

Abstract
Voltage-gated K+ channels are complexes of membrane-bound, ion-conducting α and cytoplasmic ancillary (β) subunits. The primary physiologic effect of coexpression of α and β subunits is to increase the intrinsic rate of inactivation of the α subunit. For one β subunit, Kvβ1.1, inactivation is enhanced through an N-type mechanism. A second β subunit, Kvβ1.2, has been shown to increase inactivation, but through a distinct mechanism. Here we show that the degree of enhancement of Kvβ1.2 inactivation is dependent on the amino acid composition in the pore mouth of the α subunit and the concentration of extracellular K+. Experimental conditions that promote C-type inactivation also enhance the stimulation of inactivation by Kvβ1.2, showing that this β subunit directly stimulates C-type inactivation. Chimeric constructs containing just the nonconserved N-terminal region of Kvβ1.2 fused with an α subunit behave in a similar fashion to coexpressed Kvβ1.2 and α subunit. This shows that it is the N-terminal domain of Kvβ1.2 that mediates the increase in C-type inactivation from the cytoplasmic side of the pore. We propose a model whereby the N terminus of Kvβ1.2 acts as a weakly binding “ball” domain that associates with the intracellular vestibule of the α subunit to effect a conformational change leading to enhancement of C-type inactivation.
Voltage-gated K+ channels are found in nearly all mammalian tissues where their function is to stabilize membrane potential and promote repolarization. Despite their common roles many K+ currents have been described; they are especially prominent in excitable cell types found in the nervous system, heart, and smooth muscle. The large assortment of K+ currents is probably generated by differential expression of the 18 known α subunits (1) and the 5 known β subunits (2–7).
Perhaps the most prominent difference in the physiologic properties of K+ channels are their inactivation properties, which can be extremely variable in time course and can occur by multiple mechanisms. Channel inactivation is best understood in Shaker-type K+ channels. One type of inactivation mechanism found in Shaker K+ channels is N-type or fast inactivation, caused by occlusion of the cytoplasmic side of the pore by a “ball” domain on the N terminus of some α or β subunits (3, 8, 9). A second, and possibly more common inactivation mechanism is C-type inactivation (9, 10), which may be mediated by closure of domains near the pore mouth (11). Each type of inactivation can occur independently, or N- and C-type inactivation can coexist in the same channel. Additionally, entry into the C-type inactivated state is faster in the presence of N-type inactivation (10).
A recently discovered class of ancillary (β) subunit, can dramatically increase the rate of inactivation of the membrane-bound Kv1 α subunits (3–6, 12). One β subunit, Kvβ1.1, has been shown to increase the rate of inactivation through interaction of its N terminus with the channel pore to cause N-type inactivation (3). Kvβ1.2, which except for a unique N-terminal 78-amino acid leader is identical to Kvβ1.1, can also increase the rate of inactivation, but through an undetermined mechanism (4).
We show that the increased rate of inactivation in Kvα1 subunits associated with Kvβ1.2 is due to an increase in the rate of C-type inactivation. Further, unlike the intrinsic C-type inactivation seen in Kvα subunits expressed alone, it is the unique N-terminal leader of Kvβ1.2 that is responsible for this effect. We propose that the mechanism of β subunit-stimulated C-type inactivation involves the N terminus of Kvβ1.2 binding to the inner vestibule of the channel pore with rapid kinetics and low affinity. This interaction does not generate resolvable N-type inactivation, but is sufficient to result in faster C-type inactivation through its coupling with N-type inactivation.
