

Abstract
In contrast to replicative DNA polymerases, Sulfolobus solfataricus Dpo4 showed a limited decrease in catalytic efficiency (kcat/Km) for insertion of dCTP opposite a series of N2-alkylguanine templates of increasing size from (methyl (Me) to (9-anthracenyl)-Me (Anth)). Fidelity was maintained with increasing size up to (2-naphthyl)-Me (Naph). The catalytic efficiency increased slightly going from the N2-NaphG to the N2-AnthG substrate, at the cost of fidelity. Pre-steady-state kinetic bursts were observed for dCTP incorporation throughout the series (N2-MeG to N2-AnthG), with a decrease in the burst amplitude and kpol, the rate of single-turnover incorporation. The pre-steady-state kinetic courses with G and all of the six N2-alkyl G adducts could be fit to a general DNA polymerase scheme to which was added an inactive complex in equilibrium with the active ternary Dpo4·DNA·dNTP complex, and only the rates of equilibrium with the inactive complex and phosphodiester bond formation were altered. Two crystal structures of Dpo4 with a template N2-NaphG (in a post-insertion register opposite a 3′-terminal C in the primer) were solved. One showed N2-NaphG in a syn conformation, with the naphthyl group located between the template and the Dpo4 “little finger” domain. The Hoogsteen face was within hydrogen bonding distance of the N4 atoms of the cytosine opposite N2-NaphG and the cytosine at the –2 position. The second structure showed N2-Naph G in an anti conformation with the primer terminus largely disordered. Collectively these results explain the versatility of Dpo4 in bypassing bulky G lesions.
Accurate and efficient replication of DNA is crucial for the preservation of genomic integrity and survival of organisms (1). A major obstacle for DNA replication is caused by various DNA lesions, which are inevitably formed by both endogenous sources and exogenous mutagens in cells and are usually present in replicating DNA after avoiding repair (2). DNA polymerases often produce abnormal phenomena when encountering lesions during DNA replication, including misinsertion of incorrect nucleotides, slippage, and blockage of replication, all of which can result in mutation or cell death and can lead to detrimental effects, including aging and cancer in mammals (3).
Some DNA adducts are small (e.g. abasic sites and oxidative adducts), but others are very bulky, e.g. pyrimidine dimers, photoproducts, large carcinogen-bound adducts, and cross-links (2). The bulkiness of DNA adducts may be a differentiating factor in the misincorporation and blockage of DNA polymerases. The N2 atom of guanine is susceptible to modification by various potential carcinogens, including formaldehyde (4), acetaldehyde (5–8), styrene oxide (9), N-nitrosopyrrolidine (10), oxidation products of heterocyclic aromatic amines (e.g. N-hydroxy-2-amino-3-methylimidazo[4,5-f]quinoline and N-hydroxy-2-amino-3,8-dimethylimidazo[4,5-f]quinoxaline (11)), and the oxidation products of various polycyclic aromatic hydrocarbons (e.g. benzo[a]pyrene (BPDE)3) (12, 13). N2-Et deoxyguanosine has been detected in granulocyte and lymphocyte DNA and urine of alcoholic patients (14–16). Misincorporation opposite N2-EtG by Escherichia coli DNA polymerase I Klenow fragment exo– (5) suggests possible relevance to mutation and subsequent carcinogenesis. Crotonaldehyde- and acetaldehyde-derived 1,N2-propanodeoxyguanosine adducts have been detected in DNA from human tissues (8). An N2-guanyl adduct has been detected in urine of rats treated with N-nitrosopyrrolidine (17), and pyridyloxobutyl-derived N2-guanyl adducts have been found in rats treated with the tobacco-specific nitrosamine N'-nitrosonornicotine (18).
The nature of the mutations produced during replication is not only dependent on the specific damage in the DNA but also on the DNA polymerases involved (19, 20). Replicative DNA polymerases replicate unmodified DNA bases with high fidelity and efficiency but are generally intolerant of the DNA distortions caused by many DNA lesions; thus, these polymerases are blocked and/or inefficiently misinsert bases opposite DNA adducts during replication. The “translesion synthesis” DNA polymerases have more open and larger active sites and can synthesize DNA across and beyond various replication-blocking DNA lesions (21, 22). However, synthesis can still be inhibited (or blocked) by certain DNA lesions and bypass can be error-prone or error-free, depending on the adduct (23).
We previously studied incorporation and extension past the adducts N2-MeG, N2-EtG, N2-IbG, N2-BzG, N2-AnthG, N2-(2,3,4-trihydroxy-1-butyl)G, 8-hydroxy-1,N2-propanoG, 8-hydroxy-6-methyl-1,N2-propanoG, and the styrene oxide and polycyclic hydrocarbon products formed at the guanyl-N2 atom with the replicative DNA polymerases bacteriophage pol T7– and HIV-1 reverse transcriptase (24–26). Even the relatively small N2-MeG and N2-EtG adducts were miscoded with Klenow fragment (4, 5) and pol T7– and HIV-1 reverse transcriptase (24). N2-EtG and larger N2-guanyl adducts cause strong blockage of polymerization by pol T7– and HIV-1 reverse transcriptase (24). The eukaryotic DNA polymerases pol ι, κ, and η and Rev1 still preferred to incorporate dCTP opposite the N2-guanyl adducts, but the incorporation efficiency and the fidelity with bulkier N2-guanyl adducts were decreased compared with unmodified G (27–29). Although kinetic data have been accumulated for translesion synthesis by various DNA polymerases, the structures of the N2-guanyl adducts in the enzyme active sites and mechanistic details are still not clear.
The DNA polymerase Dpo4 from Sulfolobus solfataricus P2 (polymerase IV) (30) has been investigated in considerable detail both in terms of its function (31–35) and structure (36–38). Dpo4 can bypass various DNA adducts, including 8-oxoG (39, 40), O6-MeG (41), O6-BzG (42), UV-cross-linked products (43), BPDE adducts (44, 45), and abasic lesions (38). Several crystal structures of ternary complexes (with Dpo4, DNA template-primer construct containing an adducted base, and incoming dNTP) have been determined (40–44, 46, 47), and this structural data has revealed details of misincorporation in replication bypass.
In the present work, Dpo4 was studied in detail regarding both kinetics and structure in the processing of a series of N2-guanyl adducts ranging in size, including N2-MeG, N2-EtG, N2-IbG, N2-BzG, N2-NaphG, and N2-AnthG. Two crystal structures of Dpo4 with an oligonucleotide containing an N2-NaphG adduct were determined. The complexes are identical in their composition but differ in the arrangement of the template N2-NaphG and in the pairing schemes. A kinetic model involving active and inactive DNA polymerase complexes was developed to fit the available data and is discussed in relation to the available structural data.
EXPERIMENTAL PROCEDURES
Materials—Dpo4 was expressed in E. coli and purified to electrophoretic homogeneity as described previously (46). Unlabeled dNTPs were obtained from Amersham Biosciences, (Sp)-dCTPαS and (Sp)-dTTPαS were purchased from Biolog Life Science Institute (Bremen, Germany), and [γ-32P]ATP (specific activity 3 × 103 Ci mmol–1) was from PerkinElmer Life Sciences. T4 polynucleotide kinase and restriction endonucleases were purchased from New England Biolabs (Beverly, MA). Bio-spin columns were obtained from Bio-Rad (Hercules, CA).
Oligonucleotides—Unmodified 24-mer, 25-mer, and 36-mer (Table 1) were purchased from Midland Certified Reagent Co. (Midland, TX). Six 36-mers, each containing an N2-alkyl G adduct (N2-MeG, N2-EtG, N2-IbG, N2-BzG, N2-NaphG, or N2-AnthG, Fig. 1) were prepared as previously described and characterized by capillary gel electrophoresis and matrix-assisted laser desorption ionization/time-of-flight MS (24, 27–29, 48). The extinction coefficients for the oligonucleotides, estimated by the Borer method (49), were: 24-mer, ε260 = 224 mm–1 cm–1; 25-mer, ε260 = 232 mm–1 cm–1; and 36-mer, ε260 = 310 mm–1 cm–1 (Table 1) (24, 27).
TABLE 1.
Oligodeoxynucleotides used in this study
G*: G, N2-MeG, N2-EtG, N2-IbG, N2-BzG, N2-NaphG, or N2-AnthG, Cdd, terminal dideoxycytidine.
| 24-mer | 5′-GCCTCGAGCAGCCGCAGACGAG-3′ |
| 25C-mer | 5′-GCCTCGAGCAGCCGCAGACGAGC-3′ |
| 25T-mer | 5′-GCCTCGAGCAGCCGCAGACGAGT-3′ |
| 36-mer | 3′-CGGAGCTCGTCGGCGTCTGCTCG*CTCTGCGGCT-5′ |
| 14-mer | 5′-GGGGGAAGGTTCCdd-3′ |
| 18-mer | 3′-CCCCCTTCCAAG(N2-NaphGCACT-5′ |
FIGURE 1.

N2-Alkylguanine derivatives used in this study.
Reaction Conditions for Enzyme Assays and Analysis—Standard DNA polymerase reactions were carried out in 50 mm Tris-HCl buffer (pH 7.5 at 25 °C) containing 50 mm NaCl, 5 mm dithiothreitol, 100 μg of bovine serum albumin ml–1, and 5% (v/v) glycerol) at 37 °C (24, 27–29, 48). Primer (32P-labeled at the 5′-end using T4 polynucleotide kinase/[γ-32P]ATP) was annealed to either an unmodified or modified (N2-alkyl-G) template by heating the primer and template (equimolar mixture) to 95 °C for 5 min and then slowly cooling to room temperature. All reactions were initiated by the addition of dNTP solutions containing MgCl2 (5 mm final concentration) to the preincubated enzyme/DNA mixtures. After reaction, 5-μl aliquots of the reaction mixture were quenched with EDTA-formamide solution (50 μl of 20 mm EDTA in 95% formamide (v/v) with 0.5% bromphenol blue (w/v) and 0.05% xylene cyanol (w/v)). Products were resolved using a 20% polyacrylamide (w/v) denaturing gel electrophoresis system containing 8 m urea and then visualized and quantitated by PhosphorImager analysis using a Bio-Rad Molecular Imager FX instrument and Quantity One software (Bio-Rad).
Primer Extension Assay with All Four dNTPs—Unmodified or modified DNA (100 nm) was incubated with Dpo4 (0, 0.5, 2, or 10 nm) in 50 mm Tris-HCl buffer (pH 7.5 at 25 °C) containing 50 mm NaCl, 5 mm dithiothreitol, 100 mg of bovine serum albumin ml–1, and 5% glycerol (v/v)). Reactions were initiated by adding dNTP·Mg2+ (100 μm each dNTP and 5 mm MgCl2) solution at 37 °C. At the indicated time points, reactions were quenched by the addition of EDTA-formamide solution and analyzed by gel electrophoresis.
