (blue) was stained with DAPI. Yellow spots (arrows) indicate colocalization of hDNA2 and mtHSP70 (merged views). The square box in the upper-right panel is a magnification of the area framed in white. B. Localization of exogenous hDNA2 and its deletion mutants fused with GFP at their C termini. The pEGFP-N1 vector (Clontech, Mountain View, CA) harboring the human DNA2L gene to express hDNA2-EGFP or hDNA2 deletion mutant-EGFP fusion protein was transformed into HeLa cells. Mitotracker red (mitochondria) and EGFP (green) signals in live HeLa cells (full length hDNA2) or in 2% paraformaldehyde-fixed HeLa cells (hDNA2 deletion mutants) were examined (40× objective and 10× ocular lens). C. Western blotting of hDNA2 in cytoplasm (CE), mitochondrial extract (ME), and nuclear extract (NE). 50 µg of CE, NE, and ME were resolved in 4–15% SDS-PAGE. The presence of hDNA2, mtHSP70, actin, or lamin A was determined by Western blotting using antibodies to corresponding proteins (Abcam). mtHSP70 was used as a positive mitochondria marker, Lamin A as a nuclear marker. Actin is present in both CE and NE. All of these markers were used as controls to determine the ME purity. D. Western blotting analysis of hDNA2 in protease K treated mitochondria. Purified mitochondria were treated with 1 mg/ml protease K. Proteins were extracted and analyzed by Western blotting using antibodies against hDNA2, hFEN1, or Polγ.
To further elucidate how hDNA2 localizes into mitochondria, we analyzed the hDNA2 sequence with the pSORT II program, which predicted a potential mitochondrial targeting R-2 motif (Gavel and von Heijne, 1990) at the hDNA2 N-terminus (Supplementary Figure S1A). However, the N-terminal 91 amino acid residues with EGFP (hDNA21–91-EGFP) did not localize to mitochondria in HeLa cells (Supplementary Figure S2). This suggested that the N-terminal peptide might not be the mitochondrion-targeting signal. Previous studies have shown that mitochondrial localization could be mediated by internal or C-terminal peptides (Diekert et al., 1999). To identify the region essential for mitochondrial localization of hDNA2, we expressed varying hDNA2 deletion mutants fused to the N-terminus of EGFP (Supplementary Figure S1B), and analyzed their mitochondrial localization. We discovered that the fragment covering amino acid residues 734–829 was essential and sufficient for the mitochondrial localization of EGFP fusion proteins (Figure 2B and Supplementary Figure S2).
To further validate the mitochondrial localization of hDNA2, proteins extracted from the cytoplasm, mitochondria, and nuclei were analyzed by Western blotting. Consistent with the immunostaining, hDNA2 was predominantly detected in the mitochondrial extract (ME) (Figure 2C). Moreover, similar to Polγ and FEN1, hDNA2 in mitochondria was resistant to treatment with protease K (Figure 2D), suggesting the nuclease/helicase was localized within the inner membrane or matrix of mitochondria.
hDNA2 interacts with Polγ and enhances Polγ-catalyzed DNA synthesis on double-stranded plasmid
Its mitochondrial localization suggested potential roles for hDNA2 in mtDNA replication and/or repair. This hypothesized that hDNA2 was complexed with the mitochondrial replication or repair machinery through interaction with mitochondrial DNA replication or repair proteins such as Polγ. Supporting our hypothesis, a mouse polyclonal antibody against Polγ, but not the non-specific mouse IgG, co-immunoprecipitated hDNA2 (Figure 3A). We further found that recombinant hDNA2 bound to Polγ coupled to Sepharose 4B, but not BSA-Sepharose 4B (Figure 3B), as well as treatment of the cell lysate with DNase I and RNase A, did not decrease the amount of co-precipitated hDNA2 (Figure 3B), suggesting that hDNA2 directly interacted with Polγ. In addition, the hDNA2/Polγ interaction was resistant to high salt, because most hDNA2 remained bound to Polγ-conjugated Sepharose 4B beads after sequential washes of the resin with a buffer containing 150, 300, 500, or 800 mM NaCl (Figure 3C).
Figure 3. hDNA2 interacts with and stimulates Polγ activity.
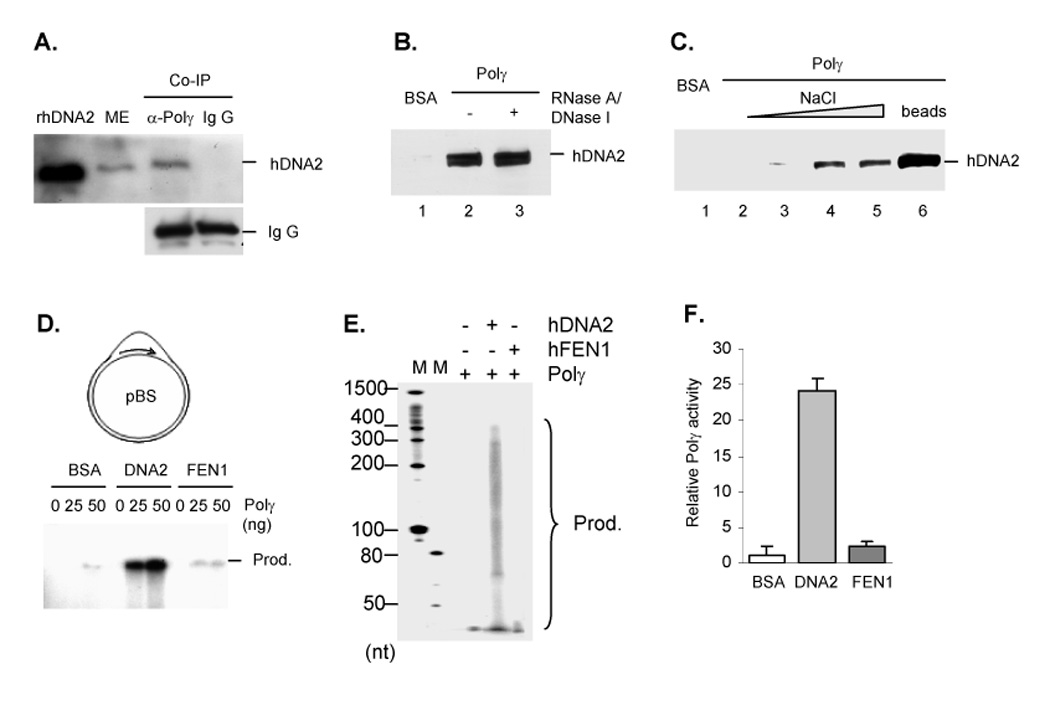
A. Co-immunoprecipitation of Polγ and hDNA2 from a ME. Lane 1 was the recombinant hDNA2 (rhDNA2), which was a positive control for Western blotting. Lane 2 was ME input, and Lane 3 and 4 was hDNA2 pulled down by anti-Polγ or non-specific mouse Ig G. B. Interaction between recombinant hDNA2 and Polγ. Polγ or BSA coated Sepharose 4B beads were incubated with 1 mg/ml insect cell lysates containing recombinant hDNA2 in the absence or presence of 100 µg/ml each of DNase I and RNase A. hDNA2 bound to the Sepharose 4B beads were analyzed with Western blotting. C. hDNA2 bound to Polγ-coated Sepharose 4B beads was sequentially eluted with a 50 mM Tris buffer (pH 7.5) containing 150, 300, 500, or 800 mM NaCl (lanes 2–5). The beads were finally incubated with the SDS-PAGE loading buffer (lane 6) to dissociate the remaining hDNA2 from the beads. D–F. hDNA2 stimulated Polγ-driven primer extension on D-loop substrates. 25 or 50 ng of purified Polγ was incubated with 100 ng DNA substrates, 5 µCi [α-32P] dATP, 1 µM dATP, and 50 µM each of the other three dNTP, in the presence of 100 ng BSA, 100 ng hDNA2 or 40 ng hFEN1. Products were analyzed with 0.5% agarose gel (D) or 5% denaturing PAGE (E). The products from reactions catalyzed by 50 ng Polγ were purified by the Qiagen plasmid purified kit and quantified by a scintillation counter (F). Relative Polγ activities were calculated as in panel E. Values were means ± SD (n = 3).