MATERIALS AND METHODS
K+ channel β subunits were named using the suggested nomenclature (6, 7). Kvβ1.2 was formerly referred to as Kvβ3 (4, 5), and Kvβ1.1 as Kvβ1 (3). Unless otherwise stated, standard molecular biology techniques were used (13). Enzymes were used according to the manufacturers recommendations. Complementary DNAs encoding ferret Kvβ1.2, FK1Δ2-146, and FK1Δ2-146[K532Y] were described elsewhere (4, 14, 15). Mutations in ShBΔ6-46 (9) were constructed by the method of Ray and Nickoloff (16) using the mutagenic oligonucleotides (mutagenic bases are underlined): TGGTGACATGAAACCCGTCGGCGT (T449K mutation) or TGGTGACATGTATCCCGTCGGCGT (T449Y) mutation. The selection oligonucleotide for both mutations was AATATGGACAGCTGGTAACGTGAT (which destroyed a unique SpeI site). Fidelity of the mutations was confirmed by sequencing the region inclusive of the PCR primers. Kvβ1.1 was isolated by reverse transcription of rat brain RNA, followed by PCR with the primers AGGATTTACTCATGCAGCTTAAGTG and CTGTGTGTGGGTTCTGAAGTCAGC. The product was cloned into the SmaI site of pGEM-HE5 [a modified version of a Xenopus oocyte expression vector pGEM-HE (17), modified to include XhoI, NotI, and SalI restriction sites downstream from the Xenopus β globin 3′ untranslated region]. DNA sequence analysis showed that this clone encoded an open reading frame identical to that previously reported (3). pNβ1.2/FK1Δ2-146 chimera was constructed by recombinant PCR (18). The first 80 amino acids of Kvβ1.2 were excised by PCR with an adapter oligonucleotide [ACATTCATCACCATGCATAGCCATGCCCGTCTCTGCTC; the first 20 nucleotides are from nucleotides 738–757 of FK1 (15), and the 3′ 20 bases are from nucleotides 281–300 of FHBK1 (4)] and BS828L (CAGGAAACAGCTATGACCATGATT; nucleotides 805–828 of pBS-SK−; Strategene). The purified PCR product was then reamplified using NcoI-digested pMC1-12 (containing FK1) (15) as template, with BS828L and MC12-34 (CTCTCTCACAAAGCCCT; nucleotides 1125–1141) primers. The PCR product was digested with SpeI and EcoRV, and ligated into the SpeI/EcoRV sites of FK1Δ2-146. The resulting construct contains the first 80 amino acids of Kvβ1.2 spliced to the N-terminus of FK1Δ2-146. The fidelity of the mutants was confirmed by DNA sequence analysis. For in vitro transcription, RNAs were linearized with HindIII (Sh constructs), NotI (Kvβ1.1), or Asp718 (others), and transcribed with T7 (Sh and Kvβ1.1) or T3 RNA polymerase as described (19) using 0.6 mM GTP, and 2.4 mM m7G(5′)ppp(5′)GTP in the transcription reactions.
In vitro transcribed RNA (up to 50 ng) was injected into defolliculated stage V–VI Xenopus laevis oocytes. Voltage-clamp experiments were conducted on oocytes within 3–6 days of injection using a two electrode “bath clamp” amplifier (OC-725A, Warner Instruments, Hamden, CT) as described (4). No correlation between current size and inactivation rate was detected for the range of currents measured in this study. Extracellular solution was 1.0 mM MgCl2/1.8 mM CaCl2/5 mM Hepes·NaOH, pH 7.4, plus 96 mM NaCl/2 mM KCl (low [K+]o) or 0 mM NaCl/98 mM KCl (high [K+]o). Electrodes were filled with 3 M KCl (resistances 0.6–1.5 MΩ) and shielded to reduce capacitive coupling. All measurements were carried out at room temperature. Data were not leakage or capacitance subtracted, unless otherwise specified. Data were filtered at 2 kHz and analyzed using an Axon Instruments (Foster City, CA) TL-1 interface and pclamp software. To control for variability in the kinetics of C-type inactivation that occurred between batches of oocytes, data comparisons were performed the same day against suitable controls from the same batch of oocytes. Inactivation rates were fit with either one or two exponentials and the dominant exponential time constants reported. Confidence levels were calculated using an unpaired t test.
