

Abstract
Emergence of antiviral drug resistance is a major challenge to human immunodeficiency virus (HIV) therapy. The archetypal example of this problem is loss of antiviral activity of the nucleoside analogue 3′-azido-3′-deoxythymidine (AZT), caused by mutations in reverse transcriptase (RT), the viral polymerase. AZT resistance results from an imbalance between rates of AZT-induced proviral DNA chain termination and RT-induced excision of the chain-terminating nucleotide. Conversion of the AZT prodrug from its monophosphorylated to diphosphorylated form by human thymidylate kinase (TMPK) is inefficient, resulting in accumulation of the monophosphorylated AZT metabolite (AZT-MP) and a low concentration of the active triphosphorylated metabolite (AZT-TP). We reasoned that introduction of an engineered, highly active TMPK into T cells would overcome this functional bottleneck in AZT activation and thereby shift the balance of AZT activity sufficiently to block replication of formerly AZT-resistant HIV. Molecular engineering was used to link highly active, engineered TMPKs to the protein transduction domain of Tat for direct cell delivery. Combined treatment of HIV-infected T cells with AZT and these cell-permeable, engineered TMPKs restored AZT-induced repression of viral production. These results provide an experimental basis for the development of new strategies to therapeutically increase the intracellular concentrations of active nucleoside analogue metabolites as a means to overcome emerging drug resistance.
INTRODUCTION
Despite the success of highly active antiretroviral therapy (HAART) in lowering viral load to undetectable levels in ~70% of human immunodeficiency virus (HIV)-infected patients (Lowe et al., 2004), treatment failure is a growing problem. A central role in HAART is played by nucleoside reverse transcriptase inhibitors (NRTIs). These are prodrugs that require intracellular conversion to triphosphorylated forms for antiviral activity. The therapeutic efficacy of NRTIs is determined, to a significant extent, by the efficiency of this activation by human cellular kinases. For 3′-azido-3′-deoxythymidine (AZT), the first NRTI approved for HIV treatment, the activation process is highly inefficient because of the low level of phosphorylation of AZT-monophosphate (AZT-MP) to AZT-diphosphate (AZT-DP) by human thymidylate kinase (TMPK) (Furman et al., 1986; Qian et al., 1994). Because of this AZT activation bottleneck, inactive AZT-MP accumulates to a high concentration, whereas the active, triphosphorylated form (AZT-TP) constitutes only ~2 % of the total intracellular AZT metabolites (Agarwal & Mian, 1991; Fridland et al., 1990).
The effectiveness of AZT blockade of HIV proviral DNA synthesis is determined by the balance between AZT-TP-production-related AZT-MP incorporation into and termination of the growing proviral DNA and mutant reverse transcriptase (RT)-induced excision of chain-terminating AZT-MP. Most strategies, to attack AZT resistance, focus on methods to reduce the effects of RT mutations. An alternative but less well-explored strategy is the development of methods to enhance the efficiency of AZT-TP production. Our previous studies on engineering TMPK to remove the bottleneck of AZT-MP conversion to AZT-DP have provided one approach to this alternative strategy (Brundiers et al., 1999; Wöhrl et al., 2005).
TMPK is essential for the conversion of thymidme monophosphate (TMP) to thymidme diphosphate (TDP), with ATP as the preferred phosphoryl donor (Jong & Campbell, 1984). Like other nucleoside monophosphate (NMP) kinases, TMPK contains a P-loop motif that binds the α- and β-phosphate groups of ATP, and a so-called LID region, defined as a flexible stretch of residues that covers the ATP-binding site (Lavie et al., 1997) (Fig. 1a). The LID region of other NMP kinases supplies several arginine residues that participate in catalysing phosphoryl transfer. Eukaryotic TMPKs do not contain these positively charged residues in the LID region and appear to partly compensate for this by incorporating a single arginine residue in the P-loop sequence (Lavie et al., 1997; Ostermann et al., 2000b). Our previous work implicated repulsion between the 3′-azido group of AZT and the conserved carboxylate group in the P-loop to explain mispositioning of the P-loop upon AZT binding (Lavie et al., 1998). We proposed that the catalytic P-loop arginine (the residue following the conserved aspartic acid) is not able to fulfil its catalytic role. This model explains the in vitro kinetic data, showing that the rate of AZT-MP phosphorylation by human TMPK is ~60-fold lower than that of the physiological substrate, TMP (Brundiers et al., 1999). We have developed engineered TMPK enzymes (TMPKEN) with much greater AZT-MP phosphorylation efficiency (Brundiers et al., 1999; Ostermann et al., 2000a, b). Transient expression of genes encoding these TMPKEN in HeLa cells shifted AZT metabolite pools to contain markedly increased proportions of AZT-TP, and AZT treatment of HIV-infected CD4+ HeLa cells stably transfected with TMPKEN reduced HIV LTR expression (Wöhrl et al., 2005).
Fig. 1.

TMPK structure, mutations and comparison of enzymic activities, (a) Ribbon diagram of a monomer of the homodimeric human TMPK enzyme. The P-loop, phenylalanine (F) 105 and LID region, indicated by arrows, are the areas where mutagenesis was carried out to generate the TMPKEN variants, (b) The specific amino acid substitutions are indicated for each TMPK variant, where residues that differ from the wild-type (WT) sequence are shown in bold.