We then tested whether hDNA2 had any effect on Polγ activity by investigating the efficiency of primer extension by Polγ in the absence or presence of hDNA2. Consistent with a previous study (Korhonen et al., 2004), our data showed that Polγ alone was not active in extending a 21 nt primer annealed to a plasmid mimicking mitochondrial DNA (Figure 3D–F). However, hDNA2 dramatically enhanced Polγ activity (Figure 3D–F). The discrete bands in the agarose gel likely represented a DNA triplex with extended products of different lengths (Figure 3D). Denaturing PAGE analyses suggested that Polγ alone resulted in products that were less than 50 nt, but in the presence of hDNA2, the extended products increased to 400 nt (Figure 3E). Purification and further quantification of the products demonstrated that, in the presence of hDNA2, there was a 60-fold increase in Polγ activity under these conditions (Figure 3E). In contrast, no stimulation was observed in the presence of hFEN1 (Figure 3D–E). Interestingly, hDNA2 did not significantly stimulate Polγ activity on an oligo-primed, single-stranded DNA template (Supplementary Figure S3).
hDNA2 in mitochondria is important for efficient processing of RNA primer intermediates
We hypothesized that one of functions of hDNA2 was to participate in RNA primer removal during mtDNA replication. To test this hypothesis, we first determined if hDNA2 contributed to the cleavage of a flap structure that might be formed during RNA primer removal, by a mitochondrial extract (ME). The ME from HeLa cells efficiently cleaved the ssDNA flap (20 nt) of a 5’ end 32P-labeled DNA substrate at multiple sites (Lanes 3–6, Figure 4A). Cleavage products were grouped into two categories: the first group included products of 1–10 nt in length resulting from endonuclease cleavage at the 5’ end or in the middle of the ssDNA flap. The second group consisted of products 20 nt in length, resulting from endonuclease cleavage at the ssDNA and dsDNA junction (Figure 4A). While depletion of hDNA2 from the mitochondrial extract diminished group 1 products (Lane 7–10, Figure 4A), supplementation of hDNA2-depleted ME with recombinant hDNA2 recovered these cleavage products (Lanes 11–14, Figure 4A). Furthermore, group 1 products resulting from the ME cleavage corresponded to those resulting from the purified hDNA2 cleavage (Lanes 15–18, Figure 4A). These data suggested that hDNA2 in mitochondria mediated the endonuclease cleavage of the ssDNA flap, generating shorter flap structures. It is possible that the 20 nt product might have been generated mainly by hFEN1, which was recently demonstrated to localize to both nuclei and mitochondria (Liu et al., 2008). While depletion of hFEN1 from the ME abolished most of the 20 nt bands (Lanes 19–22, Figure 4A), addition of recombinant hFEN1 restored the 20 nt products (Lanes 23–26, Figure 4A). Our results clearly indicated two distinct flap endonuclease activities corresponding to hDNA2 and hFEN1 in mitochondria, and depletion of either one decreased the efficiency of endonuclease cleavage of flap substrates.
Figure 4. Two flap endonuclease activities in ME.
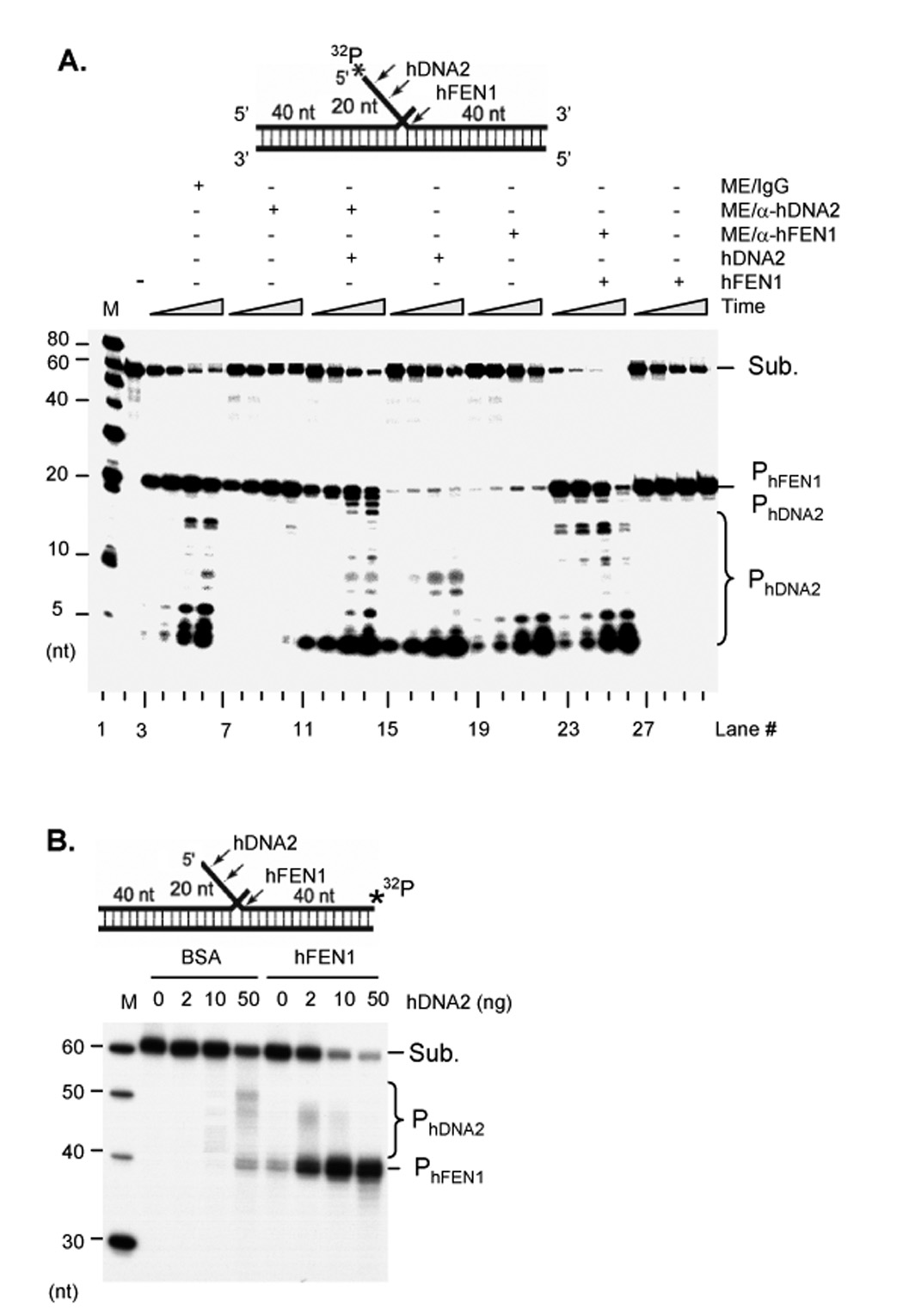
A. Flap endonuclease activities in ME. 1 µg non-depleted ME or a ME depleted of either hDNA2 or hFEN1 were incubated with 1 pmol 5’ end 32P labeled flap DNA substrates (upper panel). 100 ng hDNA2, or 10 ng hFEN1 were added to the reactions as specified. Reactions were carried out at 37°C for 5, 10, 30, and 60 min. B. hDNA2 and hFEN1 synergistically cleave flap substrates. 0, 2, 10, and 50 ng hDNA2 were incubated with 1 pmol 3’ end 32P labeled flap DNA substrates in the presence of 1 ng BSA or hFEN1. Reactions were carried out at 37°C for 60 min. The cleavage products were analyzed with 15% denaturing PAGE in both A and B.
To further characterize the cleavage of flap substrates by hDNA2 and hFEN1, we labeled the flap substrate with 32P at the 3’ end of the flap strand. Incubation of hDNA2 with the 3’ end labeled DNA substrates resulted in 40–50 nt bands (Figure 4B), consistent with the previous observation that hDNA2 cleaved flap substrates at multiple sites within the ssDNA strand. On the other hand, hFEN1 cleaved the flap substrate at the ssDNA and dsDNA junction, generating a 40 nt band suitable for ligation (Figure 4B). The 40 nt band could be further cleaved by the exonuclease activity of hFEN1 in vitro (Figure 4B). Importantly, hDNA2 stimulated hFEN1 cleavage of the flap substrate in a concentration-dependent manner. Our data revealed that 1 ng hFEN1 alone cleaved approximately 10% of the substrate, but that addition of 10 ng or 50 ng hDNA2 resulted in 50% and more than 90% excision, respectively (Figure 4B). On the other hand, 45–50 nt bands resulting from hDNA2 cleavage were not observed in the presence of 1 ng hFEN1 and 50 ng hDNA2 (Figure 4B), likely due to efficient processing of short flap intermediates by the coordinated action of the two nucleases.