RESULTS AND DISCUSSION
We have previously shown that Kvβ1.2 can accelerate inactivation of Kvα1.4 and Shaker B that lack N-type inactivation due to removal of their N-terminal ball domains (14). We have reproduced these results for N-terminal deleted mutants of Kv1.4 (FK1Δ2-146) and Shaker (ShBΔ6-46) in Fig. 1 A and B. The apparent time constant of inactivation decreases by 73% and 33% for Kv1.4 (FK1Δ2-146) and Shaker (ShBΔ6-46), respectively, when coexpressed with Kvβ1.2. However, the enhancement is much slower than would be predicted for the wild-type N-type inactivating channels. Both these mutant α subunits display a slow phase of inactivation attributable to C-type inactivation (9, 14). Unlike Kvβ1.1, Kvβ1.2 did not alter inactivation of Kvα1.1, an α subunit that shows little C-type inactivation (3, 20), despite data demonstrating that Kvβ1.2 is capable of binding Kvα1.1 (21–23).
Figure 1.
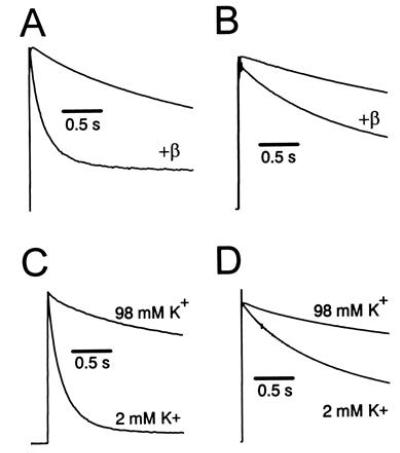
Kvβ1.2 causes a [K+]o-sensitive increase in the rate of inactivation of α subunits. (A) Effects of coexpression of Kvβ1.2 with FK1Δ2-146. Current traces are in response to a depolarizing pulse to +50 mV from a holding potential of −90 mV. Currents shown in this and subsequent figures were normalized to peak current for purposes of comparison. (B) Effects of coexpression of Kvβ1.2 with ShBΔ6-46. (C) [K+]o sensitivity of coexpression of Kvβ1.2 with FK1Δ2-146. Peak current values at +50 mV in 2 mM [K+]o were 3.9 ± 1.5 μA for FK1Δ2-146 plus Kvβ1.2. (D) [K+]o sensitivity of coexpression of Kvβ1.2 with ShBΔ6-46. Peak current value at +50 mV in 2 mM [K+]o was 8.30 ± 0.19 μA.
These results suggested that Kvβ1.2 increased the rate of inactivation through C-type inactivation instead of the N-type inactivation shown for Kvβ1.1. To test for β subunit stimulation of C-type inactivation, we used criteria described by Hoshi et al. (9): inhibition by high extracellular K+ concentration, and response to specific amino acid substitutions near the pore mouth at position 449 in ShB (see Fig. 2A). Increasing [K+]o from 2 to 98 mM reduced inactivation ≈3-fold [at 6.0 s, P < 0.05; see Fig. 1C; Table 1) of N-terminal-deleted Kvα1.4 (FK1Δ2-146 (15)] coexpressed with Kvβ1.2. Similar results are obtained when ShBΔ6-46 plus Kvβ1.2 inactivation was measured at 98 mM [K+]o, decreasing total inactivation 45% (P < 0.05) at the end of 3.0 s (Fig. 1D). The sensitivity of inactivation to high [K+]o shows that the Kvβ1.2-mediated increase in inactivation fulfills an important criterion for C-type inactivation.
Figure 2.