Steady-state Kinetic Analyses—A 32P-labeled primer/template complex was extended in the presence of varying concentrations of a single dNTP. The molar ratio of Dpo4 to DNA was <0.10. Polymerase concentrations and reaction time were adjusted to keep the extent of product formation <20% (50). All determinations were usually done at twelve dNTP concentrations (with the Km values in the center of the points). Reactions were quenched by the addition of EDTA-formamide solution, analyzed using gel electrophoresis, and then quantitated (phosphorimaging). Graphs of product formation versus dNTP concentration were fit using nonlinear regression (hyperbolic fits) using GraphPad Prism Version 3.0 (San Diego, CA) for the determination of kcat and Km values.
Pre-steady-state Reactions—Rapid quench experiments were performed using a model RQF-3 KinTek Quench Flow Apparatus (KinTek Corp., Austin, TX), with 50 mm Tris-HCl (pH 7.4, 25 °C) aqueous solution in the drive syringes. Reactions were initiated by rapid mixing of 32P-primer/template/Dpo4 mixtures (12.5 μl) with the dNTP·Mg2+ complex (10.9 μl) and were then quenched with 0.6 m EDTA after varying times. 20-μl aliquots of the reaction product solution were mixed with 100 μl of EDTA-formamide solution, and the products were separated using denaturing gel electrophoresis (see above) and quantitated. Pre-steady-state data, with excess DNA or Dpo4, were fit to Equations 1 or 2, respectively, using nonlinear regression analysis in GraphPad Prism Version 3.0,
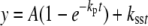 |
(Eq. 1) |
 |
(Eq. 2) |
where y is the concentration of product, A is the burst amplitude, kp is the pre-steady-state rate of nucleotide incorporation, t is time, and kss is the steady-state rate of nucleotide incorporation (51, 52).
Pre-steady-state Trap Experiments—Pre-steady-state incorporation of dCTP opposite N2-AnthG was also analyzed in a RQF-3 KinTek Quench Flow Apparatus, with excess unlabeled trap DNA added along with dCTP. Reactions were initiated by rapid mixing of a 70 nm Dpo4 plus 120 nm 32P-labeled 24-mer primer/36-mer template (containing N2-AnthG) complex in 50 mm Tris-HCl buffer (pH 7.4) with a second solution containing 1 mm dCTP, 5 mm Mg2+, and 1.2 μm unlabeled trap 24-mer/36-mer in the same buffer. Reactions were quenched with 0.6 m EDTA after varying times. The reaction products were quantified by denaturing gel electrophoresis and the data points were fit to Equation 1.
Phosphorothioate Analysis—Phosphorothioate analysis was done to compare the burst rates for the incorporation of dCTP and (Sp)-dCTPαS opposite the template bases. Reactions were initiated by rapid mixing of 70 nm 32P-primer/template and 120 nm Dpo4 in 50 mm Tris-HCl buffer (pH 7.4, 12.5 μl) with a mixture of 1 mm dNTP or (Sp)-dNTPαS and 5 mm MgCl2 solution (10.9 μl) in the quench flow apparatus and were then quenched with 0.6 m EDTA after varying reaction times. The reaction products were quantified by denaturing gel electrophoresis and the data points were fit to Equation 1.
Determination of kpol and Kd,dCTP—kpol and Kd,dCTP were estimated by performing pre-steady-state reactions in the quenched-flow apparatus with varying reaction times at different dNTP concentrations. Graphs of the burst rates (kp) versus dNTP concentration were fit to a hyperbolic equation,
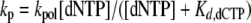 |
(Eq. 3) |
where kpol is the maximal rate of nucleotide incorporation and Kd,dCTP is the equilibrium dissociation constant for dCTP (51, 52).
LC-MS/MS Analysis of Oligonucleotide Products from Dpo4 Reactions (46, 53)—Dpo4 (5 μm) was preincubated with primer/template (10 μm), and the reaction was initiated by addition of dNTP (1 mm each) and MgCl2 (5 mm) in 50 mm Tris-HCl buffer (pH 7.4) at 37 °C for 4 h. Reactions were terminated by extraction of the remaining dNTPs using a size-exclusion chromatography column (Bio-Spin 6 chromatography column, Bio-Rad). Concentrated stocks of Tris-HCl, dithiothreitol, and EDTA were added to restore the concentrations to 50 mm, 5 mm, and 1 mm, respectively. E. coli uracil DNA glycosylase (20 units, Sigma-Aldrich) was added, and the solution was incubated at 37 °C for 6 h to hydrolyze the uracil residue on the extended primer. The reaction mixture was then heated at 95 °C for 1 h in the presence of 0.25 m piperidine, followed by removal of the solvent by centrifugation under vacuum. The dried samples were resuspended in 100 μl of H2O for MS analysis.
LC-MS/MS analysis was performed on an Acquity UPLC system (Waters, Milford, MA) connected to a Finnigan LTQ mass spectrometer (Thermo Fisher Scientific, Waltham, MA), operating in the ESI-negative ion mode. An Acquity UPLC BEH octadecylsilane (C18) column (1.7 μm, 1.0 mm × 100 mm) was used with the following LC conditions: Buffer A contained 10 mm CH3CO2NH4 plus 2% CH3CN (v/v), and buffer B contained 10 mm CH3CO2NH4 plus 95% CH3CN (v/v). The following gradient program was used, with a flow rate of 150 μl min–1: 0–2.5 min, linear gradient from 100% A to 95%A/5% B (v/v); 2.5–6.0 min, linear gradient to 75% A/25% B (v/v); 6–6.5 min, linear gradient to 100% B; 6.5–8.0 min, hold at 100% B; 8.0–9.0 min, linear gradient to 100% A; 9.0–12.0 min, hold at 100% A. The temperature of the column was maintained at 50 °C. Samples were injected with an autosampler system. ESI conditions were as follows: source voltage, 4 kV; source current, 100 mA; auxiliary gas flow rate setting, 20; sweep gas flow rate setting, 5; sheath gas flow setting, 34; capillary voltage, –49 V; capillary temperature, 350 °C; and tube lens voltage, –90 V. MS/MS conditions were as follows: normalized collision energy, 35%; activation Q, 0.250; and activation time, 30 ms. The doubly charged (negative ion) species were generally used for CID analysis. Calculations for the CID fragmentations of oligonucleotide sequences were done using a program linked to the Mass Spectrometry Group of Medicinal Chemistry at the University of Utah.
Kinetic Simulations—Kinetic simulations were performed using two mechanisms: a minimal mechanism, which has been previously defined for DNA polymerization (54, 55), and a mechanism with the presence of a non-productive ternary complex (56, 57). The fits to the experimentally determined data were developed utilizing DynaFit (BioKin, Watertown, MA) (58), run on an iMac computer using a Macintosh OS 8.5.1 operating system (Apple Computer, Cupertino, CA).
Crystallization and X-ray Diffraction Data Collection—The 18-mer template containing N2-NaphG and 14-mer primer containing a 3′-terminal dideoxycytidine (Table 1) were annealed to form a duplex. Dpo4 was mixed with DNA (1:1.2 molar ratio) in 20 mm Tris-HCl buffer (pH 8.0, 25 °C) containing 60 mm NaCl, 4% glycerol (v/v), and 5 mm β-mercaptoethanol and then placed on ice for 1 h prior to incubation with 5 mm MgCl2 and 1 mm dGTP. The final Dpo4 concentration was ∼10 mg ml–1. Crystals were grown using the sitting drop/vapor-diffusion method with the reservoir solution containing 20 mm Tris-HCl (pH 8.0 at 25 °C), 15% polyethylene glycol 3350 (w/v), 60 mm NaCl, 5 mm MgCl2, and 4% glycerol (v/v). Droplets consisted of a 1:1 (v/v) mixture of the Dpo4·DNA·Mg2+·dGTP complex and the reservoir solutions and were equilibrated against the reservoir solutions. Crystals were soaked in mother liquor containing an additional 25% polyethylene glycol 3350 (w/v) and 15% ethylene glycol (v/v) and then swiped through paratone-N (Hampton Research, Aliso Viejo, CA) and flash frozen in a stream of liquid nitrogen.
X-ray Diffraction Data Collection and Processing—X-ray diffraction data for the Dpo4-N2-NaphG-1 (Npg-1) and Dpo4-N2-NaphG-2 (Npg-2) complex crystals were collected at the Advanced Photon Source (Argonne National Laboratory, Argonne, IL) on the 21-ID (LS-CAT) and 22-ID (SER-CAT) beam lines, respectively. Both data sets were recorded from cryoprotected crystals using a wavelength of 0.98 Å at 110 K. The crystals diffracted to 3.0-Å resolution (Table 2). Individual frames were indexed and scaled with the program XDS (59) (Npg-1) or HKL2000 (60) (Npg-2). Both complex crystals belong to space group P21212. X-ray diffraction data collection and processing statistics are listed in Table 2.
TABLE 2.
Crystal data and refinement parameters for the ternary (protein·DNA·dGTP) complexes of Dpo4
| Parameter | Npg-1 | Npg-2 |
|---|---|---|
| Data collection | ||
| X-ray source | APS (LS-CAT) | APS (SER-CAT) |
| Beam line | ID-21 | ID-22 |
| Detector | MAR CCD | MAR CCD |
| Wavelength (Å) | 0.98 | 0.99 |
| Temperature (K) | 110 | 110 |
| Number of crystals | 1 | 1 |
| Space group | P21212 | P21212 |
| Unit cell (a, b, c; Å) | 95.04, 103.91, 52.32 | 95.92, 103.83, 52.49 |
| Resolution range (Å) | 19.97-3.10 | 29.25-3.0 |
| Highest resolution shella | (3.19-3.10) | (3.19-3.0) |
| Number of measurements | 63,806 (6,312) | 78,594 (1,746) |
| Number of unique reflections | 9,733 (912) | 11,751 (1,607) |
| Redundancy | 6.5 (6.9) | 6.6 (7.3) |
| Completeness (%) | 98.4 (95.1) | 98.5 (97.1) |
| Rmergeb | 9.4 (64.9) | 7.8 (60.7) |
| Signal to noise (〈I/σI〉) | 15.68 (3.22) | 23.37 (4.70) |
| Solvent content (%) | 56.3 | 59.5 |
| Model composition (asymmetric unit) | ||
| Number of amino acid residues | 342 | 342 |
| Number of water molecules | 42 | |
| Number of Mg2+ ions | 4 | 4 |
| Number of template nucleotides | 16 | 16 |
| Number of primer nucleotides | 14 | 14 |
| Number of dGTP | 1 | 1 |
| Rfc (%) | 23.3 | 22.6 |
| Rfreed (%) | 28.2 | 28.2 |
| Estimated coordinate error (Å) | ||
| From Luzatti plot | 0.45 | 0.40 |
| From Luzatti plot (c-ve) | 0.59 | 0.54 |
| From σA plot | 0.54 | 0.36 |
| From σA plot (c-v) | 0.79 | 0.50 |
| Temperature factors | ||
| From Wilson plot (Å2) | 62.4 | 57.3 |
| Mean isotropic (Å2) | 88.9 | 77.4 |
| r.m.s.d. in temperature factors | ||
| Bonded main-chain atoms (Å2) | 1.25 | 1.50 |
| Bonded side-chain atoms (Å2) | 1.53 | 2.25 |
| r.m.s.d. from ideal values | ||
| Bond lengths (Å) | 0.009 | 0.009 |
| Bond angles (°) | 1.6 | 1.6 |
| Dihedral angles (°) | 22.8 | 22.2 |
| Improper angles (°) | 1.96 | 1.77 |
Values in parentheses correspond to the highest resolution shells.