Here, we report an extension of our studies of engineered Tat–TMPK (TMPKEN) enhancement of AZT activity to T lymphocytes infected with highly drug-resistant HIV, where engineered TMPK variants were introduced into cells using protein transduction domain (PTD) technology (Schwarze & Dowdy, 2000). Direct TMPKEN delivery and AZT treatment of infected T cells resulted in a marked reduction in AZT-resistant virus production. These results support further development of strategies to enhance intracellular AZT conversion to AZT-TP to complement other approaches to the emerging problem of AZT resistance of HIV.
METHODS
Preparation of PTD-TMPK fusion proteins
Cloning, expression and purification of human TMPK has been described previously (Brundiers et al., 1999). The Tat PTD (sequence YGRKKRRQRRR) was fused to the N terminus of the TMPK variants used in these studies. Enzymes were expressed in Escherichia coli as glutathlone S-transferase (GST) fusion proteins. Following bacterial lysis by sonication, the proteins were purified using Glutathione Sepharose Fast Flow resin (Amersham Biosciences), and the GST tag was cleaved with thrombin on the GST column. The eluted protein was concentrated to ~10 mg ml−1 and stored at −80 °C.
Cell culture and studies of protein uptake
The T cell leukaemia line CEM-SS (CEM) was obtained from the AIDS Research and Reference Reagent Programme [contributed by Peter Nara (Foley et al., 1965; Nara & Fischinger, 1988; Nara et al., 1987), National Institutes of Health (Rockville, MD)] and was maintained in RPMI 1640 medium supplemented with 10 mM HEPES, 2 mM glutamine, 1 mM sodium pyruvate and 10% fetal bovine serum (FBS) (all from Biowhitaker) (complete medium). Cells were incubated at 37 °C in a humidified atmosphere with 5 % CO2, and passaged twice weekly in complete medium. For studies of cellular uptake of Tat–TMPKEN proteins, CEM cells were washed free of medium and incubated with 1 % BSA to block non-specific protein binding. Cells were then washed twice with PBS and incubated for 30 min at room temperature with either fluorescein isothiocyanate (FITC) -labelled Tat–TMPKEN or FITC-labelled TMPK as a Tat-negative control. Excess protein was removed by washing cells three times with PBS, and cells were fixed with formaldehyde and observed using fluorescence microscopy.
Cytotoxicity assays
The effects of Tat–TMPKEN proteins on CEM cell growth were evaluated using the soluble tetrazolium salt 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt (MTS) assay (Promega). Cellular cytotoxicity of Tat–TMPKEN proteins in combination with AZT was assessed using the measurement of cellular release of [51Cr] in 24 h assays. Cells were labelled for 45 min with 100 μCi(3.7 MBq) [51Cr] in 50% serum and then washed free of excess label and purged for 30 min in complete medium prior to use in cytotoxicity assays. After incubation of control, untreated or treated cells, specific [51Cr] release was used to estimate cellular lysis. Specific radiolabel release was calculated using the following formula by accounting for spontaneous release from control cells and by comparison with the total releasable counts determined by lysing cells with 1 % SDS: percentage specific [51Cr] release = [(experimental release–spontaneous release)/(total release–spontaneous release)] × 100. Survival of cells cultured under the indicated conditions was expressed as a percentage of the survival of untreated control cells cultured in complete medium.
HIV infection and virus production assay
The AZT-resistant strain, HIV-1RTMC/MT-2 (Larder & Kemp, 1989), was used for all experiments except those shown in Fig. 3(b), where AZT-sensitive virus was tested. The HIV-1RTMC/MT-2 strain is markedly less sensitive to AZT, as a result of the four prototypic mutations that are well known to convey AZT resistance, D67N, K70R, T215F and K219Q. The AZT-sensitive strain, HIV-MN, was used for control studies of comparative AZT antiviral activity (Fig. 3b). HIV-RTMC was obtained from the AIDS Research and Reference Reagent Programme, where it was contributed by B. Larder and S. Kemp (Junker et at., 1997; Larder & Kemp, 1989). HIV-MN was obtained from the AIDS Research and Reference Reagent Programme, where it was contributed by R. Gallo (Shaw et al., 1984). T cell suspensions were adjusted to a concentration of 2 × 106 cells m−1 and were infected at 37 °C on a rotating shaker for 2 h with HIV at a multiplicity of 30 ng p24 antigen per 2 × 106 cells. HIV-infected cells were pelleted and washed three times with PBS containing 2% FBS. The cell pellet was resuspended in complete medium and adjusted to a concentration of 4 × 105 cells ml−1 for testing of virus production, as assessed by detection of increasing amounts of p24 antigen in culture supernatants. HIV-infected cells were cultured in 200 μl medium at 37 °C, in the absence or presence of AZT or AZT plus Tat–TMPKEN variants. Supernatant samples (100 μl) were removed daily for p24 antigen assays and replaced with complete medium, supplemented with freshly prepared AZT or AZT plus TMPKEN. These samples were frozen at −70 °C until analysis. The p24 antigen assay was done using commercially available ELISA kits (AIDS Vaccine Programme, Frederick, MD). Results from AZT or AZT + TMPKEN-treated cells were expressed as the percentage of the control (untreated, infected cell) levels of p24 antigen production (in pg ml−1).
Fig. 3.