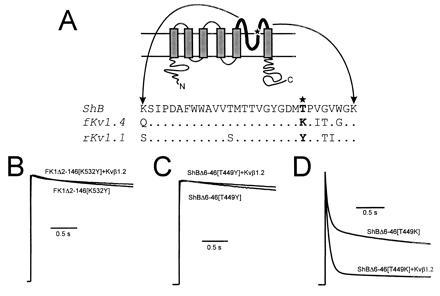
Kvβ1.2 influence on the rate of C-type inactivation rate in the presence of pore mouth mutations that affect C-type inactivation. (A Upper) Schematic representation of a voltage gated K+ channel 6-membrane spanning domain structure. The bold line represents the pore forming domain, with the star showing the approximate position of the pore mouth mutations. (Bottom) Alignments of the amino acid sequence of the pore regions from ShB (24), ferret Kv1.4 (15), and rat Kv1.1 (25). Mutations that alter the rate of C-type inactivation were constructed at position 449 in Shaker and 532 in fKv1.4 (★). (B) Coexpression of Kvβ1.2 with FK1Δ2-146[K532Y] had no effect on inactivation rate (P > 0.25). (C) Coexpression of Kvβ1.2 with ShBΔ6-46[T449Y] also had no effect on inactivation rate (P > 0.25). (D) ShBΔ6-46[T449K] mutation greatly increased the rate of C-type inactivation. Total inactivation measured at 100 ms increased 1.4 fold (Table 1; P < 0.05) in response to Kvβ1.2 coexpression. All current traces shown are in response to a depolarizing pulse to +50 mV from a holding potential of −90 mV. Peak current values measured at +50 mV in 2 mM [K+]o were 5.6 ± 1.6 μA for FK1Δ2-146[K532Y], 21 ± 4 μA for ShBΔ2-46[T449Y], 3.4 ± 1.2 μA for FK1Δ2-146[K532Y] plus Kvβ1.2, 28 ± 8 μA for ShBΔ2-46[T449Y] plus Kvβ1.2, 17 ± 1 μA for ShBΔ2-46[T449K], and 19 ± 3 μA for ShBΔ2-46[T449K] plus Kvβ1.2.
Table 1.
Effect of [K+]o or Kvβ1.2 on the development of C-type inactivation
| Channel | [K+]0, mM | dF/P, % | Time, s | n | P |
|---|---|---|---|---|---|
| 2 | 20 ± 0.4 | ||||
| FK1Δ2-146 plus Kvβ1.2 | 98 | 60 ± 0.9 | 6.0 | 3 | <0.05 |
| 2 | 42 ± 1.9 | ||||
| ShBΔ6-46 plus Kvβ1.2 | 98 | 76 ± 1.0 | 3.0 | 4 | <0.05 |
| FK1Δ2-146[K532Y] | 80 ± 0.7 | ||||
| FK1Δ2-146[K532Y] plus Kvβ1.2 | 2 | 73 ± 5.7 | 5.0 | 9 | >0.25 |
| ShBΔ6-46[T449Y] | 79 ± 1.8 | ||||
| ShBΔ6-46[T449Y] plus Kvβ1.2 | 2 | 80 ± 3.5 | 6.0 | 7 | >0.25 |
| ShBΔ6-46[T449K] | 58 ± 4.0 | ||||
| ShBΔ6-46[T449K] plus Kvβ1.2 | 2 | 42 ± 7.6 | 0.1 | 3 | <0.05 |
| 2 | 7.1 ± 3.1 | ||||
| Nβ1.2-FK1Δ2-146 | 98 | 54 ± 3.0 | 2.0 | 5 | <0.01 |
| Nβ1.2-FK1Δ2-146 | 12 ± 2.1 | ||||
| Nβ1.2-FK1Δ2-146[K5432Y] | 2 | 40 ± 2.3 | 5.0 | 4 | <0.05 |
Summary of data from Figs. 1, 2, 3. The degree of inactivation (dF/P) is expressed as the ratio of current amplitudes at the final time (shown in subsequent column) vs peak amplitude; smaller percentages represent greater extent of inactivation. Confidence levels represent the difference or similarity of matched sets.
