

Abstract
BACKGROUND
Arrhythmogenic right ventricular cardiomyopathy (ARVC), has been linked to mutations in desmosomal proteins, including plakophilin2 (PKP2). Little is known about the changes in cellular function and structure that follow expression of ARVC-relevant PKP2 mutations.
OBJECTIVE
Here, we investigated the function and distribution of an ARVC-relevant PKP2 mutant where arginine at position 79 was replaced by a stop codon (R79x).
METHODS
Results were compared to those obtained with mutation 179fs (frameshift at position 179). Mutant constructs were introduced by adenoviral infection into neonatal rat ventricular myocytes in culture.
RESULTS
Both mutant proteins failed to preferentially localize to sites of cell-cell apposition. Their expression did not disrupt localization of endogenous PKP2, connexin43 (Cx43) or desmoplakin (DP). However, we observed reduced abundance of Cx43 following R79x expression. Early truncation of PKP2 at position 79 also prevented its physical interaction with both DP and Cx43. Finally, R79x expression correlated with loss of expression of HSP90, a protein relevant to cardiomyocyte apoptosis.
CONCLUSION
These results provide the first observations of the cellular/molecular phenotype consequent to these PKP2 mutations and give insight into the possible cellular substrates that lead to ARVC.
Keywords: Connexin43, Plakophilin-2, Arrhythmogenic Right Ventricular Cardiomyopathy, ARVC
INTRODUCTION
Arrhythmogenic right ventricular cardiomyopathy (ARVC) is an inherited disease characterized by fibrofatty infiltration of right ventricular predominance, a high incidence of arrhythmias and sudden death.1,2,3 ARVC has been linked to mutations in desmosomal proteins, including plakophilin2 (PKP24,5,6). Little is known about the cellular/molecular changes that follow expression of ARVC-relevant PKP2 mutations.
In the vast majority of cases of PKP2-related ARVC, mutations are found in only one allele. As such, two conditions are present: decreased gene dose for the wild-type protein, and expression of a mutant form. Recently, we demonstrated that loss of PKP2 expression leads to remodeling of connexin43 (Cx437). The latter is consistent with results obtained in the study of Naxos disease8 and Carvajal syndrome9. Here we explored, for the first time, the structural/functional consequences of expression of two ARVC-related PKP2 mutations in neonatal rat ventricular myocytes (NRVMs).
Over 50 ARVC-related PKP2 mutations have been reported. We focused on two N-terminal mutations, R79x (stop-codon replacing arginine 794,5,6) and 179fs (a frameshift in that position6). It has been suggested that the plakophilin-desmoplakin interaction involves the amino termini of both proteins. Thus, R79x and 179fs allowed us to explore the minimal PKP2 sequence necessary for DP binding. We extended this question to the previously-reported PKP2-Cx43 interaction.7 Mutation R79x has been detected in a number of independent probands,4,5,6 suggesting important functional effects upon expression of the mutated protein. Our data support the notion that mutant R79x plays an active role in determining the ARVC phenotype, and provide the first characterization of cellular phenotype related to ARVC-relevant PKP2 mutations.
Materials and Methods
Generation of cDNA constructs
Full length (“wildtype”) human PKP2a (p915 PKP2a-pFLAG-cmv5a) and DP (p1140 DP.FLAG) C-terminal FLAG constructs were gifts from Dr. Kathleen Green (Northwestern University). Construct p915 served as template to generate PKP2a mutants. Mutation 179fs was generated by inserting bases C and T at nucleotides 534-535, respectively.6 A C235T substitution produced mutant R79x. A FLAG® tag was placed at the C terminus of both mutants. To generate adenoviruses, each p915 construct was used as template and cloned into pShuttle-IRES-hrGFP-1 (Stratagene). Expression of hrGFP was prevented by site-directed mutagenesis. Adenoviral recombinants were generated as per the manufacturer’s instructions (Stratagene AdEasy XL).