Rmerge = Σhkl Σj = 1,N |〈Ihkl〉 — Ihklj|/Σhkl Σj = 1,N|Ihklj|, where the outer sum (hkl) is taken over the unique reflections.
Rf = Σhkl Fohkl|—k|Fchkl/Σhkl|Fohkl|, where |Fohkl| and |Fchkl| are the observed and calculated structure factor amplitudes, respectively.
Rfree idem, for the set of reflections (5% of the total) omitted from the refinement process.
c-v, cross-validation.
Structure Determination and Refinement—A refined wild-type Dpo4-G complex (pdb accession code 2bqr (46)) minus solvent molecules, template residue 5, metal ions, and dGTP was used as the starting model for Npg-1. Molecular replacement was performed using MOLREP as a part of the CCP4 program suite (61). The refined structure of the Npg-1 complex (minus solvent molecules, template residue 5, metal ions, and dGTP) served as the starting model for the Npg-2 structure, and the locations of the individual models were optimized by several rounds of rigid body refinement while gradually increasing the resolution of the diffraction data.
Manual model rebuilding was done with the program TURBO-FRODO.4 The maps were computed using the sA-modified coefficients (62). Clear positive density for the Mg2+ ions and the dGTP was observed in the initial difference Fourier electron density maps of both complexes, although the positive density was sparser for dGTP in the Npg-1 structure. Positive density was observed for the N2-NaphG-modified template residue, but several rounds of modeling and refinement were required to ascertain the final orientation of the base and the naphthalene moiety in both structures. The CNS package (63) was used for the refinement of the models by performing simulated annealing, gradient minimization, and refinement of individual isotropic temperature and occupancy factors. The statistics of the refined models for all structures are summarized (Table 2). The crystallographic figures were prepared using PyMOL.5 Calculation of the DNA helical parameter descriptions was performed using CURVES (64).
RESULTS
Primer Extension by Dpo4 Using All Four dNTPs—Full-length extension beyond various N2-alkyl G adducts in the presence of all four dNTPs by Dpo4 is shown in Fig. 2. An increase in the Dpo4 concentration enhanced extension for all DNA adducts examined. Dpo4 readily extended beyond the unmodified G and N2-MeG, N2-EtG, N2-IbG, N2-BzG, and N2-NaphG templates to yield 35-, 36-, and 37-mer products, although the extensions were partially blocked opposite these adducts to yield (unelongated) 25-mer product. Other products (28-, 29-, and 31-mers) were also produced. With N2-AnthG, only a one-base extension product was observed, and subsequent extension was severely blocked under these conditions.
FIGURE 2.

Extension of 32P-labeled primers opposite G, N2-MeG, N2-EtG, N2-IbG, N2-BzG, N2-NaphG, and N2-AnthG by Dpo4 with all four dNTPs present. A primer(24-mer) was annealed with each of the seven different 36-mer templates (Table 1) containing an unmodified G or N2-alkylG placed at the 25th position from the 3′-end. Reactions were done for 20 min with increasing concentrations of Dpo4 (0, 0.5, 2, or 10 nm) and 100 nm DNA substrate (primer/template) as indicated. The 32P-labeled 24-mer primer was extended in the presence of all four dNTPs (100 μm each). Reaction products were resolved by denaturing gel electrophoresis, with subsequent phosphorimaging analysis.
Steady-state Kinetics of dNTP Incorporation by Dpo4 Opposite G and N2-Alkyl G Adducts—Steady-state kinetic parameters (kcat and Km) were measured for the single incorporation of each dNTP opposite G and N2-alkyl G adducts, with varying concentrations of dNTP (Table 3). Dpo4 preferentially incorporated dCTP opposite all of the N2-alkyl G adducts. However, the efficiencies (kcat/Km) for correct incorporation opposite N2-alkyl G adducts were decreased 3- to 125-fold compared with unmodified G, due to both the decreased catalytic rates (kcat) and increased Km,dNTP values. The efficiencies for the misincorporation of dATP, dGTP, and dTTP opposite all the N2-alkyl G adducts were similar to those measured with unmodified G.
TABLE 3.
Steady-state kinetic parameters for one-base incorporation opposite G and N2-alkyl G adducts by Dpo4
The extent of conversion of primer to the product was kept <20% by adjustment of the enzyme concentration and reaction time.
| Template | dNTP | Km,dNTP | kcat × 103 | kcat/Km × 105 | Efficiency relative to dCTP:G | Misincorporation frequency |
|---|---|---|---|---|---|---|
| μm | s-1 | μm-1 s-1 | ||||
| G | C | 1.2 ± 0.3 | 960 ± 50 | 8.2 × 104 | 1 | |
| A | 490 ± 60 | 5.7 ± 0.2 | 1.2 | 68,000-fold less | 1.4 × 10-5 | |
| G | 190 ± 20 | 6.3 ± 0.1 | 3.3 | 25,000-fold less | 4.1 × 10-5 | |
| T | 780 ± 140 | 34 ± 2 | 4.4 | 19,000-fold less | 5.4 × 10-5 | |
| N2-MeG | C | 1.9 ± 0.5 | 500 ± 21 | 2.7 × 104 | 3-fold less | |
| A | 650 ± 70 | 7.6 ± 0.3 | 1.2 | 68,000-fold less | 0.44 × 10-4 | |
| G | 380 ± 80 | 23 ± 2 | 6.1 | 13,000-fold less | 2.3 × 10-4 | |
| T | 320 ± 90 | 40 ± 5 | 12 | 6,800-fold less | 4.7 × 10-4 | |
| N2-EtG | C | 7.4 ± 1.0 | 83 ± 3 | 1100 | 75-fold less | |
| A | 360 ± 60 | 3.4 ± 0.2 | 0.93 | 88,000-fold less | 0.82 × 10-3 | |
| G | 210 ± 30 | 5.5 ± 0.3 | 2.6 | 32,000-fold less | 2.4 × 10-3 | |
| T | 78 ± 18 | 2.6 ± 0.1 | 3.4 | 24,000-fold less | 3.0 × 10-3 | |
| N2-IbG | C | 16 ± 3 | 100 ± 5 | 650 | 126-fold less | |
| A | 280 ± 50 | 3.0 ± 0.2 | 1.1 | 75,000-fold less | 1.6 × 10-3 | |
| G | 170 ± 10 | 8.8 ± 0.2 | 5.2 | 16,000-fold less | 8.0 × 10-3 | |
| T | 140 ± 10 | 1.6 ± 0.0 | 1.2 | 68,000-fold less | 1.9 × 10-3 | |
| N2-BzG | C | 21 ± 4 | 270 ± 20 | 1300 | 63-fold less | |
| A | 190 ± 30 | 2.1 ± 0.1 | 1.1 | 75,000-fold less | 0.89 × 10-3 | |
| G | 120 ± 20 | 2.2 ± 0.1 | 1.8 | 46,000-fold less | 1.4 × 10-3 | |
| T | 320 ± 53 | 4.4 ± 0.2 | 1.4 | 59,000-fold less | 1.1 × 10-3 | |
| N2-NaphG | C | 5.2 ± 1.0 | 110 ± 3 | 2200 | 37-fold less | |
| A | 320 ± 60 | 1.0 ± 0.1 | 0.33 | 248,000-fold less | 1.5 × 10-4 | |
| G | 180 ± 20 | 3.8 ± 0.1 | 2.2 | 37,000-fold less | 9.8 × 10-4 | |
| T | 120 ± 20 | 1.5 ± 0.1 | 1.3 | 63,000-fold less | 5.6 × 10-4 | |
| N2-AnthG | C | 1.3 ± 0.15 | 43 ± 1 | 3200 | 26-fold less | |
| A | 360 ± 450 | 0.78 ± 0.03 | 0.21 | 390,000-fold less | 0.67 × 10-4 | |
| G | 870 ± 140 | 6.5 ± 0.5 | 0.75 | 109,000-fold less | 2.3 × 10-4 | |
| T | 100 ± 10 | 2.3 ± 0.05 | 2.3 | 36,000-fold less | 7.1 × 10-4 |
Correct incorporation of dCTP opposite unmodified G was preferred over misincorporation for Dpo4, yielding a misincorporation frequency (defined as f = (kcat/Km)dNTP/(kcat/ Km)dCTP, where dNTP ≠ dCTP) of 10–5. The misincorporation frequency was 10–3 to 10–4 for all the N2-guanyl adducts (10- to 100-fold increase), mainly due to decreased correct incorporation efficiency (as opposed to changes in the misincorporation efficiency).
Steady-state Next-base Extension following C or T Paired with G or N2-Alkyl G Adducts—Steady-state next-base extension (i.e. incorporation of dGTP opposite the next template base, C (Table 4)) was examined; only the extension from mismatched G (or N2-alkyl G):T was studied here because all misincorporation events opposite the N2-alkyl G adducts showed similar efficiencies. Extension efficiencies (kcat/Km) were decreased 10- to 74-fold (for extension from the N2-alkyl G:C pairs, compared with the unmodified G:C pair). The N2-alkyl G adducts showed little effect on the extension efficiency from the mispairs of N2-alkyl G adducts (or the G:T mismatch). Overall, extension from the matched base pair was still more efficient than from a mismatched pair for all the N2-alkyl G adducts; the misextension ratio (fext) (defined as fext = (kcat/Km)mispair/(kcat/Km)correct pair (50, 65)) was increased 6- to 22-fold for the N2-alkyl G adducts (compared with unmodified G).
TABLE 4.