Shaker Kvα1 subunits show dramatically different rates of C-type inactivation in the presence of various amino acids at position 449 (532 in FK1; Fig. 2A) (14, 26), although other amino acids can influence the intrinsic inactivation rate. Lysine, which is present in Kvα1.4 at this position, is most favorable to C-type inactivation; threonine, found in Shaker B, supports an intermediate level of C-type inactivation. Channels that contain a tyrosine at this position, such as Kvα1.1, display very little inactivation (20). One interpretation of these observations is that Kvβ1.2-mediated acceleration of C-type inactivation should be more pronounced in the presence of α subunits with an amino acid sequence favorable to C-type inactivation. It is also possible that the observed effects might be due to some other property of these α subunits. We therefore constructed a series of pore mouth mutations in FK1Δ2-146 and ShBΔ6-46 (Fig. 2A). As predicted, Kvβ1.2 fails to increase the rate of inactivation in FK1Δ2-146[K532Y] and ShBΔ6-46[T449Y] mutants, which display little inactivation in the absence of β subunit (Fig. 2 B and C and Table 1). A mutant that promotes C-type inactivation (ShBΔ6-46[T449K]; Fig. 2D) showed a further rate increase when coexpressed with Kvβ1.2. These effects were not the result of general interaction of β subunits with the amino acid at position 449, as coexpression of Kvβ1.1 with FK1Δ2-146[K532Y] and ShBΔ6-46[T449Y] resulted in rapid, [K+]o insensitive, N-type inactivation (data not shown). The response of the α subunits to coexpression with Kvβ subunits shows that the acceleration of inactivation mediated by Kvβ1.2 is through a C-type mechanism.
All known Kvβ subunits are highly conserved in their C-terminal domains (6, 7), suggesting that unique properties are conferred by their N termini. Yet, C-type inactivation is thought to be mediated through domains at or near the extracellular mouth of the channel pore (9, 26). To determine the portion of Kvβ1.2 responsible for acceleration of C-type inactivation, we examined chimeric constructs in which the unique N-terminal domain of Kvβ1.2 was attached to the N terminus of FK1Δ2-146 in the presence and absence of the K532Y mutation (Fig. 3A). This chimeric construct (Nβ1.2-FK1Δ2-146) showed an increased rate of inactivation, which was slowed by elevation of [K+]o (Fig. 3B and Table 1). An additional mutation in the pore region (K532Y) that largely removed C-type inactivation greatly slowed the inactivation rate of this chimeric construct (Fig. 3C and Table 1). This shows that the N terminus of Kvβ1.2 is both necessary and sufficient to promote C-type inactivation in Kvα subunits.
Figure 3.
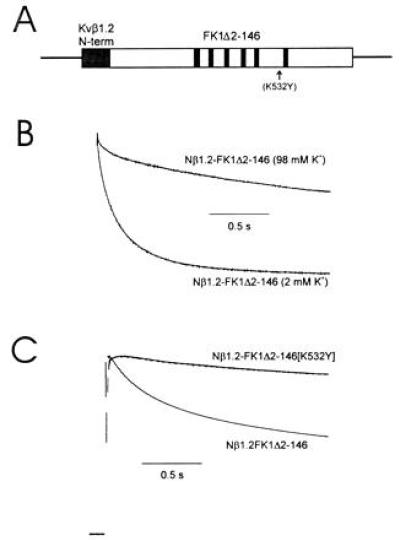
The N-terminal domain of Kvβ1.2 increases the rate of C-type inactivation of FK1. (A) Diagram of the Kvβ1.2–FK1 chimeric construct. Amino acids 1–79 were spliced to the N terminus of FK1Δ2-146 (B and C) or FK1Δ2-146[532Y] (C). (B) The chimeric construct between Kvβ1.2 and FK1 showed an increased rate of inactivation which was sensitive to an increase of [K+]o. (C) Inactivation of the chimeric construct was greatly slowed by a mutation in the pore region (K532Y) that largely removed C-type inactivation. Current traces shown are in response to a depolarizing pulse to +50 mV from a holding potential of −90 mV. Currents were normalized to peak current for purposes of comparison of inactivation rates. Peak current values measured at +50 mV in 2 mM [K+]o were 11 ± 3.5 μA for the chimera and 21 ± 4 μA for the mutant chimera.