The GST-PKP2H fusion protein was produced by cloning the first 335 amino acids of human PKP2, C-terminal to GST in pGEX-TK (Amersham). This construct was mutagenized to obtain R79x and 179fs mutations. HA-tagged DPNT was generated by PCR amplification of nucleotides coding for amino acids 1-584 using p1140 DP-FLAG as template (construct DPNT-HA).
Primary culture of NRVMs and adenoviral infection
Dissociation and culture of NRVMs has been described.7 Myocytes were plated to 70-80% confluency in either 60 mm dishes (Western blots), or coverslips in a 12-well dish (immunofluorescence). Cells were infected with adenoviruses containing FLAG-tagged wtPKP2, R79x or 179fs. Five days post-infection, cells were processed for either Western blot or immunofluorescence.
Antibodies used
Antibody against FLAG (Oct-A) and desmoplakin I/II H-300 (sc-33555) were purchased from Santa Cruz Biotechnology, Inc. Cx43 was detected either with Rabbit anti-Cx43 (AB1728, Chemicon), monoclonal anti-Cx43 C-terminus, or Cx43 N-terminus (CX43CT or Cx43NT1, Fred Hutchison Cancer Research Center, FHCRC). Antibody against ß-actin (A5441) was purchased from Sigma. HA was detected with antibody PRB-101P (Covance); PKP2 and HSP90 monoclonal antibodies were purchased from Biodesign; BD Transduction Laboratories. The anti-PKP2 antibody targeted the C-terminus of the protein, thus avoiding detection of exogenous R79x or 179fs. This antibody recognized FLAG-tagged PKP2, though with a much weaker signal, probably because of differences in epitope availability/specificity. A polyclonal antibody (AHP320; Serotec) was used for detection of desmoplakin.
Immunochemistry
Western Blot and immunolocalization experiments (including nuclear localization with Hoechst dye; blue staining) followed published procedures.7 Co-immunoprecipitation and immunoblot of FLAG-tagged proteins followed manufacturer’s instructions. NRVMs were plated in six-well dishes to 60-70% confluency, infected with adenovirus containing FLAG-tagged wt PKP2, R79x or 179fs as described, and lysed five days post infection.
GST pull-down assays
Pull-down assays were performed as described.7 Cx43 was pulled-down from adult rat heart lysate. DPNT-HA was pulled-down from transfected HEK293 cells.
Triton solubility assay
NRVMs (70-80% confluency; 6-well dishes) were infected with adenoviruses containing wtPKP2, R79x or 179fs. Five days post-infection, cells were briefly rinsed in ice cold phosphate buffer saline (pH 7.4) and harvested by scraping. Cells were pelleted by centrifugation (5 minutes at 14,000 rpm) and pellets resuspended in extraction buffer (50mM Tris -pH 8.0-, 150mM NaCl, 0.02% sodium Azide, 1% Triton X-100, 1mM PMSF, 1μg/ml Aprotinin, and Complete® protease inhibitor –Roche-). After a 30-minute incubation on ice, an aliquot was saved to determine total protein; the remaining lysate was centrifuged (14,000 rpm; 10 minutes; 4°C). Pellet and supernatant fractions are referred to as “triton-insoluble” and “triton-soluble,” respectively. All samples were solubilized in SSB prior to immunoblotting. Detection of FLAG, DP or Cx43 followed the procedures described above.
Dye transfer in NRVMs
Dye transfer through gap junctions was assessed in NRVM cell pairs using the method described previously.7
Results
Expression of exogenous constructs
We used immunochemical techniques to assess expression and distribution of exogenous, C-terminus FLAG-tagged proteins R79x-FLAG, 179fs-FLAG, and wtPKP2-FLAG, in NRVMs. Genetic material was transferred by adenoviral infection (Methods). Concentration of viral particles was adjusted for each case (12.5, 50 and 12.5 MOI for wtPKP2, R79x and 179fs, respectively) to ensure maximum expression of the exogenous product while limiting cell damage due to the infection procedure. Figure 1A shows a Western blot for FLAG-immunoreactive proteins in cells either untreated (lane labeled “Control”), or expressing PKP2-FLAG, R79x-FLAG, or 179fs-FLAG (five days post-infection). Data show that all constructs expressed a protein of the expected molecular weight. Yet, we consistently observed that the amount of R79x protein detected was significantly less than that of either wtPKP2 or 179fs. (Note that cell lysates from cells expressing R79x and 179fs were 25 and 5 times more concentrated, respectively, than that of wtPKP2.)