Relative sensitivities of HIV-1RTMC/MT-2 versus HIV-MN strains to AZT. T cells were infected with either (a) the AZT-resistant HIV-1RTMC/MT-2 strain or (b) the AZT-sensitive HIV-MN strain and were then incubated for 5–6 days in the absence (control) or presence of the indicated concentrations of AZT. p24 antigen production, in culture supernatants from infected cells treated with AZT at the indicated concentrations, was expressed as a percentage of the control p24 production levels detected in the supernatants of untreated, infected cell cultures. The results represent the mean±SEM percentage of control levels of p24 production from three assays with each virus strain. p24 production by HIV-MN-infected T cells was significantly repressed by AZT at 0.01 μM (P=0.041) and 0.1 μM (P=0.004) compared with untreated controls, whereas there was no significant AZT-induced reduction in p24 production by HIV-lRTMC/MT-2-infected cells at any AZT concentration tested (0.1, 1 or 10 μM).
RESULTS
Creation of engineered TMPK enzymes and comparison of AZT-specific activity with wild-type TMPK
In contrast to eukaryotic TMPKs, bacterial TMPKs do not contain the catalytic arginine residue in the P-loop. Instead, the catalytic role of this residue is compensated by an arginine-rich LID region, as is present in most NMP kinases (Fig. 1a). We predicted that any perturbation to the P-loop conformation caused by AZT-MP binding would be less deleterious to the activity of the bacterial enzyme than it is to the human enzyme, since the catalytic arginine in the bacterial enzyme resides in the more exposed LID sequence and not in the P-loop. In fact, we have confirmed previously that E. coli TMPK is quite efficient at AZT-MP phosphorylation (Lavie et al., 1998). Using this information, we designed a mutant human enzyme, which was predicted to be efficient at AZT-MP phosphorylation, by substituting the human TMPK LID sequence with the E. coli sequence and at the same time mutating the P-loop arginine to a glycine (as is found in E. coli TMPK) to avoid a steric clash between the P-loop and the introduced LID arginine (Brundiers et al., 1999). This variant, called BigLID (Fig. 1b), has more than a 100-fold higher rate of AZT-MP phosphorylation than the native (wild-type) form (Table 1). Another TMPK variant that was created with a single substitution of phenylalanine 105 to a tyrosine (F105Y) also exhibited higher activity and preferential specificity towards AZT-MP (Fig. 1b, Table 1). The Km values of AZT-MP are similar for wild-type and mutant TMPKs (Wöhrl et al., 2005).
Table 1. Enzyme kinetic studies comparing three engineered TMPK enzymes in their native and Tat-conjugated forms with the native (wild-type) enzyme.
Enzymes used in this study are highlighted in bold.
| Construct | Kcat dTMP* (s−1) | Kcat AZT-MP (s−1) | Ratio mutant :WT Kcat AZT-MP |
|---|---|---|---|
| WT† | 0.73 | 0.012 | 1 |
| Tat–WT | 0.76 | 0.003 | 0.25 |
| BigLID† | 1.1 | 1.7 | 142 |
| Tat–BigLID | 0.83 | 1.5 | 125 |
| F105Y† | 0.17 | 0.27 | 22.6 |
| Tat–F105Y | 0.14 | 0.17 | 14.2 |
| D15A† | <0.001 | < 0.001 | - |
| Tat–D15A | <0.001 | <0.001 | - |
Studies of the phosphorylation of the natural substrate, TMP, versus AZT-monophosphate {AZT-MP).
Linkage of TMPKEN to a PTD
We have shown previously that transient transfection of a BigLID construct into HeLa cells increased AZT conversion to AZT-TP (Wöhrl et al., 2005). Over 90% of the AZT metabolites in control cells were present in the monophosphorylated form, whereas AZT-MP levels were <20 % of the total AZT metabolites in BigLID-TMPK expressing cells. Of note is the observation that transduction with wild-type TMPK resulted in only a modest increase in the AZT-TP levels in those studies, thus underscoring the importance of using engineered TMPK for optimal effect on AZT activation. That earlier report validated the conclusion that TMPK is the rate-limiting enzyme in AZT activation, rather than conversion of AZT-DP to AZT-TP being rate-limiting, even at higher intracellular levels of TMPK activity. However, our results with TMPK-transfected cells left a critical question unanswered: could the presence of engineered TMPK enzymes in T lymphocytes repress replication of HIV that is resistant to AZT monotherapy, i.e. restore the antiviral activity of AZT against drug-resistant virus by shifting the balance of AZT metabolism towards AZT-TP overproduction?
To address this question, we developed a strategy to introduce engineered TMPK enzymes directly into T lymphocytes. It has been shown elsewhere that a highly basic amino acid sequence consisting of 11 residues in the HIV Tat protein confers on any fusion protein, or even gold particles (Tkachenko et al., 2004), the ability to traverse biological membranes (Schwarze et al., 1999). Such sequences were named PTDs. We exploited these observations by conjugating our engineered TMPK enzymes with a Tat PTD sequence to create fusion proteins that would enter T cells in culture directly. N-terminal Tat fusion proteins were produced, and preservation of the improved AZT-MP kinetics was verified for the fusion proteins (Table 1). Studies using fusion proteins labelled with a fluorescent marker (FITC) demonstrated high-efficiency entry of Tat-containing TMPK enzymes into cells, as evidenced by both nuclear uptake and cytoplasmic fluorescent staining of ⩾90 % of treated T cells (data not shown). The nuclear uptake of the Tat-conjugated proteins was predicted, considering the well-characterized nuclear import properties of Tat (Efthymiadis et al., 1998).