Steady-state kinetic parameters for next base extension from G (or N2-alkyl G):C (or T) template primer termini by Dpo4
The extent of conversion of primer to the product was kept <20% by adjustment of the enzyme concentration and reaction time.
| Template | Primer | Km, dNTP | kcat × 103 | kcat/Km × 105 | Efficiency relative to G:C |
|---|---|---|---|---|---|
| μm | s-1 | μm-1 s-1 | |||
| G | C | 23 ± 3 | 530 ± 30 | 2300 | |
| T | 180 ± 20 | 17 ± 1 | 10 | 230-fold less | |
| N2-MeG | C | 190 ± 30 | 190 ± 10 | 102 | 23-fold less |
| T | 73 ± 9 | 6.9 ± 0.2 | 9.5 | 240-fold less | |
| N2-EtG | C | 290 ± 60 | 200 ± 20 | 68 | 34-fold less |
| T | 57 ± 9 | 2.6 ± 0.2 | 4.6 | 500-fold less | |
| N2-IbG | C | 820 ± 130 | 260 ± 30 | 31 | 74-fold less |
| T | 110 ± 30 | 0.93 ± 0.05 | 0.8 | 2,900-fold less | |
| N2-BzG | C | 660 ± 140 | 220 ± 30 | 34 | 68-fold less |
| T | 26 ± 4 | 0.70 ± 0.01 | 2.8 | 820-fold less | |
| N2-NaphG | C | 190 ± 30 | 140 ± 10 | 76 | 30-fold less |
| T | 38 ± 9 | 1.4 ± 0.1 | 3.7 | 620-fold less | |
| N2-AnthG | C | 100 ± 20 | 220 ± 20 | 225 | 10-fold less |
| T | 110 ± 20 | 6.7 ± 0.3 | 6.0 | 380-fold less |
Pre-steady-state Burst Kinetics of dCTP Incorporation Opposite G or N2-Alkyl G Adducts by Dpo4—Dpo4 clearly showed burst phases for the correct incorporation of dCTP opposite unmodified G or N2-alkyl G adducts, even with the bulky adduct N2-AnthG (Fig. 3). The burst phase measures incorporation of dCTP opposite G or modified G during the initial binding event between enzyme and substrate. The linear phase of the plot results from subsequent binding events and is dependent upon a conformational relaxation from the active conformation following pyrophosphate release (66). The burst phase for dCTP incorporation opposite N2-alkyl G adducts showed a trend toward decreased amplitude as the adduct bulk increased from N2-MeG to N2-AnthG, and burst rates were also attenuated compared with the unmodified G.
FIGURE 3.

Pre-steady-state kinetics of nucleotide incorporation of dCTP or (Sp)-dCTPαS opposite G or each of the N2-alkyl G adducts by Dpo4. Dpo4 (70 nm) was incubated with each 24-mer/36-mer primer·template complex (120 nm, 32P-labeled primer) in a rapid quench-flow instrument and mixed with 1 mm dCTP (▪) or (Sp)-dCTPαS (•) to initiate single-base incorporation reactions with A, G; B, N2-MeG; C, N2-EtG; D, N2-IbG; E, N2-BzG; F, N2-NaphG; and G, N2-AnthG. Pre-steady state rates (kp) were estimated using a burst equation (Equation 1), and the solid lines represent the best fits. The following rates and burst concentrations were estimated: A, dCTP, kp = 1.8 ± 0.4 s–1/42 nm and (Sp)-dCTPαS, kp = 1.8 ± 0.4 s–1; B, dCTP, kp = 1.7 ± 0.2 s–1/71 nm and (Sp)-dCTPαS, kp = 1.5 ± 0.1 s–1; C, dCTP, kp = 0.73 ± 0.08 s–1/52 nm and (Sp)-dCTPαS, kp = 0.51 ± 0.09 s–1; D, dCTP, kp = 0.11 ± 0.01 s–1/43 nm and (Sp)-dCTPαS, no burst, linear rate k = 0.0085 ± 0.0003 s–1; E, dCTP, kp = 0.27 ± 0.02 s–1/39 nm and (Sp)-dCTPαS, no burst, linear rate k = 0.011 ± 0.001 s–1; F, dCTP, kp = 0.19 ± 0.03 s–1/17 nm and (Sp)-dCTPαS, no burst, linear rate k = 0.013 ± 0.0004 s–1; and G, dCTP, kp = 0.46 ± 0.04 s–1/13 nm and (Sp)-dCTPαS, kp = 0.83 ± 0.04 s–1.
Steady-state and Pre-steady-state Burst Kinetics of dTTP Incorporation Opposite G and N2-Alkyl G Adducts by Dpo4—No bursts were detected for the incorporation of dTTP opposite G or any N2-alkyl G adducts, except for N2-MeG (Fig. 4A). The incorrect incorporation of dTTP opposite N2-AnthG yielded a higher misincorporation rate (Fig. 4A). Misincorporation of both dTTP and (Sp)-dTTPαS opposite N2-MeG showed small bursts (Fig. 4B), with much lower burst amplitudes (∼3% of the enzyme concentration), burst rates, and steady-state rates.
FIGURE 4.

Steady-state and pre-steady-state kinetics of incorporation of dTTP opposite G and N2-alkyl G adducts by Dpo4. Dpo4 (70 nm) was incubated with 24-mer/36-mer primer·template complex (120 nm) in a rapid quench-flow instrument and mixed with 1 mm dTTP/5 mm MgCl2 to initiate reactions. A, no bursts were observed for G (▪), N2-EtG (♦), N2-IbG (•), N2-BzG (□), N2-NaphG (▵), or N2-AnthG (▿). The linear rates (slope) were estimated: G, 0.40 ± 0.01 s–1; N2-EtG, 0.10 ± 0.00 s–1, N2-IbG, 0.047 ± 0.001 s–1; N2-BzG, 0.030 ± 0.001 s–1; N2-NaphG, 0.011 ± 0.000 s–1; and N2-AnthG, 0.076 ± 0.003 s–1). B, the presence of burst for incorporation of dTTP (▪) or (Sp)-dTTPαS (•) opposite N2-MeG. The data for N2-MeG were fit to a burst equation (Equation 1), and the others were fit to a linear equation. The rates for the incorporation of dTTP (▪) or (Sp)-dTTPαS opposite N2-MeG were, respectively, kobs = 0.59 ± 0.11 s–1 and kobs = 0.61 ± 0.09 s–1.
Determination of kpol and Kd,dCTP Values for dCTP Incorporation by Dpo4—Analysis of the changes in the pre-steady-state burst rates as a function of increasing dNTP concentration (supplemental Fig. S1) yielded maximum rates of nucleotide incorporation (kpol) and apparent binding affinity of the dCTP to the Dpo4·DNA binary complex to form a ternary complex poised for catalysis (Kd,dCTP) (51, 52). The incorporation of dCTP opposite unmodified G or N2-MeG showed similar kpol (∼3.0 s–1) and Kd,dCTP (∼10 μm) values (Fig. 5, A and B). For the incorporation opposite bulkier N2-alkyl G adducts (N2-EtG to N2-NaphG, Fig. 5, C–F) the kpol values were decreased 4- to 32-fold compared with G, but the kpol value for N2-AnthG (Fig. 5G) increased unexpectedly (compared with N2-NaphG). The Kd,dCTP values for all the N2-alkyl G adducts (8 to 29 μm) were similar to that of unmodified G (10 μm) (3-fold variation compared with G).
FIGURE 5.

Estimation of kpol and Kd,dCTP for Dpo4 by pre-steady-state burst rate dependence on dCTP concentration. Dpo4 (200 nm) was incubated with 100 nm 24-mer/36-mer primer·template complexes (32P-labeled) in a rapid quench-flow instrument and mixed with varying dCTP concentrations (2–300 μm) to initiate reactions. Plots of burst rates (kobs, fit by Equation 2) versus [dCTP] were fit to a hyperbolic equation (Equation 3). A, G (unmodified); B, N2-MeG; C, N2-EtG; D, N2-IbG; E, N2-BzG; F, N2-NaphG; and G, N2-AnthG. The calculated kpol and Kd values are shown in each panel of the figure.
Phosphorothioate Analysis of dCTP Incorporation Opposite G and N2-Alkyl G Adducts by Dpo4—Incorporation of dCTP or (Sp)-dCTPαS opposite G, N2-MeG, and N2-EtG (Fig. 3, A–C) showed no obvious thio effects (burst rate ratios of 1.0, 1.1, and 1.4, respectively). In contrast, the incorporation of dCTP or (Sp)-dCTPαS opposite N2-IbG, N2-BzG, or N2-NaphG (Fig. 3, D–F) resulted in different rates and thio effects (ratios higher than 13, 25, and 15, respectively). Unexpectedly, incorporation of (Sp)-dCTPαS opposite N2-AnthG (Fig. 3G) showed a similar burst rate as in the incorporation of dCTP, without an obvious thio effect.
Determination of kpol and Kd,dCTP for dTTP Incorporation Opposite N2-MeG by Dpo4—The incorporation of dTTP opposite N2-MeG also showed a burst phase, and the corresponding kpol and Kd,dTTP were determined to be 0.95 (± 0.06) s–1 and 132 (± 25) μm, respectively (supplemental Fig. S2). Compared with correct dCTP incorporation (Fig. 4B), dTTP incorporation had a decreased amplitude (23-fold), decreased burst rate (3-fold), and weakened dNTP binding affinity (15-fold).
Pre-steady-state Incorporation of dCTP Opposite N2-AnthG with Excess Trap DNA—A 10-fold molar excess of unlabeled unmodified DNA was used to trap free Dpo4 after Dpo4 dissociated from the 32P-labeled DNA containing N2-AnthG; this experiment was done to establish whether a burst was indeed observed, because only Dpo4 that was still bound to the labeled DNA could extend the (radiolabeled) primer during the single binding event (24). Incorporation of dCTP opposite N2-AnthG with or without trap DNA showed a burst phase, and the burst amplitudes were similar (Fig. 6). A slow linear phase is observed in the trap experiment, most likely due to the relatively low burst in product formation (∼17% of [Dpo4]) and the fact that the protein trap is not 100% efficient. The major point of the trapping experiment was to be certain that the small burst amplitude observed with the N2-AnthG-containing substrate is representative of true pre-steady-state conditions.
FIGURE 6.

Pre-steady-state kinetics of incorporation of dCTP opposite N2-AnthG by Dpo4 in the absence and presence of excess unlabeled DNA trap. Dpo4 (70 nm) was incubated with 24-mer/36-mer primer·template complex containing N2-AnthG (120 nm) in a rapid quench-flow instrument and mixed with a mixture of either (i) 5 mm MgCl2/1 mm dCTP or (ii) 5 mm MgCl2/1 mm dCTP/1200 nm unlabeled trap 24-mer/36-mer DNA to initiate reactions. All polymerization reactions were quenched with 0.6 m EDTA at the indicated time intervals. The data were fit to the burst equation, y = A(1 – exp(–kpt)) + ksst (Equation 1). The burst rates were estimated to be 0.46 (± 0.04) s–1 for the case with dCTP only and 1.7 (± 0.2) s–1 for the case with the DNA trap.
Analysis of Primer Incorporation/Extension Products using LC-MS/MS—Full-length extension assays showed that all of the adducts, except for N2-AnthG, can be readily extended (Fig. 2). LC-MS/MS was utilized for the sequence analysis of the oligonucleotide extension products (46, 67). A key element for the success of this method is the placement of uracil residues in the primer strand and the use of uracil DNA glycosylase to hydrolyze at the introduced uracil sites, to reduce the product length for MS sequence analysis (46). Each product in the oligonucleotide product mixtures could be readily identified and quantified by LC-MS/MS (Table 5, supplemental Figs. S3, S4, and S5, and Tables S1–S4). From unmodified G to the N2-NaphG template, all of the detected products corresponded to the correct incorporation of dCTP opposite G or the N2-alkyl G adduct, with accurate extension following (as well as some blunt-end addition of A or C) (Table 5). For the bulkiest adduct, N2-AnthG, 79% of the products resulted from (correct) dCTP incorporation and the other 21% corresponded to misincorporation of dATP opposite N2-AnthG, with error-free extension following. No frameshift products were detected in any case.
TABLE 5.