We have shown that the mechanism of action of the Kvβ1.2 subunit is to accelerate the rate of C-type inactivation in both Kv1.4 and Shaker B channels. The region which is both necessary and sufficient to promote this acceleration of inactivation is localized to the N-terminal portion of the β subunit. The chimeric construct which attaches the N terminus of Kvβ1.2 to FK1Δ2-146 confirms that N terminus is the active domain and reaches its binding site from the intracellular side of the channel. Hydropathy analysis indicates that the Kvβ1.2 N terminus is largely hydrophilic (data not shown), suggesting that it does not bind to the transmembrane spanning regions. Thus, direct interaction of the N terminus with the external mouth of the pore is unlikely to occur.
We propose that the N terminus of Kvβ1.2 acts at as a rapid but relatively low affinity inactivation ball that interacts with an intracellular binding site to promote C-type inactivation. This model explains why channels where C-type inactivation is apparently absent, such as Kvα1.1, would fail to show an effect when coexpressed with Kvβ1.2 (4), even though they are capable of association (23). N-type inactivation increases the rate at which C-type inactivation proceeds (10, 14). In Kv1.4 channels, binding of the N-terminal inactivation domain promotes C-type inactivation so that 100% of the inactivated channels are driven into the C-type inactivated state (14).
Fig. 4 illustrates our proposed mechanism. We propose that the N-terminal ball domain of Kvβ1.2 binds rapidly to the intracellular vestibule, orienting the channel into a conformation favorable for C-type inactivation. Unbinding then occurs before the C-type inactivated conformation is attained because of its relatively low affinity compared with N-terminal domains capable of inducing fast inactivation. The result is an acceleration of C-type inactivation similar to that seen in the presence of intact α subunit ball domains (9, 14). The diagram suggests that binding of the β-subunit N terminus blocks the channel. Although our data do not address that issue, it is also possible that the N terminus binds to the intracellular side and increases the rate of C-type inactivation without occluding the pore. It may also bind without any dependence on the activation state of the channel. However, at least two other previous observations suggest that the Kvβ1.2 N-terminus can at least partially block the pore. When the membrane is depolarized to very positive potentials using fast voltage clamp methods, activation is very fast and a rapid transient inactivation component (τ ≈ 2.5 ms at +50 mV) was observed (27). Rapid low affinity open channel block also causes a slowing of deactivation tail currents, (e.g., a cross-over phenomenon) and Kvβ1.2 has been shown to result in an ≈3-fold increase in the time constant of deactivation of FK1Δ2-146 (27).
Figure 4.
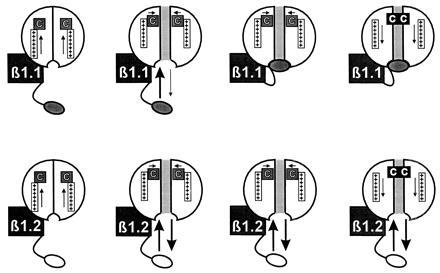
Proposed mechanism of action of Kvβ1.2. The N-terminal domain of Kvβ1.2 promotes C-type inactivation by binding rapidly to the intracellular vestibule, resulting in conformational changes favorable to C-type inactivation and/or reducing K+ occupancy in the pore. Unbinding may occur before attainment of the C-type inactivated conformation because it is relatively low affinity compared with conventional N-terminal domains.