Next, we employed a nick-translation assay on a gapped flap substrate to resemble gap-filling, RNA primer removal, and DNA ligation, which occur during Okazaki fragment maturation or during the final maturation process of displacement mitochondrial DNA replication. Incorporation of [α-32P] dCTP into the upstream primer (32 nt) by Polγ, filled the gap and resulted in extended 32P-labeled bands, which were visualized with a phosphorimager. Polγ mainly produced the 40 nt band, indicating that the polymerase was very active in filling the gap (Figure 5A). Interestingly, hDNA2 but not hFEN1 enhanced the displacement of DNA synthesis by Polγ (Figure 5A), possibly due to its helicase activity. In the absence of DNA ligase IIIα, no 80 nt band was observed, indicating that generation of the full length product required the involvement of DNA ligase and could not be explained simply by continued strand-displacement (Lanes 3–6, Figure 5A). Reaction with Polγ, Lig IIIα, and hDNA2 did not generate a visible ligated product of 80 nt (Lane 9, Figure 5A), consistent with previous observations that hDNA2 mainly cleaved the flap substrate in the middle of the flap strand, producing a shorter flap substrate not suitable for ligation. However, the presence of hFEN1, Polγ, and Lig IIIα produced a considerable amount of ligated products (Lane 10, Figure 5A). Furthermore, the presence of both hDNA2 and hFEN1 also resulted in a significant increase in the amount of ligated products, compared to hFEN1 alone (Figure 5A). Similar results were observed utilizing a substrate with an RNA-DNA flap (17 nt RNA and 3 nt DNA), which better resembled the intermediate structure during RNA primer removal (Figure 5B). Altogether, our data suggested that both hDNA2 and FEN1 were important for efficient RNA primer removal.
Figure 5. hFEN1 and hDNA2 are critical for removal of RNA primers in mitochondria.
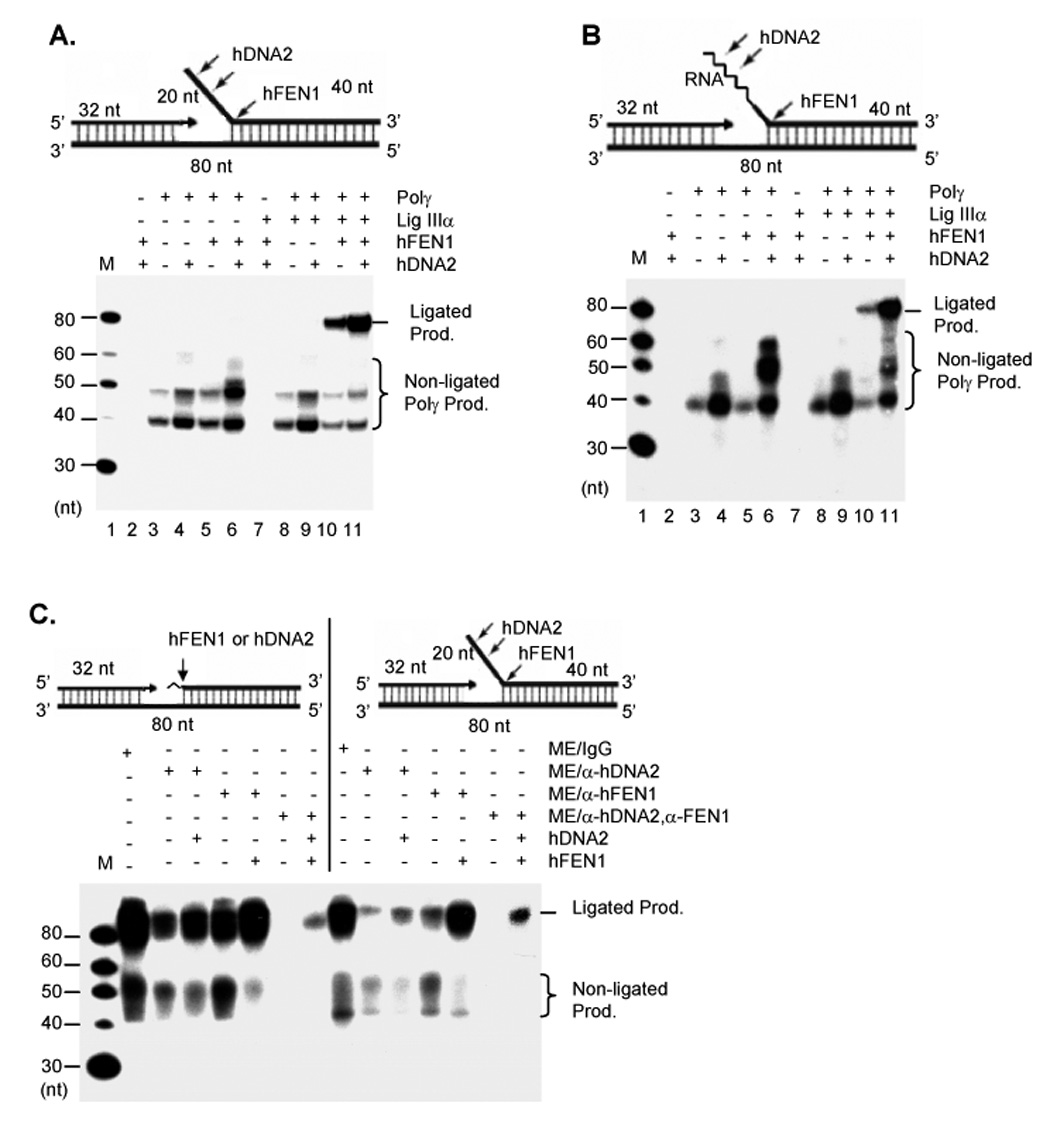
A and B. Nick translation by purified recombinant mitochondrial proteins with a gap substrate containing (A) a 20 nt DNA flap or (B) with a gap substrate with a 20 nt RNA-DNA flap, which consists of 17 ribonucleotides and 3 deoxyribonucleotides and is represented by the zigzag line. C. Nick translation by ME and ME depleted of hDNA2 or/and hFEN1 with either a gap substrate containing a ribonucleotide (left) or a 20 nt DNA flap (right). The zigzag line represents the single ribonucleotide in the gapped duplex (left). 2 µg each of ME was incubated with 1 pmol of indicated DNA substrates. All reactions in A–C were carried out in a buffer containing 5 µCi [α-32P] dCTP and 50 µM each of dATP, dGTP, and dTTP.at 37°C for 1 hour and analyzed with a 15% denaturing PAGE.
We further investigated the impact of hDNA2 and hFEN1 deficiency on the effectiveness of RNA primer removal by ME preparations. We immunodepleted hDNA2 or hFEN1 from ME and assayed nick-translation activity on the gapped duplex with a ribonucleotide or a flap at the 5’ end of the downstream oligomer. Non-depleted ME efficiently filled the gap, processed the ribonucleotide or flap, and ligated the DNA nicks, generating a full length 80 nt band (Figure 5C). Depletion of hDNA2 significantly decreased Polγ activity and generation of the ligated product, but addition of the recombinant hDNA2 partially restored production of the 80 nt product. Similarly, depletion of hFEN1 also decreased the overall efficiency of generation of the ligated product, but the nick-translation activity was restored following addition of recombinant hFEN1. In the absence of hDNA2 or hFEN1 in the ME, there was a significant increase in levels of non-ligated products relative to ligated products, indicating that hDNA2 or hFEN1 deficiency resulted in defective removal of ribonucleotides or flaps. This in turn affected the conversion of non-ligatable products into ligated ones. Immunodepletion of both hDNA2 and FEN1 completely shut down the gap-filling activity and generation of the ligated product. Re-addition of hDNA2 and hFEN1 only recovered about 10% of the activity. This might have resulted from the removal of Polγ that had been complexed with hDNA2, or other replication factors bound to hDNA2 and hFEN1.
hDNA2 is important for long-patch base excision repair
A flap structure also occurs in long-patch base excision repair (LP-BER), and hDNA2 could also contribute to the processing of such intermediates. We previously showed that hFEN1 played an important role in LP-BER in mitochondria (Liu et al., 2008). Depletion of hFEN1 from a ME led to an approximate 60% decrease in LP-BER efficiency (Liu et al., 2008), suggesting that FEN1 was vital in mitochondrial LP-BER. These results did not, however, rule out contributions from additional nucleases in LP-BER, as suggested by recent studies implicating hFEN1-independent flap endonuclease activity in ME (Akbari et al., 2008; Szczesny et al., 2008).