Products of extension of N2-alkylG template·primer complexes by Dpo4
Reaction conditions: 2.5 μm Dpo4, 5 μm 24/36 oligonucleotide complex, 1 mm each dNTP, 5 mm Mg2+, 4 h. LC-MS: tR 2.90–2.96 min; charge: –4; area: m/z value ± 0.2. The position of incorporation is indicated in italics.
| 5′-GCCUCGAGUCAGCCGUAGACGU | AG | |||||
| 3′-CGGAGCTCGGTCGGCG TCT GCG | TCG | CTC | CTG | CGG | CT | |
| Unmodified G | AGC | GAG | GAC | GCC | GA | 17% |
| AGC | GAG | GAC | GCC | GAC | 24% | |
| AGC | GAG | GAC | GCC | GAA | 59% | |
| N2-MeG | AGC | GAG | GAC | GCC | GA | 27% |
| AGC | GAG | GAC | GCC | GAC | 23% | |
| AGC | GAG | GAC | GCC | GAA | 50% | |
| N2-EtG | AGC | GAG | GAC | GCC | GA | 93% |
| AGC | GAG | GAC | GCC | GAA | 7% | |
| N2-IbG | AGC | GAG | GAC | GCC | GA | 45% |
| AGC | GAG | GAC | GCC | GAC | 11% | |
| AGC | GAG | GAC | GCC | GAA | 44% | |
| N2-BzG | AGC | GAG | GAC | GCC | GA | 35% |
| AGC | GAG | GAC | GCC | GAC | 20% | |
| AGC | GAG | GAC | GCC | GAA | 45% | |
| N2-NaphG | AGC | GAG | GAC | GCC | GA | 96% |
| AGC | GAG | GAC | GCC | GAA | 4% | |
| N2-AnthG | AGC | GAG | GAC | GCC | GA | 56% |
| AGC | GAG | GAC | GCC | GAC | 7% | |
| AGC | GAG | GAC | GCC | GAA | 16% | |
| AGA | GAG | GAC | GCC | GA | 21% |
Crystal Structures of an N2-NaphG·C Complex—Two x-ray structures were determined for Dpo4 in complex with a DNA duplex in which the template strand contains an N2-NaphG residue. A 14-base primer was designed such that the dideoxycytosine (Cdd) at the terminus should pair with the N2-NaphG template residue. Both structures contained three Mg2+ ions total (two coordinated in the active site) and an incoming dGTP, putting N2-NaphG in the post-insertion (–1) position. Each of the crystals diffracted to ∼3 Å, with reasonable Rmerge values (9.4% and 7.8%, for Npg-1 and Npg-2, respectively). Both crystals belong to the space group P21212, with a single complex per asymmetric unit.
The two structures are very similar overall (Fig. 7A). Analysis of the helical parameters of the DNA substrates reveals perturbations that are observed in both complexes. Similar to results obtained with O6-BzG-modified templates (42), each of the two refined structures containing N2-NaphG possesses a distinct arrangement of the DNA in the active site. The naphthyl moiety exhibits well defined electron density for both orientations (Fig. 7). A simulated annealing omit map (excluding template N2-NaphG, primer terminal ddC, incoming dGTP, and active site Mg2+) further supports the correct positioning of the residues bound in the active site of both complexes (supplemental Fig. S6). The N2-NaphG residue is found in the syn orientation in the Npg-1 complex, which allows hydrogen bonding to occur between the exocyclic amino groups of the last two cytosines of the primer with the O6 atom of N2-NaphG (Fig. 7C). The α-phosphate of the incoming dGTP is positioned ∼5 Å from the primer terminus, and two of the Mg2+ ions are coordinated in the active site of the enzyme by residues Asp-7, Asp-105, Glu-106, and Asp-156. Overall, Npg-1 would appear to be an “active” complex.
FIGURE 7.

Comparison of the Npg-1 and Npg-2 structures. A, superimposition of the refined Npg-1 (cyan) and Npg-2 (purple) structures reveals similar overall topology (r.m.s.d. 0.52). B, the DNA substrates from Npg-1 and Npg-2 were superimposed with the DNA from the ternary complex of Dpo4 inserting dCTP opposite 8-oxoG (orange, pdb ID code 2c2e). A more pronounced bending of the helical axis was observed in the N2-NaphG-modified DNA. Other perturbations in the N2-NaphG DNA, e.g. buckling of the bases and widening of the major groove near the primer·template junction, are also evident. C, the active site of the Npg-1 crystal structure is shown in schematic form. The 3Fo – 2Fc electron density map (cyan mesh) is contoured at the 1σ level around the N2-NaphG residue. The incoming dGTP (red) is shown along with the magnesium ions (green spheres). D, base pairing and stacking interactions observed in the Npg-1 structure. E, the active site of the Npg-1 crystal structure is shown in schematic form. The 3Fo – 2Fc electron density map (purple mesh) is contoured at the 1σ level around the N2-NaphG residue. The incoming dGTP (red) is shown along with the magnesium ions (green spheres). F, base pairing and stacking interactions observed in the Npg-2 structure.
The N2-NaphG residue is found in the anti orientation in the Npg-2 complex but the terminal C of the primer is largely disordered. Based on the density surrounding the terminal phosphate and sparse density in the region of the base, the dideoxycytosine was flipped into the minor groove, as has been observed in other non-productive Dpo4 structures (38, 42).
The N2-NaphG residue is found in the anti orientation in the Npg-2 complex. The terminal dideoxycytosine of the primer is flipped into the minor groove, based on the density surrounding the terminal phosphate and sparse density in the region of the pyrimidine ring system. Such a conformation has been observed in other non-productive Dpo4 structures (38, 42). There is considerable distortion of the DNA duplex in both Npg-1 and Npg-2 structures when compared with the structure with a DNA lesion that replicated with high efficiency (dCTP opposite 8-oxoG by Dpo4; pdb ID code 2c2e; Fig. 7B). In both instances the base pairs to the 3′-side of the N2-NaphG residue are severely buckled (35° and 24° for Npg-1 and Npg-2, respectively), with large tilts for the cytosine at the –2 position (t = 27° and t = 19° for Npg-1 and Npg-2, respectively). Furthermore, the base pairing distance between hydrogen bond donors/acceptors is increased several residues upstream from N2-NaphG, indicating that the lesion can distort the helix several base pairs away from the actual site of damage.
Kinetic Simulation to a Mechanism—A general minimal mechanism is shown in Fig. 8A (51, 54, 55, 68–72), modified with the addition of step 8 (56, 66). The binding affinity and a burst rate for the incorporation of dCTP opposite G and N2-alkyl G adducts by Dpo4 were initially set at 15 μm and 3 s–1, respectively, based on the pre-steady-state kinetic determinations (Fig. 5). Recently, our laboratory utilized mutant Dpo4 containing Trp as a fluorescent probe to rigorously examine the kinetics describing Dpo4-catalyzed polymerization (66). It was concluded that a slow conformational relaxation following pyrophosphate release is the major rate-limiting determinant in the Dpo4 catalytic cycle. The kinetic mechanism used in our modeling studies reflects these new insights into Dpo4 catalysis (66).
FIGURE 8.

Kinetic mechanisms for DNA polymerase incorporation and fitting of progress curves for incorporation of dCTP opposite G or N2-alkyl G. A, general mechanism. The mechanism without step 8 and the non-productive complex is termed the “minimal mechanism” in the text. Step 8 was used in fitting (Table 6). E, polymerase; Dn, DNA substrate; E*, conformationally modified polymerase; Dn+1, DNA extended by one base (product); E#, inactive polymerase complex; and PPi, pyrophosphate. Forward and reverse rate constants for each step were developed on the basis of simulations of pre-steady-state nucleotide incorporation reactions with unmodified substrates. Parts B–H, the values shown in Part A and the values of k4, k8, and k–8 presented in Table 5 were used in fitting burst experiment progress curves for each of the templates (data points from Fig. 3).
The minimal mechanism fit generally well for the DNA polymerization with a stoichiometric burst amplitude. In this study, the incorporation of dCTP opposite G and N2-alkyl G adducts (except N2-MeG) by Dpo4 resulted in reduced burst amplitudes (Fig. 5). Simulation of experimental results for partial bursts cannot yield a reasonable fit for G itself or the other N2-alkyl G adducts using the minimal mechanism (except for N2-MeG), as expected (56), even with adjustment of the rates of steps after phosphodiester bond formation (step 4 in Fig. 8A). An alternate mechanism with an additional non-productive E-DNA-dNTP ternary complex (56, 66) (Fig. 8A) yielded good fits to the experimental results for the incorporation of dCTP opposite G (Fig. 8B) and all of the N2-alkyl G adducts (except for N2-MeG, Fig. 8C), using the experimentally measured enzyme concentration (Fig. 8, C–H). The same rate constants shown in the scheme of Fig. 8A were used in the simulations to generate values of k4, k8, and k–8 (Table 6), and the ratios for the conversion of intermediate to the non-productive complex versus to the product (k8/k4) varied from 0.23 (N2-MeG) to 4.8 (for N2-AnthG). It should be noted that any product arising from non-productive complexes that escape the kinetic side reaction (k8/k–8) does not appear to greatly alter the progress curve, because all of the burst experiments are best fit by single-exponential and not double-exponential equations.
TABLE 6.
Kinetic modeling of pre-steady-state rate constants in a DNA polymerase mechanism
Indicated rates and errors were estimated using the program Dynafit. See Fig. 8A for mechanism, Fig. 8 (B–H) for fits to progress curves, and supplemental Table S5 for Dynafit script and sample results.
|
Oligonucleotide base
|
EDN⇆XDN, k4
|
XDN⇆ODN
|
k8/k4
|
|
|---|---|---|---|---|
| k8 | k-8 | |||
| s-1 | s-1 | s-1 | ||
| G | 3.2 ± 0.7 | 2.1 ± 1.2 | 0.07 ± 0.19 | 0.66 |
| N2-MeG | 6.1 ± 0.6 | 1.4 ± 0.2 | 10-5 | 0.23 |
| N2-EtG | 1.6 ± 0.2 | 0.96 ± 0.23 | 0.053 ± 0.022 | 0.60 |
| N2-IbG | 0.18 ± 0.01 | 0.14 ± 0.02 | 0.025 ± 0.004 | 0.78 |
| N2-BzG | 0.40 ± 0.03 | 0.52 ± 0.05 | 10-5 | 1.3 |
| N2-NaphG | 0.15 ± 0.02 | 0.32 ± 0.10 | 0.06 ± 0.015 | 2.1 |
| N2-AnthG | 0.21 ± 0.01 | 1.0 ± 0.1 | 0.0070 ± 0.0030 | 4.8 |
DISCUSSION
Although the Y-family DNA polymerase Dpo4 has been used extensively as a model for translesion polymerization, systematic studies on its ability to incorporate past lesions of different sizes have not been done previously. We had a set of oligonucleotides differing only in their N2-substitution at a single G site available and used these in this study with Dpo4. As in the cases of the human Y-family polymerases we have analyzed (24, 27–29, 48), the catalytic efficiency (steady-state kcat/Km) showed relatively limited attenuation with increasing size of the lesion at the G N2 atom, in contrast to replicative DNA polymerases (e.g. human pol δ, HIV-1 reverse transcriptase, and bacteriophage pol T7–) (24). Very recently RNA polymerases have been found to vary in their abilities to insert and copy past N2-EtG (16). With Dpo4 and this series of N2-guanyl derivatives, the fidelity showed only a limited decrease until the size increased to that of N2-AnthG (Table 5). Of interest was the maintenance of a pre-steady-state kinetic burst throughout the entire series of N2-guanyl substitutions, with decreasing burst amplitude. The Kd,dCTP did not change very much with increasing bulk at the guanyl N2 atom.