There are several potential explanations for the enhancement of C-type inactivation observed in the presence of N-terminal inactivation domains. The effect may be caused by a β subunit reducing K+ occupancy of the channel pore, which when combined with prolonged opening of the channel’s activation gate, could prolong the period in which C-type inactivation could take place (28). Another possible explanation is that N-terminal domain binding either immobilizes the channel or otherwise induces a conformational change in the channel that permits C-type inactivation to proceed more rapidly (14, 29). Neither mechanism necessarily excludes the other, as there is variability in kinetic behavior for C-type inactivation in different channel types. Some channels may be more sensitive to enhancement of C-type inactivation through an immobilization mechanism, while in others permeation may be the dominant mechanism. Bi-exponential C-type inactivation suggests multiple conformational changes are involved in C-type inactivation, and it may be possible that certain of these steps are more susceptible to N-terminal binding than others. Our data suggest that the fast component of C-type inactivation is less strongly influenced by Kvβ1.2 than the slower component.
This study demonstrates that Kvβ subunits can modulate voltage-gated K+ channel inactivation by both known mechanisms of channel inactivation. For both forms of inactivation, this requires participation of β subunit N-terminal domains on the cytoplasmic side of the pore. This suggests that determination of the specific associations of the N-terminal domains of K+ channel β subunits with α subunit pore domains will be important for understanding the role of K+ channels in determination of action potential duration and firing rate of excitable cells. β subunit modulation of C-type inactivation has important implications for understanding the molecular pharmacology of use-dependent K+ channel inhibitors that are important as antiarrhythmic, anticonvulsive, and immunosuppressive medications.
Acknowledgments
We wish to thank Anne L. Crews, Robert C. Castellino, and Douglas J. Opel for excellent technical assistance; Donald L. Campbell for useful discussions; and Ligia Toro for the gift of Shaker B. This work was supported by the National Heart, Lung, and Blood Institute of the National Institutes of Health (HL-19216 and HL-52874); the North Carolina Affiliate of the American Heart Association (M.J.M.); and the Lucille P. Markey Charitable Trust (J.O.W.).
References
- 1.Chandy K G, Gutman G A. In: Handbook of Receptors and Channels: Ligand and Voltage-Gated Ion Channels. North R A, editor. Boca Raton, FL: CRC; 1995. pp. 1–71. [Google Scholar]
- 2.Scott V E S, Rettig J, Parcej D N, Keen J N, Findlay J B C, Pongs O, Dolly J O. Proc Natl Acad Sci USA. 1994;91:1637–1641. doi: 10.1073/pnas.91.5.1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rettig J, Heinemann S H, Wunder F, Lorra C, Parcej D N, Dolly J O, Pongs O. Nature (London) 1994;369:289–294. doi: 10.1038/369289a0. [DOI] [PubMed] [Google Scholar]
- 4.Morales M J, Castellino R C, Crews A L, Rasmusson R L, Strauss H C. J Biol Chem. 1995;270:6272–6277. doi: 10.1074/jbc.270.11.6272. [DOI] [PubMed] [Google Scholar]
- 5.Majumder K, De Biasi M, Wang Z, Wible B A. FEBS Lett. 1995;361:13–16. doi: 10.1016/0014-5793(95)00120-x. [DOI] [PubMed] [Google Scholar]