To test whether hDNA2 was also involved in processing nicked or flapped LP-BER intermediates, we assayed hDNA2 cleavage of a nicked substrate or a short flap substrate with an abasic tetrahydrofuran (THF) residue at the 5’ end of the downstream oligomer, which resembled the intermediate resulting from AP endonuclease 1 (APE1) cleavage of a LP-BER substrate. hDNA2 efficiently cleaved both the nicked and the flap substrates, resulting in a cleavage pattern that was identical to that obtained with ME (Figure 6A), supporting our hypothesis that both hDNA2 and hFEN1 processed DNA intermediates in LP-BER.
Figure 6. hDNA2 and hFEN1 are required for efficient LP-BER in mitochondria.
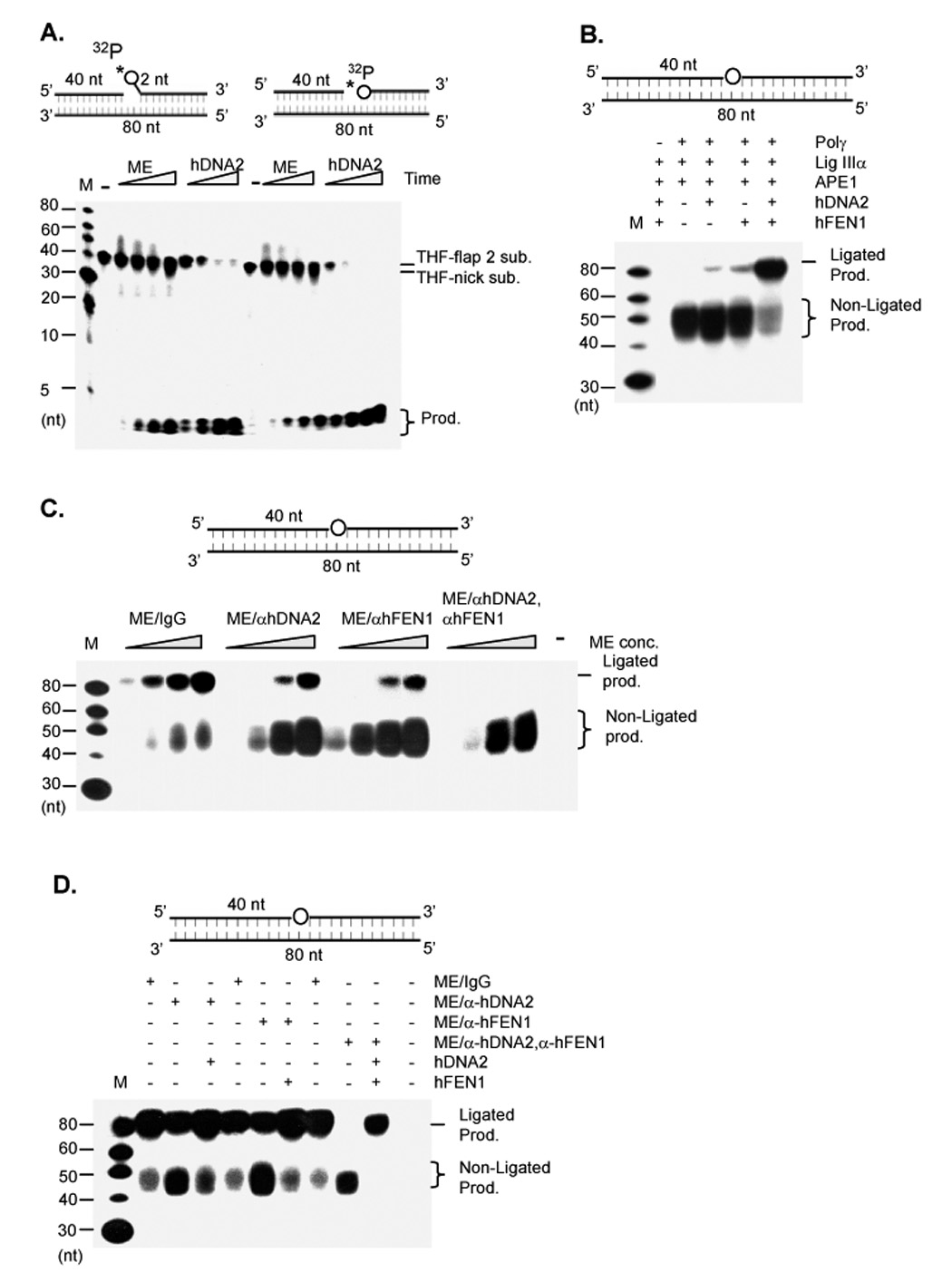
A. Cleavage of nick or flap substrates with a 5’ THF AP site, which was represented by a circle (upper panels). 2 µg ME or 100 ng hDNA2 were incubated with 1 pmol 5’ end 32P labeled DNA substrates in a total volume of 15 µl at 37°C for 0, 5, 10, 30, and 60 min. B. Reconstitution of LP-BER with recombinant mitochondrial proteins. 20 ng APE1, 50 ng Polγ, 200 ng Lig IIIα were mixed with no flap endonuclease or with 50 ng hDNA2 or/and 20 ng hFEN1. The mixture was then incubated with 500 fmol of the THF model LP-BER substrate in the reaction buffer containing 5 µCi dCTP and 50 µM each of dATP, dGTP, and dTTP. The reactions were carried out at 37°C for 30 minutes. The THF residue was represented by a circle in the middle of the substrate (upper panel). C. LP-BER on hDNA2 or hFEN1-depleted ME. 0.5, 1, 2, and 4 µg ME or ME depleted of hDNA2 or/and hFEN1 were incubated with 500 fmol LP-BER model substrates in a total volume of 20 µl. D. LP-BER on hDNA2 or hFEN1-depleted ME supplemented with corresponding recombinant nucleases. 4 µg each of indicated ME preparation were mixed with 500 fmol of the LP-BER substrate. 50 ng hDNA2 or 20 ng hFEN1 was added to the reaction as specified. In each panel, reactions were carried out at 37°C for 30 min. Intermediates or ligated products were analyzed with 15% denaturing PAGE.
To further elucidate the role of hDNA2 and hFEN1 in LP-BER, we reconstituted the reaction to repair an abasic THF residue as a model base and sugar damage in a DNA duplex substrate. In the absence of hDNA2 and hFEN1, all resulting products were non-ligated intermediates and were 40–55 nt in length (Lane 3, Figure 6B). Addition of either human hDNA2 or hFEN1 generated a weak ligated 80 nt band. However, inclusion of both hDNA2 and hFEN1 in the reaction dramatically (> 10 fold) elevated the level of 80 nt ligated product, while significantly decreasing the levels of non-ligated intermediates (Figure 4B). These data demonstrated the synergistic effect of hDNA2 and hFEN1 in processing LP-BER intermediates for DNA ligation.
We then determined if deficiency of hDNA2 and FEN1 resulted in defective LP-BER in mitochondrial extracts. The non-depleted ME efficiently repaired the THF damage (Figure 6C). However, removal of hDNA2 from the ME resulted in significant accumulation of non-ligated product and a marked decrease in the formation of ligated product (Figure 6C). A similar phenomenon was observed with hFEN1-depleted ME (Figure 6C). Depletion of both human DNA2 and FEN1 completely blocked the formation of ligated products (Figure 6C). Furthermore, addition of purified recombinant hDNA2 or hFEN1 to hDNA2-or hFEN1-depleted ME fully restored the repairing activity (Figure 6D), suggesting that the inefficient LP-BER observed in the hDNA2- or hFEN1-depleted ME was solely due to deficiency of DNA2 or hFEN1, not due to the incidental co-immunoprecipitation of other proteins with hDNA2 or hFEN1.
We then tested whether deficiency of hDNA2 resulted in defective LP-BER in vivo. We employed siRNA to nock down the expression of hDNA2 in HeLa cells. After 48 h incubation post-transfection, the levels of hDNA2 in HeLa cells treated with siRNA oligos targeting hDNA2 (siRNA-hDNA2) decreased to less than 20% of untreated cells, whereas HeLa cells treated with control siRNA oligos (siRNA-control) did not affect hDNA2 levels (Figure 7A). We found that in response to H2O2 treatment, significantly more oxidative damage accumulated in mtDNA of siRNA-hDNA2 HeLa cells than in siRNA-control cells (Figure 7B). However, knockdown of hDNA2 in HeLa cells did not affect the amount of oxidative damage in nuclear DNA (Figure 7C). Consistently, we found that the recovery of mtDNA from H2O2-induced DNA damage was delayed in hDNA2 knockdown HeLa cells (Figure 7D). However, the recovery time-course of nuclear DNA in siRNA hDNA2 transfected cells was similar to that in control cells (Figure 7E). These results suggested that hDNA2 deficiency selectively impaired repair of oxidative damage in mtDNA.