Two types of initial experiments, cytostasis and cytolysis assays, were used to define non-toxic concentrations of the highly active Tat–BigLID fusion protein that could be used to test the enhancing effects of engineered TMPK on AZT antiviral activity in T lymphocytes. Using the MTS cell proliferation assay, CEM T cells were found to continue to divide at normal control rates when incubated in Tat–BigLID concentrations of up to 5 μg ml−1 (Fig. 2a). Cytostasis was observed at 10 μg ml−1. Using 51Cr-release assays, it was observed that Tat–BigLID concentrations of up to 20 μg ml−1 did not cause any detectable T cell lysis, in either the absence or presence of 10 μM AZT (Fig. 2b). Light microscopic examination of T cells revealed no detectable changes in cell morphology under the above culture conditions. As a result of these observations, subsequent studies were done using a low Tat–BigLID concentration (2 μg ml−1) to ensure that any TMPKEN-induced enhancement of AZT antiviral activity was independent of adverse effects of the engineered enzyme, or the enzyme plus AZT, on the T lymphocytes in which viral replication and production was measured.
Fig. 2.
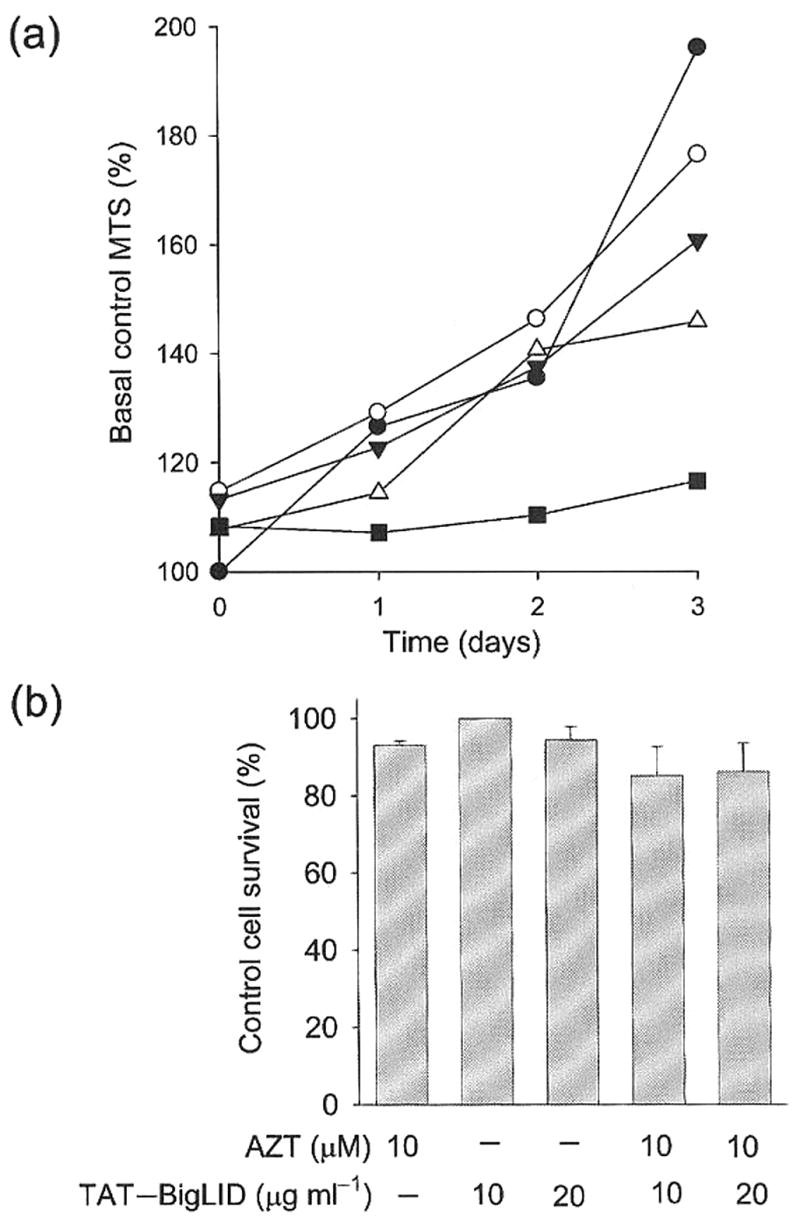
Dose–response studies of the growth-inhibitory and cytotoxic effects of Tat–BigLID and Tat–BigLID+AZT on T cells, (a) Effect of Tat–BigLID on T cell growth. T cells were incubated in complete medium alone (control) or with increasing concentrations of Tat–BigLID (●, 0 μg ml−1; ○, 1 μg ml−1; ▼, 2 μg ml−1; △, 5 μg ml−1; ■, 10 μg ml−1). Three-day cell growth curves were defined by daily measurements of increasing cell number using the MTS assay. Data from a representative experiment are shown as the daily percentages of initial (basal) control levels of MTS signals for each set of replicate cultures. (b) Cytolytic effects of Tat–BigLID and Tat–BigLID+AZT. 51Cr-labelled T cells were incubated in complete medium with either AZT, Tat–BigLID or the combination of AZT+Tat–BigLID at the indicated concentrations. Cytolysis was measured by radiolabel release, and cell survival was compared with that of untreated control cells. The data presented are the mean ± SEM of results of two assays. There was no cytolytic effect of either AZT alone (10 μM) or AZT+Tat–BigLID at 10 or 20 μg ml−1.