- 6.Heinemann S H, Rettig J, Wunder F, Pongs O. FEBS Lett. 1995;377:383–389. doi: 10.1016/0014-5793(95)01377-6. [DOI] [PubMed] [Google Scholar]
- 7.England S K, Uebele V N, Koidali J, Bennett P B, Tamkun M M. J Biol Chem. 1995;270:28531–28534. doi: 10.1074/jbc.270.48.28531. [DOI] [PubMed] [Google Scholar]
- 8.Zagotta W N, Hoshi T, Aldrich R W. Science. 1990;250:568–571. doi: 10.1126/science.2122520. [DOI] [PubMed] [Google Scholar]
- 9.Hoshi T, Zagotta W N, Aldrich R W. Science. 1990;250:533–538. doi: 10.1126/science.2122519. [DOI] [PubMed] [Google Scholar]
- 10.Hoshi T, Zagotta W N, Aldrich R W. Neuron. 1991;7:547–556. doi: 10.1016/0896-6273(91)90367-9. [DOI] [PubMed] [Google Scholar]
- 11.Liu Y, Jurman M E, Yellen G. Neuron. 1996;16:859–867. doi: 10.1016/s0896-6273(00)80106-3. [DOI] [PubMed] [Google Scholar]
- 12.Uebele V N, England S K, Chaudhary A, Tamkun M M, Snyders D J. J Biol Chem. 1996;271:2406–2412. doi: 10.1074/jbc.271.5.2406. [DOI] [PubMed] [Google Scholar]
- 13.Sambrook J, Fritsch E F, Maniatis T. Molecular Cloning: A Laboratory Manual. 2nd Ed. Plainview, NY: Cold Spring Harbor Lab. Press; 1989. [Google Scholar]
- 14.Rasmusson R L, Morales M J, Castellino R C, Zhang Y, Campbell D L, Strauss H C. J Physiol (London) 1995;489:709–721. doi: 10.1113/jphysiol.1995.sp021085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Comer M B, Campbell D L, Rasmusson R L, Lamson D R, Morales M J, Zhang Y, Strauss H C. Am J Physiol. 1994;267:H1383–H1395. doi: 10.1152/ajpheart.1994.267.4.H1383. [DOI] [PubMed] [Google Scholar]
- 16.Ray F A, Nickoloff J A. BioTechniques. 1992;13:342–348. [PubMed] [Google Scholar]
- 17.Liman E R, Tytgat J, Hess P. Neuron. 1992;9:861–871. doi: 10.1016/0896-6273(92)90239-a. [DOI] [PubMed] [Google Scholar]
- 18.Higuchi R. In: PCR Protocols: A Guide to Methods and Applications. Innis M A, Gelfand D H, Sninsky J J, White T J, editors. San Diego: Academic; 1990. pp. 177–183. [Google Scholar]
- 19.Pokrovskaya I D, Gurevich V V. Anal Biochem. 1994;220:420–423. doi: 10.1006/abio.1994.1360. [DOI] [PubMed] [Google Scholar]
- 20.Stühmer W, Stocker M, Sakmann B, Seeburg P, Baumann A, Grupe A, Pongs O. FEBS Lett. 1988;242:199–206. doi: 10.1016/0014-5793(88)81015-9. [DOI] [PubMed] [Google Scholar]
- 21.Yu W, Xu J, Li M. Neuron. 1996;16:441–453. doi: 10.1016/s0896-6273(00)80062-8. [DOI] [PubMed] [Google Scholar]
- 22.Sewing S, Roeper J, Pongs O. Neuron. 1996;16:455–463. doi: 10.1016/s0896-6273(00)80063-x. [DOI] [PubMed] [Google Scholar]
- 23.Nakahira K, Shi G Y, Rhodes K J, Trimmer J S. J Biol Chem. 1996;271:7084–7089. doi: 10.1074/jbc.271.12.7084. [DOI] [PubMed] [Google Scholar]
- 24.Papazian D M, Schwarz T L, Tempel B L, Jan Y N, Jan L Y. Science. 1987;237:749–753. doi: 10.1126/science.2441470. [DOI] [PubMed] [Google Scholar]
- 25.Baumann A, Grupe A, Ackermann A, Pongs O. EMBO J. 1988;7:2457–2463. doi: 10.1002/j.1460-2075.1988.tb03092.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.López-Barneo J, Hoshi T, Heinemann S H, Aldrich R W. Recept Channels. 1993;1:61–71. [PubMed] [Google Scholar]
- 27.Castellino R C, Morales M J, Strauss H C, Rasmusson R L. Am J Physiol. 1995;268:H385–H391. doi: 10.1152/ajpheart.1995.269.1.H385. [DOI] [PubMed] [Google Scholar]
- 28.Baukrowitz T, Yellen G. Neuron. 1995;15:951–960. doi: 10.1016/0896-6273(95)90185-x. [DOI] [PubMed] [Google Scholar]
- 29.Roux M J, Olcese R, Toro L, Stefani E. Biophys J. 1996;70:A189. (abstr.). [Google Scholar]