Figure 7. hDNA2 deficiency affects the efficiency of repairing oxidative DNA damage in mitochondria.
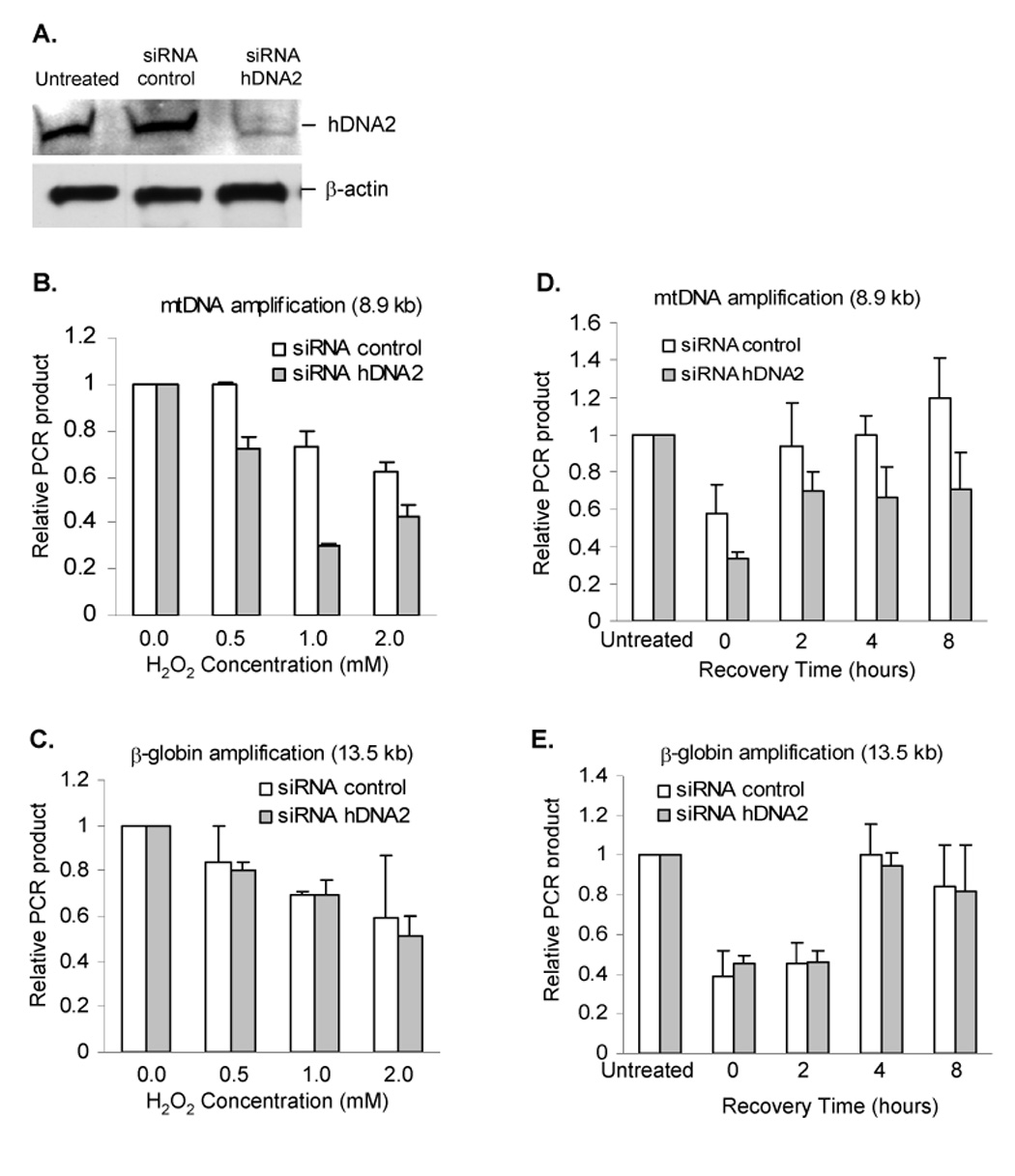
A. SiRNA knockdown of hDNA2. Knockdown efficiency was determined by Western blotting analysis of whole cell extract from HeLa cells not treated with RNA oligos (Lane 1, untreated) or transfected with control siRNA oligos (Lane 2, siRNA control) or siRNA specific to hDNA2 (Lane 3, siRNA-hDNA2). B. and C. H2O2-induced oxidative DNA damage in mitochondria (B) nuclei (C). A long-range QPCR protocol was used to evaluate the oxidative damage in mtDNA or nuclear DNA as induced by H2O2 treatment. The relative PCR amplification of 8.9 kb mtDNA fragment or 13.5 kb nuclear DNA fragment was normalized to mtDNA copy number or total amount of nuclear DNA. D. and E. Time-course of recovery from H2O2 treatment. HeLa cells were treated with 2 mM H2O2 and allowed 0, 2, 4, or 8 h to repair the damage before harvesting for DNA isolation and QPCR analysis of oxidative damage in mtDNA (D) and nuclear DNA (E). All values were mean ± SD (n=3).
Discussion
In the current study, we have shown that hDNA2 predominantly localizes to the mitochondria and the cytoplasma. This observation has significant implications regarding the in vivo function of hDNA2, which has not been fully elucidated. Because DNA2 nuclease, helicase, and ATPase activities are conserved from yeast to humans, it has been speculated that hDNA2, like its yeast homologue, may also be important in Okazaki fragment maturation (Kim et al., 2006; Masuda-Sasa et al., 2006). Our current subcellular localization studies using immunofluorescence staining, transient expression of c-myc-tagged hDNA2, and Western blotting analysis clearly show that hDNA2 does not localize to the nucleus, possibly due to the lack of a NLS. Thus, hDNA2 is unlikely to be the primary player in nuclear Okazaki fragment maturation as is yDna2. The observation that hFEN1 but not hDNA2 is recruited to nuclear DNA replication sites further suggests that FEN1 is the major nuclease in nuclear RNA primer removal in mammalian cells. Previous studies have indicated that nuclear Okazaki fragment maturation in mammals is quite different from that in yeast. While DNA2 and FEN1 in yeast are both involved in RNA primer removal, DNA2 is essential and FEN1 is not (Ayyagari et al., 2003; Bae et al., 2001; Budd et al., 1995; Reagan et al., 1995; Sommers et al., 1995). In contrast, Fen1 gene knockout mice displayed cellular death and embryonic lethality (Kucherlapati et al., 2002; Larsen et al., 2003). Depletion of FEN1 from a nuclear extract of mouse embryonic fibroblast cells eliminates most activity in processing RNA primers in a nick-translation assay, implicating FEN1 as the primary player in RNA primer removal in mammalian cells (Zheng et al., 2007a). In addition, the FEN1 point mutation E160D, which eliminates the exonuclease activity for removal of the last ribonucleotides in RNase H1-mediated RNA primer removal pathway (Turchi et al., 1994), resulted in frequent spontaneous mutations and the development of cancers (Zheng et al., 2007b). Nevertheless, our study does not exclude the possibility that hDNA2 may be co-transported into the nucleus via NLS-independent mechanisms (Xu and Massague, 2004) in response to external stimuli. Future studies will clarify if hDNA2 plays a role in nuclear DNA repair, as does its yeast homologue.
The mitochondrial localization of hDNA2 suggests a role for hDNA2 in mitochondrial DNA replication and repair. As well, hDNA2 may provide helicase activity to unwind the DNA duplex during mtDNA replication. mtDNA is typically replicated via a strand-displacement mode (Shadel and Clayton, 1997), which requires helicase activity to unwind the DNA duplex. It is generally thought that the T7-like helicase Twinkle, the product of the PEO gene, functions as a mtDNA replicative helicase (Korhonen et al., 2003; Korhonen et al., 2004; Spelbrink et al., 2001). In the current study, we show that hDNA2 forms a complex with Polγ and significantly stimulates its activity on a double strand plasmid-based D-loop substrate, but it does not enhance Polγ activity on a short oligonucleotide DNA template. We also observed that hDNA2 endonuclease activity did not cleave a D-loop substrate in vitro (data not shown). Thus, displacement of the DNA duplex mediated by the helicase activity of hDNA2 is likely responsible for hDNA2 stimulation of the Polγ activity. Both hDNA2 and Twinkle may be important in mtDNA replication as replicative helicases. It will be necessary to resolve the relative roles of hDNA2 and Twinkle in future work.