Tat-TMPKEN-induced restoration of the antiviral activity of AZT against drug-resistant HIV
We tested the prediction that direct cellular delivery of TMPKEN would enhance the antiviral effect of AZT. The critical test of this infected-cell co-treatment was whether it would repress the replication and production of AZT-resistant virus. We therefore did these studies with the prototypic AZT-resistant virus strain, HIV-lRTMC/MT-2 (Larder & Kemp, 1989), that contains four key amino acid mutations in the RT gene (see Methods). We confirmed that, under our culture conditions with infected T lymphocytes, this virus strain was > 1000-fold more resistant to AZT than the AZT-sensitive control strain, HIV-MN (Fig. 3). Viral production at representative critical AZT concentrations is shown in Fig. 3. Analysis using dose-response studies in repeated experiments using CEM T lymphocytes revealed that the AZT concentration that resulted in 50 % inhibition of HIV production (IC50) was consistently ≤ 0.01 μM with the AZT-sensitive virus strain, HIV-MN, and ⩾10 μM with the AZT-resistant virus, HIV-lRTMC/MT-2. This > 1000-fold increase in AZT resistance of HIV-lRTMC/MT-2 provided a good test of the TMPKEN, which were engineered to increase the intracellular, phosphorylation-induced activation of AZT.
The hypothesis was that delivery of TMPKEN, which is highly active in converting AZT-MP into AZT-DP, would overcome high-level viral resistance to AZT. The results of experiments testing this hypothesis using the Tat–BigLID TMPK variant are shown in Fig. 4. Treatment with Tat–BigLID TMPK in the absence of AZT had no significant effect on viral replication (results not shown); therefore, Tat-TMPKEN lacked any detectable antiviral activity in the absence of the nucleoside analogue substrate, AZT. However, when used in combination, Tat–BigLID markedly enhanced the antiviral effect of AZT, resulting in an approximately a 2000-fold reduction in the concentration of AZT required for 50 % reduction in viral production [EC50: AZT, 180 (51–630) μM vs AZT + Tat–BigLID, 0.09 (0.06–0.14) μM; mean (95% confidence interval); n = 3].
Fig. 4.

Tat–TMPKEN enhancement of AZT antiviral activity against AZT-resistant virus, HIV-1RTMC/MT-2-infected T cells were treated with either AZT alone (AZT only) at the indicated concentrations or AZT plus the indicated, TMPKEN proteins (Tat–BigLID, Tat–F105Y or Tat–D15A, all at 2 μg ml−1). HIV replication was measured by p24 antigen production in culture supernatants after 6 days of infection. The antiviral effects of AZT alone or AZT+TMPKEN were expressed as the percentage of control p24 antigen production by the same populations of HIV-infected T cells incubated without either AZT orTMPKEN treatment. The data presented are from nonlinear regression curve fitting of the mean±SEM of results from three different virus infection assays. Curve fitting data were analysed using GraphPad Prism 4 software to estimate AZT concentrations at which p24 antigen production was reduced by 50% (EC50 values) compared with p24 production levels from untreated, HIV-infected control cells. The TMPKEN-induced reductions in AZT EC50 for cells treated with the combination of AZT plus either Tat–BigLID or Tat–F105Y were highly significant (P<0.001). In contrast, there was no AZT-enhancing effect observed with cells co-treated with the enzymically inactive TMPK mutant protein, Tat–D15A.
We next tested the activity-enhanced Tat–F105Y variant of TMPK, which has a lower rate of AZT-MP phosphorylation than the Tat–BigLID variant (Kcat=0.17 vs 1.5 s−1, respectively; Table 1). As observed for Tat–BigLID enzyme treatment, the Tat–F105Y protein had no antiviral effect in the absence of AZT (results not shown). However, this TMPKEN restored the dose-dependent antiviral activity of AZT against the drug-resistant HIV strain (Fig. 4). In fact, dose–response analysis indicated that the Tat–F105Y construct was almost as effective as the Tat–BigLID variant of TMPK in this regard, inducing a ⩾500-fold reduction in the EC50 of AZT (Fig. 4) [AZT, 180 (51–630) μM vs AZT+Tat–F105Y, 0.31 (0.12–0.79) μM; mean (95% confidence interval); n=3]. The relative specificities of the BigLID and F105Y enzymes for AZT-MP and thymidine-MP are similar (Table 1). Both have Kcat values for AZT-MP that are ~50% faster than that for the endogenous cellular substrate, TMP (BigLID = 1.7 vs 1.1 s−1, F105Y=0.27 vs 0.17 s−1, respectively; Table 1). However, the absolute rate of AZT-MP phosphorylation by BigLID is sixfold faster than that of F105Y (1.7 vs 0.27 s−1; Table 1). Therefore, since AZT-TP must ultimately compete with cellular thymidine-TP for incorporation into newly synthesized DNA, these results indicated that it may be the ratio, and not the absolute rate, of substrate phosphorylation that is the most important determinant of TMPKEN enhancement of the antiviral effect of AZT.
To confirm the requirement of catalytic activity for the engineered enzymes for restoration of AZT-induced antiviral effect against AZT-resistant virus, we included studies of the effects of a catalytically inactive TMPK enzyme variant. Mutation of the P-loop aspartic acid to alanine resulted in an inactive enzyme, which lacked catalytic activity in both its native and Tat-conjugated form (D15A and Tat–D15A, respectively; Table 1). This inactive enzyme maintained a normal three dimensional structure (data not shown). Tat–D15A was tested in the same HIV-infected T cell assay for the effect of AZT on productive infection with AZT-resistant virus (Fig. 4). In contrast to the AZT-enhancing Tat–BigLID and Tat–F105Y enzymes, the enzymically inactive mutant enzyme, Tat–D15A, did not induce any detectable restoration of antiviral activity of AZT against AZT-resistant virus production (combined EC50 >200 μM).