Two crystal structures of Dpo4 with template DNA containing N2-NaphG were solved. In both cases, the N2-NaphG adduct was positioned opposite dideoxycytidine at the 3′-end of a primer (at the –1 position, i.e. post-insertion), but two different active site orientations were observed. The N2-NaphG moiety adopts the syn orientation in the Npg-1 structure and the anti conformation in the active site of Npg-2. The DNA duplex suffers some perturbation in both structures, including a slight widening of the major groove near the primer·template junction and buckling of the base pairs upstream from the adduct site. Of the two structures, the Npg-1 complex appears to be productive, whereas the Npg-2 complex is considered non-productive.
In the structure Npg-2, the anti form of N2-NaphG has the primer 14-C flipped out of the active site and distorted (Fig. 7F), indicating that incorporation of dCTP opposite the bulkier N2-guanyl adducts will be very unfavorable, with a tendency to form less productive complexes. In comparison to the Watson-Crick base pairing presumably used for dCTP with template G and small N2-alkyl G adducts, the fraction of non-productive complex(es) is presumably increased with N2-guanyl adduct bulkiness, and incorporation burst amplitudes are decreased (Fig. 3). Kinetic modeling also yielded an increase in the residence time in the non-productive complex with N2-adduct bulkiness (Table 6 and Fig. 8). The bulkier N2-NaphG can also be in a syn configuration (Npg-1), which we conclude can incorporate dCTP opposite the Hoogsteen face of a modified G (Fig. 7). Thus the N2-alkyl G adducts may be incorporating dCTP via either of two (or more) mechanisms: with the small N2-alkyl G adducts, mainly via the usual anti configuration/Watson-Crick pairing mode; for the bulkier N2-aralkyl G adducts, via a syn/Hoogsteen mode. Changes in the use of these mechanisms with increasing bulkiness of the adducts may be reflected in the variations of the observed kinetic parameters (Fig. 3).
Of the two structures solved here, the complex that contains syn N2-NaphG in a Hoogsteen-type pairing with ddC appears to be the more productive. However, the use of a syn-oriented template base would not be expected to yield the level of fidelity observed for Dpo4 (Table 3). The Y-family member pol ι has been shown to rely upon Hoogsteen base pairing even with unmodified DNA (73, 74). In biochemical terms, pol ι has an unusual incorporation spectrum in that it incorporates opposite template dA with much greater efficiency and fidelity than other bases (75). When incorporating opposite template dG, pol ι does not possess the level of fidelity observed for any of the N2-alkyl G adducts tested with Dpo4. Crystal structures of pol ι have shown that the Hoogsteen pairing mode may arise, because the template sugar is embedded in a “cavity” formed primarily by residues from the finger domain (specifically Leu-62, Lys-60, and Gln-59), which shortens the C1′–C1′ distance from 10.65 Å (anti) to 8.6 Å (syn) (74). Such a shortening of the C1′–C1′ distance is not observed in the Npg-1 structure, but a direct comparison cannot be made because the syn-oriented G in the pol ι structure is in the insertion context, while syn N2-NaphG is in the post-insertion context of Dpo4. Nevertheless, some differences in this region are worth noting. Of the three residues postulated to influence the C1′–C1′ distance in pol ι, only Gln-59 is distinctive from the Val-32 of Dpo4, because the amide side chain of Gln-59 protrudes into the region of the active site that contains the template base. The Lys-60 of pol ι contacts the phosphate backbone of the template DNA, whereas the analogous residue in Dpo4 (Phe-33) marks the beginning of a loop between β-sheets 2 and 3 in the finger domain. The loop is comprised of eight amino acids residues in Dpo4, which contrast to the short three-residue β-hairpin observed in pol ι. Previous work has shown that pol ι is not particularly tolerant of bulk at the N2 atom of guanine and suffers a greater loss of fidelity during bypass of N2-dG adducts compared with either Dpo4 or pol κ (28, 48). The fidelity observed for pol ι bypass of N2-alkyl G adducts is ∼100- to 1000-fold more error-prone than the results obtained with Dpo4. Based on the crystal structures reported here and previous kinetic analysis it appears that the molecular features resulting in Dpo4-catalyzed bypass of N2-alkyl G adducts are not homologous to the ones that influence the ability of pol ι to preferentially utilize Hoogsteen pairs. Comparison to pol ι suggests that perhaps we have not captured the true active complex representing Dpo4-catalyzed bypass of N2-NaphG.
The presence of two substrate orientations in the Dpo4 active site is reminiscent of a study in which the trans N2-(10S) BPDE-G adduct was observed in two different orientations when bound to Dpo4 (45). Modeling studies of Dpo4-catalyzed bypass of a bulky N2-BPDE-G adduct had predicted that the adduct could be accommodated in both syn and anti orientations during insertion opposite the lesion (76). The resulting orientations observed in the N2-BPDE-G crystal structures were distinct from the models, but it should be noted here that the modeling study focused on insertion, whereas in the crystal structures the N2-BPDE-G adduct is in the post-insertion context. In the “productive” crystal structure (BPG-1B) (45), the N2-BPDE-G moiety is flipped into a gap between the finger and little finger domains, which was not predicted by molecular modeling. The BPG-1B complex of Bauer et al. (45) appears to be associated with –1 frameshift events, as judged by the available gel data (i.e. the predominant dNTP incorporation is the one corresponding to the residue 5′ of the BPDE adduct in the template). These events would argue that the “Type II” crystal structure is most relevant to the observed function during bypass of N2-BPDE-G. No quantitative kinetic results were reported, so comparisons to the N2-guanyl adducts used here are not possible. The syn orientation of the N2-BPDE-G adduct in the Dpo4 active site results in a complex that is non-productive, because the BPDE ring system is stacked between the last two bases in the primer and this distortion prevents optimal binding of the incoming dATP. By way of comparison, Dpo4 places the naphthyl group between the template DNA and the little finger domain when the N2-NaphG moiety in the syn orientation, not into the “gap” observed for the BPDE adduct (supplemental Fig. S6). With the placement of the N2-NaphG moiety in the syn orientation, Dpo4 can achieve what appears to be a productive post-insertion complex. However, there is significant distortion to the base pairs adjacent to the lesion and the major groove is widened by ∼3–4 Å starting three base pairs upstream of the adduct site. Superimposition of the Npg-1 complex with BPG-1B (45) reveals that, in contrast to BP-dG, the purine ring of N2-NaphG remains stacked in the template strand, perhaps due to the added flexibility of a methylene group between the exocyclic amino group and the naphthalene moiety (supplemental Fig. S6). The major similarity between the N2-NaphG structures solved here and the N2-BPDE-G structures is the ability of Dpo4 to allow several substrate conformations without greatly altering the polymerase structure.
It is of interest to compare the Npg-1 and Npg-2 structures solved here with the reported structure of pol κ bound to unmodified DNA (77), because Dpo4 and pol κ are both members of the DinB subfamily and can be considered homologues. Superimposition of the complexes reveals that, not surprisingly, the enzymes are quite similar in domain arrangement and orientation of the DNA substrate (Fig. 9A). Closer inspection of the polymerase active site shows that pol κ can clearly accommodate the N2-NaphG adduct in the syn orientation (Fig. 9B). Furthermore, the N-terminal portion of pol κ, which forms a “clasp” that encircles the DNA, has several aromatic side chains that could in theory promote the syn orientation of aromatic N2-aralkyl G adducts during translesion synthesis by way of π -π interactions. Such a hypothesis seems somewhat counter-intuitive. One might initially predict (as we did) that minor groove adducts could be housed in the larger active sites of Y-family DNA polymerases, thereby allowing normal Watson-Crick geometry to occur during bypass. A computational study of pol κ insertion opposite N2-BPDE-dG and N6-BPDE-dA adducts predicted that the enzyme can effectively bypass N2-BPDE-dG in the anti orientation (78). The same study postulates that the N-clasp is important for “near error-free” bypass of N2-BPDE-dG and blockage of pol κ by the N6-BPDE-dA adduct. The authors propose that the N-clasp promotes the anti orientation during accurate bypass of N2-BPDE-dG. Even though a productive configuration is not observed in the Npg-2 structure, we cannot rule out the possibility that accurate bypass of the larger N2-alkyl G adducts proceeds through some orientation that is closer to normal Watson-Crick geometry than the pairing mode observed in the Npg-1 complex. Because the N-clasp is unique to pol κ, it may play a role in promoting accurate bypass of minor groove lesions. Indeed, several lines of evidence are consistent with the idea that human pol κ is an important enzyme during translesion synthesis opposite bulky minor groove adducts and cross-links, both at the insertion and extension steps (27, 48, 79, 80).
FIGURE 9.

Structural comparison between Dpo4-catalyzed bypass of N2-NaphG and human pol κ. A, superimposition of the Npg-1 complex (cyan) and the human pol κ ternary complex (purple; pdb ID code 2oh2; r.m.s.d. = 5.35). B, the active site DNA residues from the Npg-1 complex (cyan) are shown along with the superimposed pol κ structure (purple, space-filling). Hydrophobic and/or aromatic residues near the naphthyl moiety are highlighted for both enzymes.
Although we did not obtain structures of the short alkyl derivatives (e.g. N2-MeG and N2-EtG), we would anticipate that these are probably very similar to the normal G complex, in that Watson-Crick pairing is very feasible, no misincorporations occur, and the bursts and rates for incorporation (of dCTP) were very similar to those observed with G (Tables 3 and 5 and Fig. 3). The base orientation observed for the larger adducts is more difficult to predict. Based on kinetic and LC-MS analysis, Dpo4 appears to favor accurate bypass of the bulky adducts over –1 frameshift deletions. Analysis of the crystal structures suggests the possibility that, as the bulk at the N2 position increases, accurate bypass of the N2-NaphG adduct occurs when the lesion adopts the syn orientation. Still, it is difficult to explain the limited decrease in Dpo4 fidelity observed with the N2-NaphG adduct (decreased ∼20-fold relative to unmodified DNA) if the Hoogsteen face is indeed being presented to the incoming dNTP.