Our data suggest that hDNA2 may play a vital role in RNA primer removal during mtDNA replication. RNA primer removal is a critical process in mtDNA replication, regardless of whether replication occurs by a strand-displacement or by a strand-coupled replication mode (Holt et al., 2000; Shadel and Clayton, 1997). The major pathway for RNA primer removal is via displacement and cleavage of the RNA primer. Our current studies suggest that hDNA2 may function in different ways during the maturation process to remove RNA primers in mitochondria. Firstly, hDNA2 forms a complex with Polγ, serving as an alternative to the Twinkle helicase. hDNA2 helicase activity enhances Polγ-driven displacement DNA synthesis, resulting in the formation of an RNA primer flap. A similar mechanism may be employed by yDNA2 during Okazaki fragment maturation in nuclear DNA replication (Bae et al., 2002; Budd and Campbell, 1997). Secondly, hDNA2 participates in cleavage of the flap structure, which consists of both RNA and DNA portions and is typically 20 to 25 nt. While such flap structures can be processed by hFEN1 alone to generate DNA ends for ligation, the cooperative action of hDNA2 and hFEN1 results in significantly enhanced efficiency. Alternatively, RNA primers may be removed by an RNase H1-mediated pathway. RNase H1, which has been demonstrated to localize to mitochondria and is important for mtDNA replication (Cerritelli et al., 2003), may remove all ribonucleotides, with the exception of the last one in the RNA primer (Turchi et al., 1994). Both hDNA2 and hFEN1 have 5’ exonuclease activity and may play a role in this pathway. Furthermore, immunodepletion of hDNA2 or hFEN1 from HeLa ME results in a decrease in ligated products, accompanied by the accumulation of non-ligated intermediates in an in vitro nick translation assay using a gapped DNA substrate containing a ribonucleotide at the 5’ end of the downstream oligomer. This suggests that both hDNA2 and FEN1 are involved in processing the intermediate resulting from RNase H1 cleavage of the mtRNA primer.
It has been demonstrated that mtDNA is consistently exposed to DNA damaging agents due to byproducts of oxidative phosphorylation in this organelle (Harman, 1972; Sohal and Weindruch, 1996). Thus, robust DNA repair, particularly BER, is important to maintain the integrity of the mitochondrial genome. Short-patch BER has been reported in mitochondria (Bohr et al., 2002; Pinz and Bogenhagen, 1998). We have recently found that the LP-BER is also important in mitochondria, because damaged nucleotides with modified base and sugar groups such as 2-deoxyribonolactone (dL) can be only repaired through this pathway (Liu et al., 2008). In LP-BER, displacement DNA synthesis results in a flap structure bearing the abasic residue (Klungland and Lindahl, 1997). Such a DNA intermediate structure must be removed before the DNA ligase can seal the nick to complete the repair process. We and others have previously suggested that in mitochondria, FEN1 is partially responsible for processing of LP-BER intermediates. Our current studies implicate hDNA2 as another nuclease capable of processing nick or flap LP-BER intermediates in mitochondria. Furthermore, hDNA2 and FEN1 have synergistic effects in this process, most likely due to mutual stimulation of hDNA2 and FEN1. We suggest that hDNA2 helicase activity may enhance the formation of flaps for FEN1 to cleave. It has been demonstrated that the flap substrate is a far better substrate for FEN1 compared to the nick substrate (Harrington and Lieber, 1994). Thus, conversion of the nick substrate into a flap substrate could certainly enhance the efficiency of FEN1 cleavage.
Experimental Procedures
Purification of nuclei and mitochondria and preparation of mitochondrial extracts
Nuclei and mitochondria were fractionated using 1M/1.5M sucrose step gradients centrifugation as previously described (Liu et al., 2008). The integrity of purified mitochondria was tested by treatment with 1 mg / ml protease K (37°C, 30 min). To prepare nuclear extract, nuclei were incubated with a NE buffer (20 mM Hepes-KOH, pH 8.0, 1.5 mM MgCl2, 400 mM NaCl, 0.2 mM EDTA, 1 mM DTT, and 17% glycerol). To prepare the mitochondrial extract (ME), purified mitochondria were incubated in NE buffer containing 0.25% CHAPS. The ME was dialyzed against the NE buffer without CHAPS.
Protein expression and purification
hFEN1, APE1, DNA ligase III, and Polγ were expressed and purified as previously described (Frank et al., 2001; Guo et al., 2008; Yakubovskaya et al., 2006). hDNA2 was expressed and purified following a published protocol, with modifications (Masuda-Sasa et al., 2006). Briefly, the full length cDNA of hDNA2 corresponding to the DNA2L (NM_001080449) mRNA sequence was successfully amplified by RT-PCR using mRNA extracted from the MCF-7 breast cancer cell line. The hDNA2 cDNA sequence, which was confirmed by DNA sequencing, was subcloned into pBlueBacHis2 to express 6His- and Xpress tagged DNA2 in Sf9 insect cells. hDNA2 was first purified by FPLC using a Ni2+-chelating column. The eluted hDNA2 was further purified by anti-Xpress antibody conjugated agarose beads. hDNA2 bound to agarose beads was eluted with 1 mg/ml Xpress peptide and dialyzed against a buffer containing 50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 1 mM DTT, and 50% glycerol.
Nuclease assay
32P-labeled nick or flap DNA substrates were prepared as described previously (Zheng et al., 2005). To assay nuclease activity, mitochondrial extract, DNA2, or FEN1 were incubated (37°C, 30 min, or as specified) with 1 pmol DNA substrates in reaction buffer A containing 50 mM Hepes-KOH (pH 7.5), 5 mMMgCl2, 2 mM DTT, and 0.25 mg/ml BSA.
Polγ activity assay
To prepare plasmid-based D-loop DNA substrates, T3 primers (21 nt) were incubated with pBluescript plasmid at 95 °C for 10 min followed by 55°C for 10 min. The reaction was cooled to room temperature. The D-loop substrate was purified with a Qiagen plasmid purification kit (Qiagen, Valencia, CA). To assay Polγ activity, the polymerase was incubated with indicated DNA substrates in the reaction buffer B containing 50 mM Hepes-KOH (pH 7.5), 45 mM KCl, 5 mM MgCl2, 1 mM DTT, 0.1 mM EDTA, 2 mM ATP, 200 unit creatine-phosphokinase, 0.5 mM NAD, and 5 mM phosphocreatine. Reactions (37°C, 30 min) were carried out in the presence of 5 µci [α-32P] dATP, 1 µM dATP, and 50 µM each of dCTP, dGTP, and dTTP. To quantify the DNA synthesis by Polγ, DNA products were purified by Dynabeads M-280 Streptavidin (Invitrogen, Carlsbad, CA), or the Qiagen plasmid purification kit. The incorporation of [α-32P] dATP was determined by a scintillation counter. Background counts from reactions without Polγ were subtracted from the corresponding Polγ-driven reactions.
Nick translation assay
Gap filling, RNA/DNA primer removal, and DNA ligation reactions were assayed by nick translation reaction with gapped substrates, following a modified version of published protocols (Zheng et al., 2007a). Briefly, purified proteins or mitochondrial extracts were incubated with specific gapped substrates in the reaction buffer B containing 5 µCi [α-32P]dCTP and 50 µM each of dATP, dGTP, and dTTP. Reactions were carried out at 37°C for 60 min.
In vitro LP-BER assay
LP-BER was assayed using a synthetic DNA duplex harboring a tetrahydrofuran (THF) AP site at residue 40. Reactions were conducted according to published procedures, with slight modifications (Klungland and Lindahl, 1997; Sung et al., 2005). Briefly, ME or purified BER proteins were mixed with 1 pmol DNA substrates in reaction buffer B. Each reaction also contained 5 µCi [α-32P] dCTP and 50 µM each of dATP, dGTP, and dTTP. Reactions were carried out for 30 min at 37°C.