DISCUSSION
AZT resistance of HIV has been attributed to gain-of-function RT mutations that result in excision of AZT-MP-terminated proviral DNA by a process termed phosphorolysis (Arion et al., 1998; Arion & Parniak, 1999; Meyer et al., 1999). For this type of viral resistance, phosphorolysis must be more efficient than AZT-MP incorporation into proviral DNA, resulting in a balance that shifts away from chain termination and towards continued proviral replication. Since AZT-MP incorporation into replicating proviral DNA is a direct function of intracellular AZT-TP concentration, it is predicted that higher AZT-TP concentrations should overcome the newly gained phosphorolysis activity of AZT-resistant RT. Our results are consistent with this prediction.
The results of this study extend our previous work on engineered TMPK effects on AZT activity (Wöhrl et al., 2005) in several important ways. Previous studies tested TMPKEN effects on retrovirus-transduced or enzyme-gene-transfected HeLa cells. In the current work, we examined whether TMPKEN would enhance AZT antiviral activity in T lymphocytes, a natural target of HIV infection. In the HeLa cell studies, HIV LTR-reporter activity was used as a surrogate indicator of HIV replication, whereas in the current work we measured virus production from infected T cells. Our previous experiments tested antiviral effects of TMPKEN+AZT on AZT-sensitive virus. The most important advance in the present study was that the experiments were focused on the question of whether TMPKEN delivered directly to T lymphocytes could restore AZT antiviral activity against highly AZT-resistant viruses. The key conclusion from this work is that strategies that overcome the AZT-MP-to-AZT-DP bottleneck of AZT metabolism in T cells, such as increasing TMPK activity as done in our studies, can restore the antiviral activity of AZT against what was previously a highly AZT-resistant HIV-1 strain. This suggests that novel therapeutic methods that can elevate the level of intracellular triphosphorylated AZT species can be used to complement other methods being developed to attack mutant-RT activity.
These observations raise the question of whether a similar strategy of combined enzyme + prodrug treatment could be applied to combat HIV RT mutation-related resistance to other prodrugs. In some cases, however, RT mutation(s) create viral enzymes whose drug resistance might not be overcome by increased intracellular phosphorylation of the prodrug. A case in point is the Ml84V RT mutation that is found in HIV strains isolated from patients treated with the cytidine analogue, 2′-deoxy-3′-thiacytidine (3TC, Lamivudine, Epivir) (Sarafianos et al., 1999). This RT variant sterically excludes the nucleoside analogue by discriminating against the bulky sulfur atom present in the sugar ring of 3TC (Sarafianos et al., 1999). For that reason, it is unlikely that a higher intracellular concentration of 3TC-triphosphate would restore the antiviral activity of this prodrug against 3TC-resistant virus. This contrast in the RT-mutation-induced HIV resistance to two different NRTIs reveals the importance of understanding the basic mechanisms of NRTI resistance when considering strategies to attack drug resistance. Furthermore, other resistance mechanisms against nucleoside analogue prodrugs, such as decreased transport into the cell, increased export out of the cell or reactions that deactivate the prodrug (e.g. deamination of cytosine-based nucleoside analogues), might be more difficult to reverse by increased intracellular prodrug activation. In such cases, a careful analysis of the balance between the rates and net effects of the counteracting mechanisms would have to be considered in the context of the most important final indicator of the therapeutic strategy, the net effect on antiviral drug-induced HIV production by infected cells.
To our knowledge, there is no other method available at present to overcome the AZT-MP-to-AZT-DP bottleneck of AZT activation. Two theoretical possibilities could be considered as alternative strategies to direct delivery of engineered enzymes, which has practical limitations for clinical application. One alternative that would bypass the rate-limiting drug activation step is direct delivery of AZT-DP to infected cells. Since such phosphorylated compounds are relatively cell-impermeable, masking the phosphate charges would be required for adequate drug uptake. To date, such masking of phosphorylated prodrugs (Wagner et al., 2000) has been accomplished only with nucleoside analogue monophosphate, not diphosphate, molecules (Meier et al., 1998). A second alternative is to develop small-molecule inducers of endogenous TMPK activity to enhance AZT-MP phosphorylation. It might be possible to deliver directly small molecules that allosterically activate TMPK. Such allosteric activators could work by binding at a site other than the active site to improve enzymic activity. Our work may provide further impetus for the development of these alternative methods for increased AZT phosphorylation activation.
A problem that would have to be addressed before any AZT-activation strategies could be useful for future application is the question of how such approaches would affect AZT toxicity. There was no TMPKEN-induced increase in AZT toxicity detected in vitro in our previous study of HeLa cells at AZT concentrations of up to 30 μM (Wöhrl et al., 2005) or in our current study of CEM T lymphocytes at AZT concentrations up to 10 μM (Fig. 2b). However, the AZT toxicity issue is more complex than can be revealed by in vitro studies. Further studies of the effects of enhanced AZT phosphorylation on pharmacokinetics and toxicity in HIV-infected patients compared with uninfected controls (Veal & Back, 1995) would be required.