Our conclusion from the structural results with N2-NaphG is that this is a very distorted complex and the Npg-2 complex is catalytically inactive. We utilized a variation of the DNA polymerase catalytic scheme in which an inactive ternary complex is in equilibrium with the active complex (56). This model was used to fit the data for the burst plots from incorporation experiments with all of the N2-guanyl adducts (Fig. 8), with only the rate constants k4, k8, and k–8 allowed to vary. (The “trap” experiment results with N2-AnthG presented in Fig. 6 argue that the systems are operating under single-turnover conditions and that the course of each of these sets of assays occurs without dissociation of DNA from Dpo4.) The fits in Fig. 8 were good and support the model with the inactive complex. The partial-burst results cannot be accommodated with a minimal mechanism in which there is no alternate complex (56). In the model, the inactive complex is not necessarily a single one, e.g. the structure described in Fig. 7 (E and F) but could be a set of inactive complexes. The model shows an increased partitioning of an initial ternary Dpo4·DNA·dNTP complex into the inactive form, as reflected in the ratio k8/k4 (Table 6). Although we have discussed the Npg-2 structure (Fig. 7) in the context of being inactive, we cannot rule out the possibility that limited pairing with C has some productive nature and occurs at a slow rate. However, we do not believe that this is a dominant complex that explains the preferential incorporation of dCTP (Table 3).
Exactly why the catalytic efficiency with N2-AnthG is higher than with N2-NaphG is unclear. The trend is more obvious with the pre-steady-state results than those for the steady state (Fig. 10). We attempted to crystallize N2-AnthG complexes but have not been unsuccessful. The structures of the N2-NaphG complexes are suggestive that adding an even larger adduct to the N2 atom might drive the conformational equilibrium toward the syn orientation, simply because the added bulk cannot be accommodated easily in the Dpo4 active site when in the anti position. Such an occurrence was certainly true of the N2-BPDE-G structures in which the BPDE moiety was either in the syn configuration or flipped out of the active site completely (45). The efficiency of incorporation opposite N2-AnthG is still diminished relative to the smaller adducts, but “stabilizing” the syn conformation could allow for greater efficiency during nucleotide insertion (with an additional effect being downstream perturbation of the DNA helix). Alternatively, N2-AnthG could be accommodated in a manner similar to N2-BPDE-dG by moving into the gap between domains.
FIGURE 10.

Changes in Dpo4 catalytic properties as a function of bulk of N2-alkyl G. A, kcat/Km values (Table 3) for dCTP (▪), dATP (•), dGTP (▴), and dTTP (□) incorporation opposite various N2-G adducts by Dpo4 as a function of molecular volume of N2-alkyl G adducts. The volumes of the adducts were calculated according to Connolly (81). B, kcat/Km values (Table 4) for next-base extension (with dGTP) from matched G (or N2-alkyl G adduct):C template·primer termini (▪) or mismatched G (or N2-alkyl G adducts):T template·primer termini (•) by Dpo4. C, kpol/Kd values (Fig. 5) for dCTP incorporation by Dpo4 as a function of molecular volume of N2-alkyl G adducts.
In the course of this work we examined the “thio” effects for incorporation of dCTP opposite the N2-alkyl G derivatives (Fig. 3). Little effect was seen with G, N2-MeG, and N2-EtG, but major attenuations of the burst phases were seen with N2-IbG, N2-BzG, and N2-NaphG. A simplistic view of the results is that the phosphodiester bond-formation step is not rate-limiting in the case of G and the two shorter alkyl derivatives (whereas step 3 of Fig. 8A is rate-limiting), and the bond formation step (step 4, Fig. 8A) occurs in large derivatives. The modeling provides some support for this view (Table 6). However, the thio effect was not seen with N2-AnthG, which does have a faster kpol value (Fig. 3). However, the modeling does not dictate that step 4 (of Fig. 8A) is rate-limiting. The difficulties in interpretation of the thio effect experiments have been discussed elsewhere (82), and the work in Fig. 3 should not be interpreted in the context of a simple competition between the rate constants k3 and k4 (Fig. 8A).
In conclusion, we have shown that the catalytic efficiency of the Dpo4 is largely preserved with increasing adduct size, using a homologous series of N2-guanyl derivatives (Figs. 1 and 10). The results have general similarity to those described for some mammalian Y-family DNA polymerases (27–29, 48), although in this case burst kinetics persists throughout the entire series. Also, the fidelity of incorporation was highly preserved through the series. The structures of two bulky adduct complexes were determined (N2-NaphG, Fig. 7), the first of which we believe may represent an active complex (Npg-1). A second complex (Npg-2) is not predicted to be active, but we cannot rule out the possibility that some alternate form of the Npg-2 complex allows Watson-Crick pairing between dCTP and N2-NaphG. The kinetic courses of dCTP incorporation opposite all of the N2-guanyl adducts could be described using a minimal model with an inactive ternary complex in equilibrium with a catalytically competent form, although the caveat must be included that this inactive complex may be more than one and the mathematical representation may be more complex. These structure-function relationships add to our general knowledge of how Y-family DNA polymerases use their structural features to process a large variety of DNA lesions.
Supplementary Material
Acknowledgments
We thank K. C. Angel for technical assistance and K. Trisler for assistance in preparation of the manuscript. We are grateful to Dr. Z. Wawrzak, Northwestern University, for assistance with x-ray diffraction data collection. Vanderbilt University is a member institution of the Life Sciences Collaborative Access Team at sector 21 of the Advanced Photon Source (APS), Argonne National Laboratory, Argonne, IL. Additional x-ray diffraction data were collected on the 22-ID beamline of the Southeast Regional Collaborative Access Team (SER-CAT) at APS. SER-CAT supporting institutions may be found on-line.
This work was supported, in whole or in part, by National Institutes of Health Grants R01 ES010375 (to F. P. G.), F32 CA119776 (to R. L. E.), P01 ES05355 (to I. D. K., C. J. R., and M. E.), and P30 ES000267 (to F. P. G., C. J. R., and M. E.). Use of the Advanced Photon Source was supported by the U.S. Department of Energy, Basic Energy Sciences, Office of Science, under Contract W-31-109-Eng-38. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The atomic coordinates and structure factors (codes 2W8K and 2W8L) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S6 and Tables S1–S5.
Footnotes
The abbreviations used are: BPDE, benzo[a]pyrene diol epoxide; Anth, (9-anthracenyl)methyl; Bz, benzyl; CID, collision-induced dissociation; αCTPαS, 2′-deoxycytidine 5′-O-(1-thiotriphosphate); αTTPαS, 2′-deoxythymidine 5′-O-(1-thiotriphosphate); Dpo4, DNA polymerase IV; ESI, electrospray ionization; Et, ethyl; Ib, isobutyl; LC, liquid chromatography; Me, methyl; MS, mass spectrometry; MS/MS, tandem mass spectrometry; Naph, (2-naphthyl) methyl; 8-oxoG, 7,8-dihydro-8-oxo-dexoyguanosine; pol, (DNA) polymerase; r.m.s.d., root mean square deviation; HIV, human immunodeficiency virus. For simplicity the N2-guanyl adducts are usually collectively referred to as “alkyl,” although some are aralkyl.
C. Cambillau and A. Roussel (1997) Turbo Frodo, Version Open GL.1, Université Aix-Marseille II, Marseille, France.
W. L. DeLano (2002) The PyMOL Molecular Graphics System, DeLano Scientific LLC, San Carlos, CA.
References
- 1.Kornberg, A., and Baker, T. A. (1992) DNA Replication, 2nd ed., W. H. Freeman, New York
- 2.Searle, C. E. (1984) Chemical Carcinogens, 2nd Ed., Am. Chem. Soc., Washington, DC
- 3.Friedberg, E. C., Walker, G. C., Siede, W., Wood, R. D., Schultz, R. A., and Ellenberger, T. (2006) DNA Repair and Mutagenesis, 2nd Ed., ASM Press, Washington, DC
- 4.Yasui, M., Matsui, S., Ihara, M., Laxmi, Y. R., Shibutani, S., and Matsuda, T. (2001) Nucleic Acids Res. 29 1994–2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Terashima, I., Matsuda, T., Fang, T.-W., Suzuki, N., Kobayashi, J., Kohda, K., and Shibutani, S. (2001) Biochemistry 40 4106–4114 [DOI] [PubMed] [Google Scholar]
- 6.Liu, X., Lao, Y., Yang, I. Y., Hecht, S. S., and Moriya, M. (2006) Biochemistry 45 12898–12905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lao, Y., and Hecht, S. S. (2005) Chem. Res. Toxicol. 18 711–721 [DOI] [PubMed] [Google Scholar]
- 8.Zhang, S., Villalta, P. W., Wang, M., and Hecht, S. S. (2006) Chem. Res. Toxicol. 19 1386–1392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Forgacs, E., Latham, G., Beard, W. A., Prasad, R., Bebenek, K., Kunkel, T. A., Wilson, S. H., and Lloyd, R. S. (1997) J. Biol. Chem. 272 8525–8530 [DOI] [PubMed] [Google Scholar]
- 10.Wang, M., Lao, Y., Cheng, G., Shi, Y., Villalta, P. W., and Hecht, S. S. (2007) Chem. Res. Toxicol. 20 625–633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Turesky, R. J., Rossi, S. C., Welti, D. H., Lay, J. O., Jr., and Kadlubar, F. F. (1992) Chem. Res. Toxicol. 5 479–490 [DOI] [PubMed] [Google Scholar]