Immunodepletion of human DNA2 and FEN1
The polyclonal antibody against FEN1 was raised in a rabbit against purified recombinant hFEN1 (Zheng et al., 2005). The polyclonal antibody against hDNA2 was raised in a rabbit against a purified peptide TDKVPAPEQVEKGG, corresponding to the amino acid sequence from 983–996, at the C-terminus of hDNA2. The peptide synthesis and anti-hDNA2 antibody production were completed by Genscript Corp (Piscataway, NJ). Reactivity and specificity to the hDNA2 C-terminal polypeptide was verified by Elisa in Genscript (Piscataway, NJ). Effectiveness of the antibody for immunoblotting analysis of hDNA2 was confirmed by Western blotting analysis of recombinant hDNA2 (data not shown). To immunodeplete hDNA2 or hFEN1, 100 µg of HeLa mitochondrial extract was incubated with protein A agarose beads (50 µl) coated with rabbit polyclonal antibodies to hDNA2 or hFEN1 in the NE buffer at 4°C for 8 hours. After brief centrifugation (5,000 g), the supernatant was analyzed by Western blot analysis to confirm depletion efficiency.
Co-immunoprecipitation and affinity-precipitation assays
Non-specific mouse antibodies or a mouse polyclonal antibody against Polγ (Novus Biologicals, Inc. Littleton, CO) were conjugated to protein A agarose beads. Beads were incubated (4°C, overnight) with ME and washed with PBS buffer containing 0.2% Tween 20. hDNA2 associated with beads was analyzed with a rabbit polyclonal antibody against hDNA2. To co-precipitate recombinant hDNA2 with recombinant Polγ, purified Polγ or BSA were coated onto Sepharose 4B beads and incubated (4°C, overnight) with insect cell lysates containing recombinant hDNA2 in the absence or presence of 100 µg/ml each of DNase I and RNase A. Beads were washed with PBS buffer containing 0.2% Tween 20. hDNA2 bound to Sepharose 4B beads was eluted with 50 mM Tris buffer (pH 7.5) containing an indicated concentration of NaCl or SDS-PAGE sample buffer and was analyzed by Western blotting using a rabbit polyclonal antibody to hDNA2.
Immunofluorescence microscopy
HeLa cells were fixed with 4% paraformaldehyde and stained with an antibody against specified proteins. Nuclei were stained with DAPI as previously described (Zheng 2007a) and slides were examined by fluorescence microscopy, (AX70, Olympus, Center Valley, PA or Zeiss LSM510 confocal microscope, Carl Zeiss, Thornwood, NY). To observe the localization of exogenous hDNA2-GFP, HeLa cells were transfected with vectors encoding hDNA2-GFP using Lipofectamine 2000 (Invitrogen, Carlsbad, CA), cultured (24–48 h), then incubated (10 min) with Mitotracker red dye and washed with PBS. Cells were left untreated or fixed with 2% paraformaldehyde (10 min). The hDNA2-GFP and Mitotracker red signals were detected by fluorescence microscopy. All images were processed using Adobe Photoshop 7.0 (Adobe, San Jose, CA).
SiRNA
siRNA oligos (a mixture of three oligos) against hDNA2 (catalog# sc-90458) or control siRNA oligos (catalog# sc-37007) were purchased from Santa Cruz Biotechnologies (Santa Cruz, CA). siRNA oligos were transfected into HeLa cells using an siRNA transfection kit (Santa Cruz Biotechnologies, Santa Cruz, CA) according to manufacturer’s instructions. The inhibition of hDNA2 expression was confirmed by Western blotting utilizing a polyclonal antibody against hDNA2.
H2O2 treatment and Quantitative PCR
HeLa cells (2 × 105) were transfected with control siRNA oligos or siRNA specific to hDNA2. After transfection, cells were incubated (48 h) in fresh DMEM, treated with H2O2 (30 min), washed twice with fresh DMEM and harvested immediately or incubated for a specific time to allow recovery from H2O2 treatment. Nuclear or cytoplasmic DNA was isolated with DNeasy Blood and Tissue Kit (Qiagen, Valencia, CA). The relative amount of H2O2-induced oxidative DNA damage in control and siRNA-hDNA2 knockdown HeLa cells was assayed by quantitative PCR as previously described (Liu et al., 2008; Santos et al., 2002). Briefly, an 8.9 kb mitochondrial DNA fragment or a 13.5 kb nuclear DNA fragment was amplified using primers and PCR conditions as specified previously (Liu et al., 2008; Santos et al., 2002). A 221 bp small fragment was amplified to determine the copy number of mitochondria. The relative PCR product, which represents the relative level of oxidative DNA damage, was calculated by dividing the raw fluorescence value of a H2O2-treated sample by that of the corresponding H2O2-untreated sample. The relative PCR product of mtDNA was normalized to the copy number of mtDNA.
Supplementary Material
Acknowledgements
We thank the microscopy core-facility of COH for technical assistance of immunofluorescence staining of HeLa cells. We thank P. Liu for technical advice on the measurement of H2O2-induced DNA damage in nuclei and mitochondria. We thank S. R. da Costa for editorial assistance. This work was supported by an NIH grant R01 CA085344 to B.H.S as well as partly by GM040000 to B.D, and ES012039 to D.F.B.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Akbari M, Visnes T, Krokan HE, Otterlei M. Mitochondrial base excision repair of uracil and AP sites takes place by single-nucleotide insertion and long-patch DNA synthesis. DNA Repair (Amst) 2008;7:605–616. doi: 10.1016/j.dnarep.2008.01.002. [DOI] [PubMed] [Google Scholar]
- Ayyagari R, Gomes XV, Gordenin DA, Burgers PM. Okazaki fragment maturation in yeast. I. Distribution of functions between FEN1 AND DNA2. J Biol Chem. 2003;278:1618–1625. doi: 10.1074/jbc.M209801200. [DOI] [PubMed] [Google Scholar]
- Bae SH, Bae KH, Kim JA, Seo YS. RPA governs endonuclease switching during processing of Okazaki fragments in eukaryotes. Nature. 2001;412:456–461. doi: 10.1038/35086609. [DOI] [PubMed] [Google Scholar]
- Bae SH, Kim DW, Kim J, Kim JH, Kim DH, Kim HD, Kang HY, Seo YS. Coupling of DNA helicase and endonuclease activities of yeast Dna2 facilitates Okazaki fragment processing. J Biol Chem. 2002;277:26632–26641. doi: 10.1074/jbc.M111026200. [DOI] [PubMed] [Google Scholar]
- Bae SH, Seo YS. Characterization of the enzymatic properties of the yeast dna2 Helicase/endonuclease suggests a new model for Okazaki fragment processing. J Biol Chem. 2000;275:38022–38031. doi: 10.1074/jbc.M006513200. [DOI] [PubMed] [Google Scholar]
- Bohr VA, Stevnsner T, de Souza-Pinto NC. Mitochondrial DNA repair of oxidative damage in mammalian cells. Gene. 2002;286:127–134. doi: 10.1016/s0378-1119(01)00813-7. [DOI] [PubMed] [Google Scholar]
- Budd ME, Campbell JL. A yeast gene required for DNA replication encodes a protein with homology to DNA helicases. Proc Natl Acad Sci U S A. 1995;92:7642–7646. doi: 10.1073/pnas.92.17.7642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budd ME, Campbell JL. A yeast replicative helicase, Dna2 helicase, interacts with yeast FEN-1 nuclease in carrying out its essential function. Mol Cell Biol. 1997;17:2136–2142. doi: 10.1128/mcb.17.4.2136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budd ME, Campbell JL. The pattern of sensitivity of yeast dna2 mutants to DNA damaging agents suggests a role in DSB and postreplication repair pathways. Mutat Res. 2000;459:173–186. doi: 10.1016/s0921-8777(99)00072-5. [DOI] [PubMed] [Google Scholar]
- Budd ME, Choe WC, Campbell JL. DNA2 encodes a DNA helicase essential for replication of eukaryotic chromosomes. J Biol Chem. 1995;270:26766–26769. doi: 10.1074/jbc.270.45.26766. [DOI] [PubMed] [Google Scholar]
- Cerritelli SM, Frolova EG, Feng C, Grinberg A, Love PE, Crouch RJ. Failure to produce mitochondrial DNA results in embryonic lethality in Rnaseh1 null mice. Mol Cell. 2003;11:807–815. doi: 10.1016/s1097-2765(03)00088-1. [DOI] [PubMed] [Google Scholar]