One approach to minimize the toxicity of AZT-enhancing agents would be to target delivery of the engineered TMPK enzymes into CD4+ cells, which are the targets of HIV infection. This might be achieved by chemically conjugating TMPKEN to a monoclonal antibody against the CD4 receptor. Using such CD4+-cell targeting, AZT-phosphorylation-related activities would continue at their baseline levels in CD4− cells but would be increased in CD4+ cells, in which AZT activation would be greatly increased by the enhancing effect of TMPKEN. This type of approach could be used as a strategy to block the replication of AZT-resistant virus during initial or subsequent rounds of acute infection.
In summary, in this proof-of-concept work, we have used the Tat PTD sequence as a vehicle to ferry engineered TMPK enzymes into T lymphocytes infected with AZT-resistant HIV co-treated with AZT. Our work demonstrates the potential for developing strategies to bypass the bottleneck in AZT activation as a means to attack the problem of AZT resistance.
Acknowledgments
A. L., Y. S. and J. L. C. were supported by a NIH grant (CA113843). The Max Planck Society supported C. M. and M. K. We thank the UIC Campus Research Board for initial support of this work. Work in the J. L. C. laboratory was supported by the James and Marion Grant Fund. None of the authors have any conflicts of interest.
References
- Agarwal RP, Mian AM. Thymidine and zidovudine metabolism in chronically zidovudine-exposed cells in vitro. Biochem Pharmacol. 1991;42:905–911. doi: 10.1016/0006-2952(91)90052-7. [DOI] [PubMed] [Google Scholar]
- Arion D, Parniak MA. HIV resistance to zidovudine: the role of pyrophosphorolysis. Drug Resist Updat. 1999;2:91–95. doi: 10.1054/drup.1999.0076. [DOI] [PubMed] [Google Scholar]
- Arion D, Kaushik N, McCormick S, Borkow G, Parniak MA. Phenotypic mechanism of HIV-1 resistance to 3′-azido-3′-deoxythymidine (AZT): increased polymerization processivity and enhanced sensitivity to pyrophosphate of the mutant viral reverse transcriptase. Biochemistry. 1998;37:15908–15917. doi: 10.1021/bi981200e. [DOI] [PubMed] [Google Scholar]
- Brundiers R, Lavie A, Veit T, Reinstein J, Schlichting I, Ostermann N, Goody RS, Konrad M. Modifying human thymidylate kinase to potentiate azidothymidine activation. J Biol Chem. 1999;274:35289–35292. doi: 10.1074/jbc.274.50.35289. [DOI] [PubMed] [Google Scholar]
- Efthymiadis A, Briggs LJ, Jans DA. The HIV-1 Tat nuclear localization sequence confers novel nuclear import properties. J Biol Chem. 1998;273:1623–1628. doi: 10.1074/jbc.273.3.1623. [DOI] [PubMed] [Google Scholar]
- Foley GE, Lazarus H, Farber S, Uzman BG, Boone BA, McCarthy RE. Continuous culture of human lymphoblasts from peripheral blood of a child with acute leukemia. Cancer. 1965;18:522–529. doi: 10.1002/1097-0142(196504)18:4<522::aid-cncr2820180418>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- Fridland A, Connelly MC, Ashmun R. Relationship of deoxynucleotide changes to inhibition of DNA synthesis induced by the antiretroviral agent 3′-azido-3′-deoxythymidine and release of its monophosphate by human lymphoid cells (CCRF-CEM) Mol Pharmacol. 1990;37:665–670. [PubMed] [Google Scholar]
- Furman PA, Fyfe JA, St Clair MH, Weinhold K, Rideout JL, Freeman GA, Lehrman SN, Bolognesi DP, Broder S, et al. Phosphorylation of 3′-azido-3′-deoxythymidine and selective interaction of the 5′-triphosphate with human immunodeficiency virus reverse transcriptase. Proc Natl Acad Sci U S A. 1986;83:8333–8337. doi: 10.1073/pnas.83.21.8333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jong AY, Campbell JL. Characterization of Saccharomyces cerevisiae thymidylate kinase, the CDC8 gene product. General properties, kinetic analysis, and subcellular localization. J Biol Chem. 1984;259:14394–14398. [PubMed] [Google Scholar]
- Junker U, Baker J, Kalfoglou CS, Veres G, Kaneshima H, Bohnlein E. Antiviral potency of drug-gene therapy combinations against human immunodeficiency virus type 1. AIDS Res Hum Retroviruses. 1997;13:1395–1402. doi: 10.1089/aid.1997.13.1395. [DOI] [PubMed] [Google Scholar]
- Larder BA, Kemp SD. Multiple mutations in HIV-1 reverse transcriptase confer high-level resistance to zidovudine (AZT) Science. 1989;246:1155–1158. doi: 10.1126/science.2479983. [DOI] [PubMed] [Google Scholar]
- Lavie A, Vetter IR, Konrad M, Goody RS, Reinstein J, Schlichting I. Structure of thymidylate kinase reveals the cause behind the limiting step in AZT activation. Nat Struct Biol. 1997;4:601–604. doi: 10.1038/nsb0897-601. [DOI] [PubMed] [Google Scholar]