- 12.Meehan, T., and Straub, K. (1979) Nature 277 410–412 [DOI] [PubMed] [Google Scholar]
- 13.Cheng, S. C., Hilton, B. D., Roman, J. M., and Dipple, A. (1989) Chem. Res. Toxicol. 2 334–340 [DOI] [PubMed] [Google Scholar]
- 14.Fang, J. L., and Vaca, C. E. (1997) Carcinogenesis 18 627–632 [DOI] [PubMed] [Google Scholar]
- 15.Matsuda, T., Terashima, I., Matsumoto, Y., Yabushita, H., Matsui, S., and Shibutani, S. (1999) Biochemistry 38 929–935 [DOI] [PubMed] [Google Scholar]
- 16.Cheng, T. F., Hu, X., Gnatt, A., and Brooks, P. J. (2008) J. Biol. Chem. 283 27820–27828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang, M., and Hecht, S. S. (1997) Chem. Res. Toxicol. 10 772–778 [DOI] [PubMed] [Google Scholar]
- 18.Lao, Y., Yu, N., Kassie, F., Villalta, P. W., and Hecht, S. S. (2007) Chem. Res. Toxicol. 20 246–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Goodman, M. F. (1997) Proc. Natl. Acad. Sci. U. S. A. 94 10493–10495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guengerich, F. P. (2006) Chem. Rev. 106 420–452 [DOI] [PubMed] [Google Scholar]
- 21.Friedberg, E. C., Wagner, R., and Radman, M. (2002) Science 296 1627–1630 [DOI] [PubMed] [Google Scholar]
- 22.Goodman, M. F. (2002) Annu. Rev. Biochem. 71 17–50 [DOI] [PubMed] [Google Scholar]
- 23.Prakash, S., Johnson, R. E., and Prakash, L. (2005) Annu. Rev. Biochem. 74 317–353 [DOI] [PubMed] [Google Scholar]
- 24.Choi, Y.-J., and Guengerich, F. P. (2004) J. Biol. Chem. 279 19217–19229 [DOI] [PubMed] [Google Scholar]
- 25.Zang, H., Harris, T. M., and Guengerich, F. P. (2005) Chem. Res. Toxicol. 18 389–400 [DOI] [PubMed] [Google Scholar]
- 26.Zang, H., Harris, T. M., and Guengerich, F. P. (2005) J. Biol. Chem. 280 1165–1178 [DOI] [PubMed] [Google Scholar]
- 27.Choi, J.-Y., and Guengerich, F. P. (2005) J. Mol. Biol. 352 72–90 [DOI] [PubMed] [Google Scholar]
- 28.Choi, J.-Y., and Guengerich, F. P. (2006) J. Biol. Chem. 281 12315–12324 [DOI] [PubMed] [Google Scholar]
- 29.Choi, J.-Y., and Guengerich, F. P. (2008) J. Biol. Chem. 283 23645–23655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Boudsocq, F., Iwai, S., Hanaoka, F., and Woodgate, R. (2001) Nucleic Acids Res. 29 4607–4616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kokoska, R. J., McCulloch, S. D., and Kunkel, T. A. (2003) J. Biol. Chem. 278 50537–50545 [DOI] [PubMed] [Google Scholar]
- 32.Fiala, K. A., and Suo, Z. (2004) Biochemistry 43 2106–2115 [DOI] [PubMed] [Google Scholar]
- 33.Fiala, K. A., and Suo, Z. (2004) Biochemistry 43 2116–2125 [DOI] [PubMed] [Google Scholar]
- 34.Vaisman, A., Ling, H., Woodgate, R., and Yang, W. (2005) EMBO J. 24 2957–2967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Eoff, R. L., Egli, M., and Guengerich, F. P. (2009) in The Chemical Biology of DNA Damage (Geacintov, N. E., and Broyde, S., eds) Wiley-VCH, Weinheim, Germany, in press
- 36.Ling, H., Boudsocq, F., Woodgate, R., and Yang, W. (2001) Cell 107 91–102 [DOI] [PubMed] [Google Scholar]
- 37.Trincao, J., Johnson, R. E., Wolfle, W. T., Escalante, C. R., Prakash, S., Prakash, L., and Aggarwal, A. K. (2004) Nat. Struct. Mol. Biol. 11 457–462 [DOI] [PubMed] [Google Scholar]
- 38.Ling, H., Boudsocq, F., Woodgate, R., and Yang, W. (2004) Mol. Cell 13 751–762 [DOI] [PubMed] [Google Scholar]
- 39.Rechkoblit, O., Malinina, L., Cheng, Y., Kuryavyi, V., Broyde, S., Geacintov, N. E., and Patel, D. J. (2006) PLoS Biol. 4 e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zang, H., Irminia, A., Choi, J.-Y., Angel, K. C., Loukachevitch, L. V., Egli, M., and Guengerich, F. P. (2006) J. Biol. Chem. 281 2358–2372 [DOI] [PubMed] [Google Scholar]
- 41.Eoff, R. L., Irimia, A., Egli, M., and Guengerich, F. P. (2007) J. Biol. Chem. 282 1456–1467 [DOI] [PubMed] [Google Scholar]
- 42.Eoff, R. L., Angel, K. C., Egli, M., and Guengerich, F. P. (2007) J. Biol. Chem. 282 13573–13584 [DOI] [PubMed] [Google Scholar]
- 43.Ling, H., Boudsocq, F., Plosky, B. S., Woodgate, R., and Yang, W. (2003) Nature 424 1083–1087 [DOI] [PubMed] [Google Scholar]
- 44.Ling, H., Sayer, J. M., Plosky, B. S., Yagi, H., Boudsocq, F., Woodgate, R., Jerina, D. M., and Yang, W. (2004) Proc. Natl. Acad. Sci. U. S. A. 101 2265–2269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bauer, J., Xing, G., Yagi, H., Sayer, J. M., Jerina, D. M., and Ling, H. (2007) Proc. Natl. Acad. Sci. U. S. A. 104 14905–14910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zang, H., Goodenough, A. K., Choi, J.-Y., Irminia, A., Loukachevitch, L. V., Kozekov, I. D., Angel, K. C., Rizzo, C. J., Egli, M., and Guengerich, F. P. (2005) J. Biol. Chem. 280 29750–29764 [DOI] [PubMed] [Google Scholar]
- 47.Eoff, R. L., Irimia, A., Angel, K., Egli, M., and Guengerich, F. P. (2007) J. Biol. Chem. 282 19831–19843 [DOI] [PubMed] [Google Scholar]
- 48.Choi, J.-Y., Angel, K. C., and Guengerich, F. P. (2006) J. Biol. Chem. 281 21062–21072 [DOI] [PubMed] [Google Scholar]
- 49.Borer, P. N. (1975) in Handbook of Biochemistry and Molecular Biology, 3rd Ed. (Fasman, G. D., ed) pp. 589–590, CRC Press, Cleveland, OH
- 50.Goodman, M. F., Creighton, S., Bloom, L. B., and Petruska, J. (1993) Crit. Rev. Biochem. Mol. Biol. 28 83–126 [DOI] [PubMed] [Google Scholar]
- 51.Patel, S. S., Wong, I., and Johnson, K. A. (1991) Biochemistry 30 511–525 [DOI] [PubMed] [Google Scholar]
- 52.Johnson, K. A. (1995) Methods Enzymol. 249 38–61 [DOI] [PubMed] [Google Scholar]
- 53.Choi, J.-Y., Chowdhury, G., Zang, H., Angel, K. C., Vu, C. C., Peterson, L. A., and Guengerich, F. P. (2006) J. Biol. Chem. 281 38244–38256 [DOI] [PubMed] [Google Scholar]
- 54.Mizrahi, V., Henrie, R. N., Marlier, J. F., Johnson, K. A., and Benkovic, S. J. (1985) Biochemistry 24 4010–4018 [DOI] [PubMed] [Google Scholar]
- 55.Kuchta, R. D., Mizrahi, V., Benkovic, P. A., Johnson, K. A., and Benkovic, S. J. (1987) Biochemistry 26 8410–8417 [DOI] [PubMed] [Google Scholar]
- 56.Furge, L. L., and Guengerich, F. P. (1999) Biochemistry 38 4818–4825 [DOI] [PubMed] [Google Scholar]
- 57.Woodside, A. M., and Guengerich, F. P. (2002) Biochemistry 41 1039–1050 [DOI] [PubMed] [Google Scholar]
- 58.Kuzmic, P. (1996) Anal. Biochem. 237 260–273 [DOI] [PubMed] [Google Scholar]
- 59.Kabsch, W. (1988) J. Appl. Crystallogr. 21 916–924 [Google Scholar]
- 60.Otwinowski, Z., and Minor, W. (1997) Methods Enzymol. 276 307–326 [DOI] [PubMed] [Google Scholar]
- 61.Vagin, A., and Teplyakov, A. (1997) J. Appl. Crystallogr. 30 1022–1025 [Google Scholar]
- 62.Vellieux, F. M. D., and Dijkstra, B. W. (1997) J. Appl. Crystallogr. 30 396–399 [Google Scholar]
- 63.Brunger, A. T., Adams, P. D., Clore, G. M., DeLano, W. L., Gros, P., Grosse-Kunstleve, R. W., Jiang, J. S., Kuszewski, J., Nilges, M., Pannu, N. S., Read, R. J., Rice, L. M., Simonson, T., and Warren, G. L. (1998) Acta Crystallogr. Sect. D Biol. Crystallogr. 54 905–921 [DOI] [PubMed] [Google Scholar]
- 64.Lavery, R., and Sklenar, H. (1989) J. Biomol. Struct. Dynam. 6 655–667 [DOI] [PubMed] [Google Scholar]
- 65.Mendelman, L. V., Petruska, J., and Goodman, M. F. (1990) J. Biol. Chem. 265 2338–2346 [PubMed] [Google Scholar]
- 66.Beckman, J. W., Wang, Q., and Guengerich, F. P. (2008) J. Biol. Chem. 283 36711–36723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ni, J., Pomerantz, S. C., Rozenski, J., Zhang, Y., and McCloskey, J. A. (1996) Anal. Chem. 68 1989–1999 [DOI] [PubMed] [Google Scholar]
- 68.Wong, I., Patel, S. S., and Johnson, K. A. (1991) Biochemistry 30 526–537 [DOI] [PubMed] [Google Scholar]
- 69.Tan, H. B., Swann, P. F., and Chance, E. M. (1994) Biochemistry 33 5335–5346 [DOI] [PubMed] [Google Scholar]
- 70.Wöhrl, B. M., Krebs, R., Goody, R. S., and Restle, T. (1999) J. Mol. Biol. 292 333–344 [DOI] [PubMed] [Google Scholar]
- 71.Tsai, Y. C., and Johnson, K. A. (2006) Biochemistry 45 9675–9687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lee, H. R., and Johnson, K. A. (2007) J. Biol. Chem. 282 31982–31989 [DOI] [PubMed] [Google Scholar]
- 73.Nair, D. T., Johnson, R. E., Prakash, S., Prakash, L., and Aggarwal, A. K. (2004) Nature 430 377–380 [DOI] [PubMed] [Google Scholar]
- 74.Nair, D. T., Johnson, R. E., Prakash, L., Prakash, S., and Aggarwal, A. K. (2005) Structure (Cambridge) 13 1569–1577 [DOI] [PubMed] [Google Scholar]
- 75.Tissier, A., McDonald, J. P., Frank, E. G., and Woodgate, R. (2000) Genes Dev. 14 1642–1650 [PMC free article] [PubMed] [Google Scholar]
- 76.Perlow-Poehnelt, R. A., Likhterov, I., Scicchitano, D. A., Geacintov, N. E., and Broyde, S. (2004) J. Biol. Chem. 279 36951–36961 [DOI] [PubMed] [Google Scholar]
- 77.Lone, S., Twonson, S. A., Uljon, S. N., Johnson, R. E., Brahma, A., Nair, D. T., Prakash, S., Prakash, L., and Aggarwal, A. K. (2007) Mol. Cell 25 601–614 [DOI] [PubMed] [Google Scholar]
- 78.Jia, L., Geacintov, N. E., and Broyde, S. (2008) Nucleic Acids Res. 36 6571–6584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Minko, I. G., Harbut, M. B., Kozekov, I. D., Kozekova, A., Jakobs, P. M., Olson, S. B., Moses, R. E., Harris, T. M., Rizzo, C. J., and Lloyd, R. S. (2008) J. Biol. Chem. 283 17075–17082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lemee, F., Bavoux, C., Pillaire, M. J., Bieth, A., Machado, C. R., Pena, S. D., Guimbaud, R., Selves, J., Hoffmann, J. S., and Cazaux, C. (2007) Oncogene 26 3387–3394 [DOI] [PubMed] [Google Scholar]
- 81.Connolly, M. L. (1993) J. Mol. Graph. 11 139–141 [DOI] [PubMed] [Google Scholar]
- 82.Herschlag, D., Piccirilli, J. A., and Cech, T. R. (1991) Biochemistry 30 4844–4854 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.