- Diekert K, Kispal G, Guiard B, Lill R. An internal targeting signal directing proteins into the mitochondrial intermembrane space. Proc Natl Acad Sci U S A. 1999;96:11752–11757. doi: 10.1073/pnas.96.21.11752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank G, Qiu J, Zheng L, Shen B. Stimulation of eukaryotic flap endonuclease-1 activities by proliferating cell nuclear antigen (PCNA) is independent of its in vitro interaction via a consensus PCNA binding region. J Biol Chem. 2001;276:36295–36302. doi: 10.1074/jbc.M103397200. [DOI] [PubMed] [Google Scholar]
- Gavel Y, von Heijne G. Cleavage-site motifs in mitochondrial targeting peptides. Protein Eng. 1990;4:33–37. doi: 10.1093/protein/4.1.33. [DOI] [PubMed] [Google Scholar]
- Guo Z, Chavez V, Singh P, Finger LD, Hang H, Hegde ML, Shen B. Comprehensive mapping of the C-terminus of flap endonuclease-1 reveals distinct interaction sites for five proteins that represent different DNA replication and repair pathways. J Mol Biol. 2008;377:679–690. doi: 10.1016/j.jmb.2007.10.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harman D. The biologic clock: the mitochondria? J Am Geriatr Soc. 1972;20:145–147. doi: 10.1111/j.1532-5415.1972.tb00787.x. [DOI] [PubMed] [Google Scholar]
- Harrington JJ, Lieber MR. The characterization of a mammalian DNA structure-specific endonuclease. EMBO J. 1994;13:1235–1246. doi: 10.1002/j.1460-2075.1994.tb06373.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt IJ, Lorimer HE, Jacobs HT. Coupled leading- and lagging-strand synthesis of mammalian mitochondrial DNA. Cell. 2000;100:515–524. doi: 10.1016/s0092-8674(00)80688-1. [DOI] [PubMed] [Google Scholar]
- Kim JH, Kim HD, Ryu GH, Kim DH, Hurwitz J, Seo YS. Isolation of human Dna2 endonuclease and characterization of its enzymatic properties. Nucleic Acids Res. 2006;34:1854–1864. doi: 10.1093/nar/gkl102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klungland A, Lindahl T. Second pathway for completion of human DNA base excision-repair: reconstitution with purified proteins and requirement for DNase IV (FEN1) Embo J. 1997;16:3341–3348. doi: 10.1093/emboj/16.11.3341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korhonen JA, Gaspari M, Falkenberg M. TWINKLE Has 5' -> 3' DNA helicase activity and is specifically stimulated by mitochondrial single-stranded DNA-binding protein. J Biol Chem. 2003;278:48627–48632. doi: 10.1074/jbc.M306981200. [DOI] [PubMed] [Google Scholar]
- Korhonen JA, Pham XH, Pellegrini M, Falkenberg M. Reconstitution of a minimal mtDNA replisome in vitro. Embo J. 2004;23:2423–2429. doi: 10.1038/sj.emboj.7600257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kucherlapati M, Yang K, Kuraguchi M, Zhao J, Lia M, Heyer J, Kane MF, Fan K, Russell R, Brown AM, et al. Haploinsufficiency of Flap endonuclease (Fen1) leads to rapid tumor progression. Proc Natl Acad Sci USA. 2002;99:9924–9929. doi: 10.1073/pnas.152321699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen E, Gran C, Saether BE, Seeberg E, Klungland A. Proliferation failure and gamma radiation sensitivity of Fen1 null mutant mice at the blastocyst stage. Mol Cell Biol. 2003;23:5346–5353. doi: 10.1128/MCB.23.15.5346-5353.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P, Qian L, Sung JS, de Souza-Pinto NC, Zheng L, Bogenhagen DF, Bohr VA, Wilson DM, 3rd, Shen B, Demple B. Removal of Oxidative DNA Damage via FEN1-Dependent Long-Patch Base Excision Repair in Human Cell Mitochondria. Mol Cell Biol. 2008;28:4975–4987. doi: 10.1128/MCB.00457-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuda-Sasa T, Imamura O, Campbell JL. Biochemical analysis of human Dna2. Nucleic Acids Res. 2006;34:1865–1875. doi: 10.1093/nar/gkl070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakai K, Horton P. PSORT: a program for detecting sorting signals in proteins and predicting their subcellular localization. Trends Biochem Sci. 1999;24:34–36. doi: 10.1016/s0968-0004(98)01336-x. [DOI] [PubMed] [Google Scholar]
- Pinz KG, Bogenhagen DF. Efficient repair of abasic sites in DNA by mitochondrial enzymes. Mol Cell Biol. 1998;18:1257–1265. doi: 10.1128/mcb.18.3.1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reagan MS, Pittenger C, Siede W, Friedberg EC. Characterization of a mutant strain of Saccharomyces cerevisiae with a deletion of the RAD27 gene, a structural homolog of the RAD2 nucleotide excision repair gene. J Bacteriol. 1995;177:364–371. doi: 10.1128/jb.177.2.364-371.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos JH, Mandavilli BS, Van Houten B. Measuring oxidative mtDNA damage and repair using quantitative PCR. Methods Mol Biol. 2002;197:159–176. doi: 10.1385/1-59259-284-8:159. [DOI] [PubMed] [Google Scholar]
- Shadel GS, Clayton DA. Mitochondrial DNA maintenance in vertebrates. Annu Rev Biochem. 1997;66:409–435. doi: 10.1146/annurev.biochem.66.1.409. [DOI] [PubMed] [Google Scholar]
- Sohal RS, Weindruch R. Oxidative stress, caloric restriction, and aging. Science. 1996;273:59–63. doi: 10.1126/science.273.5271.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommers CH, Miller EJ, Dujon B, Prakash S, Prakash L. Conditional lethality of null mutations in RTH1 that encodes the yeast counterpart of a mammalian 5'- to 3'-exonuclease required for lagging strand DNA synthesis in reconstituted systems. J Biol Chem. 1995;270:4193–4196. doi: 10.1074/jbc.270.9.4193. [DOI] [PubMed] [Google Scholar]
- Spelbrink JN, Li FY, Tiranti V, Nikali K, Yuan QP, Tariq M, Wanrooij S, Garrido N, Comi G, Morandi L, et al. Human mitochondrial DNA deletions associated with mutations in the gene encoding Twinkle, a phage T7 gene 4-like protein localized in mitochondria. Nat Genet. 2001;28:223–231. doi: 10.1038/90058. [DOI] [PubMed] [Google Scholar]
- Sung JS, DeMott MS, Demple B. Long-patch base excision DNA repair of 2-deoxyribonolactone prevents the formation of DNA-protein cross-links with DNA polymerase beta. J Biol Chem. 2005;280:39095–39103. doi: 10.1074/jbc.M506480200. [DOI] [PubMed] [Google Scholar]
- Szczesny B, Tann AW, Longley MJ, Copeland WC, Mitra S. Long patch base excision repair in mammalian mitochondrial genomes. J Biol Chem. 2008 doi: 10.1074/jbc.M803491200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turchi JJ, Huang L, Murante RS, Kim Y, Bambara RA. Enzymatic completion of mammalian lagging-strand DNA replication. Proc Natl Acad Sci USA. 1994;91:9803–9807. doi: 10.1073/pnas.91.21.9803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L, Massague J. Nucleocytoplasmic shuttling of signal transducers. Nat Rev Mol Cell Biol. 2004;5:209–219. doi: 10.1038/nrm1331. [DOI] [PubMed] [Google Scholar]
- Yakubovskaya E, Chen Z, Carrodeguas JA, Kisker C, Bogenhagen DF. Functional human mitochondrial DNA polymerase gamma forms a heterotrimer. J Biol Chem. 2006;281:374–382. doi: 10.1074/jbc.M509730200. [DOI] [PubMed] [Google Scholar]
- Zheng L, Dai H, Qiu J, Huang Q, Shen B. Disruption of the FEN-1/PCNA interaction results in DNA replication defects, pulmonary hypoplasia, pancytopenia, and newborn lethality in mice. Mol Cell Biol. 2007a;27:3176–3186. doi: 10.1128/MCB.01652-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng L, Dai H, Zhou M, Li M, Singh P, Qiu J, Tsark W, Huang Q, Kernstine K, Zhang X, et al. Fen1 mutations result in autoimmunity, chronic inflammation, and cancers. Nature Medicine. 2007b;13:812–819. doi: 10.1038/nm1599. [DOI] [PubMed] [Google Scholar]
- Zheng L, Zhou M, Chai Q, Parrish J, Xue D, Patrick SM, Turchi JJ, Yannone SM, Chen D, Shen B. Novel function of the flap endonuclease 1 complex in processing stalled DNA replication forks. EMBO Rep. 2005;6:83–89. doi: 10.1038/sj.embor.7400313. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.