- Lavie A, Ostermann N, Brundiers R, Goody RS, Reinstein J, Konrad M, Schlichting I. Structural basis for efficient phosphorylation of 3′-azidothymidine monophosphate by Escherichia coli thymidylate kinase. Proc Natl Acad Sci U S A. 1998;95:14045–14050. doi: 10.1073/pnas.95.24.14045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe SH, Prins JM, Lange JM. Antiretroviral therapy in previously untreated adults infected with the human immunodeficiency virus type I: established and potential determinants of virological outcome. Neth ] Med. 2004;62:424–440. [PubMed] [Google Scholar]
- Meier C, Lorey M, De Clercq E, Balzarini J. Cyclosal-2′,3′-dideoxy-2′,3′-didehydrothymidine monophosphate (cyclosal-d4TMP) — synthesis and antiviral evaluation of a new d4TMP delivery system. J Med Chem. 1998;41:1417–1427. doi: 10.1021/jm970664s. [DOI] [PubMed] [Google Scholar]
- Meyer PR, Matsuura SE, Mian AM, So AG, Scott WA. A mechanism of AZT resistance: an increase in nucleotide-dependent primer unblocking by mutant HIV-1 reverse transcriptase. Mol Cell. 1999;4:35–43. doi: 10.1016/s1097-2765(00)80185-9. [DOI] [PubMed] [Google Scholar]
- Nara PL, Fischinger PJ. Quantitative infectivity assay for HIV-1 and -2. Nature. 1988;332:469–470. doi: 10.1038/332469a0. [DOI] [PubMed] [Google Scholar]
- Nara PL, Hatch WC, Dunlop NM, Robey WG, Arthur LO, Gonda MA, Fischinger PJ. Simple, rapid, quantitative, syncytium-forming microassay for the detection of human immunodeficiency virus neutralizing antibody. AIDS Res Hum Retroviruses. 1987;3:283–302. doi: 10.1089/aid.1987.3.283. [DOI] [PubMed] [Google Scholar]
- Ostermann N, Lavie A, Padiyar S, Brundiers R, Veit T, Reinstein J, Goody RS, Konrad M, Schlichting I. Potentiating AZT activation: structures of wild-type and mutant human thymidylate kinase suggest reasons for the mutants’ improved kinetics with the HIV prodrug metabolite AZTMP. J Mol Biol. 2000a;304:43–53. doi: 10.1006/jmbi.2000.4175. [DOI] [PubMed] [Google Scholar]
- Ostermann N, Schlichting I, Brundiers R, Konrad M, Reinstein J, Veit T, Goody RS, Lavie A. Insights into the phosphoryltransfer mechanism of human thymidylate kinase gained from crystal structures of enzyme complexes along the reaction coordinate. Structure. 2000b;8:629–642. doi: 10.1016/s0969-2126(00)00149-0. [DOI] [PubMed] [Google Scholar]
- Qian M, Bui T, Ho RJ, Unadkat JD. Metabolism of 3′-azido-3′-deoxythymidine (AZT) in human placental trophoblasts and Hofbauer cells. Biochem Pharmacol. 1994;48:383–389. doi: 10.1016/0006-2952(94)90111-2. [DOI] [PubMed] [Google Scholar]
- Sarafianos SG, Das K, Clark AD, Jr, Ding J, Boyer PL, Hughes SH, Arnold E. Lamivudine (3TC) resistance in HIV-1 reverse transcriptase involves steric hindrance with β-branched amino acids. Proc Natl Acad Sci U S A. 1999;96:10027–10032. doi: 10.1073/pnas.96.18.10027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarze SR, Dowdy SF. In vivo protein transduction: intracellular delivery of biologically active proteins, compounds and DNA. Trends Pharmacol Sci. 2000;21:45–48. doi: 10.1016/s0165-6147(99)01429-7. [DOI] [PubMed] [Google Scholar]
- Schwarze SR, Ho A, Vocero-Akbani A, Dowdy SF. In vivo protein transduction: delivery of a biologically active protein into the mouse. Science. 1999;285:1569–1572. doi: 10.1126/science.285.5433.1569. [DOI] [PubMed] [Google Scholar]
- Shaw GM, Hahn BH, Arya SK, Groopman JE, Gallo RC, Wong-Staal F. Molecular characterization of human T-cefl leukemia (lymphotropic) virus type III in the acquired immune deficiency syndrome. Science. 1984;226:1165–1171. doi: 10.1126/science.6095449. [DOI] [PubMed] [Google Scholar]
- Tkachenko AG, Xie H, Liu Y, Coleman D, Ryan J, Glomm WR, Shipton MK, Franzen S, Feldheim DL. Cellular trajectories of peptide-modified gold particle complexes: comparison of nuclear localization signals and peptide transduction domains. Bioconjug Chem. 2004;15:482–490. doi: 10.1021/bc034189q. [DOI] [PubMed] [Google Scholar]
- Veal GJ, Back DJ. Metabolism of zidovudine. Gen Pharmacol. 1995;26:1469–1475. doi: 10.1016/0306-3623(95)00047-x. [DOI] [PubMed] [Google Scholar]
- Wagner CR, Iyer VV, Mclntee EJ. Pronucleotides: toward the in viva delivery of antiviral and anticancer nucleotides. Med Res Rev. 2000;20:417–451. doi: 10.1002/1098-1128(200011)20:6<417::aid-med1>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- Wöhrl BM, Loubiere L, Brundiers R, Goody RS, Klatzmann D, Konrad M. Expressing engineered thymidylate kinase variants in human cells to improve AZT phosphorylation and human immunodeficiency virus inhibition. J Gen Virol. 2005;86:757–764. doi: 10.1099/vir.0.80529-0. [DOI] [PubMed] [Google Scholar]