Figure 1.
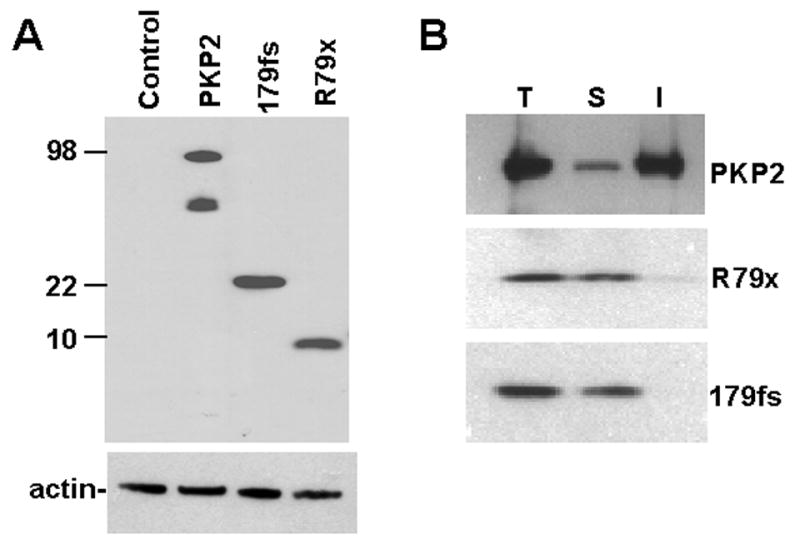
Panel A: Western blot of FLAG-tagged PKP2 proteins expressed in neonatal rat ventricular myocytes. Sample in lane labeled “R79x” was 25X and 5X more concentrated than those in lanes labeled “PKP2” and “179fs,” respectively. Actin was used as control for endogenous protein. In this case, samples were kept at an even concentration, thus showing that total protein content was the same for all samples. Notice a secondary band in the PKP2 lane, perhaps a degradation product of PKP2-FLAG consequent to overexpression of the exogenous gene.
Panel B: Western blot of FLAG-tagged PKP2 proteins before and after segregation of Triton-soluble and insoluble fractions. T, S and I: total, soluble and insoluble fractions.
As a first approach to assess the subcellular distribution of these mutants, a triton solubility experiment was conducted. It is generally accepted that cytoplasmic proteins become soluble in the presence of small triton concentrations, whereas membrane-associated proteins, or large macromolecular complexes, remain insoluble. Figure 1B shows a Western blot probed with an anti-FLAG antibody. NRVMs were expressing exogenous wtPKP2, R79x or 179fs. wtPKP2 was primarily triton-insoluble, in accordance with previous studies.11 In contrast, R79x and 179fs were found mostly in the triton-soluble fraction, suggesting a significant redistribution of these proteins into the intracellular space, away from junctional complexes.
To further study the subcellular localization of these proteins, immunolocalization experiments were conducted. Figure 2A shows the localization of endogenous PKP2 in untreated cells. Panel B displays a picture obtained from NRVMs infected with wtPKP2-FLAG. Examples of cells expressing R79x-FLAG and 179fs-FLAG are shown in panels C and D, respectively. The full-length FLAG-tagged protein localized primarily to sites of cell-cell apposition, in a manner similar to that of the endogenous PKP2 (see also refs7,10). In contrast, truncated proteins were abundantly found in the intracellular space. Overall, our data indicate that the very N-terminal end of PKP2 lacks the necessary sequences for proper membrane localization. These results led us to question whether expression of either R79x or 179fs would cause redistribution of endogenous PKP2, or of the PKP2 partner molecules DP and Cx43.
Figure 2. Immunolocalization of PKP2 in NRVMs.
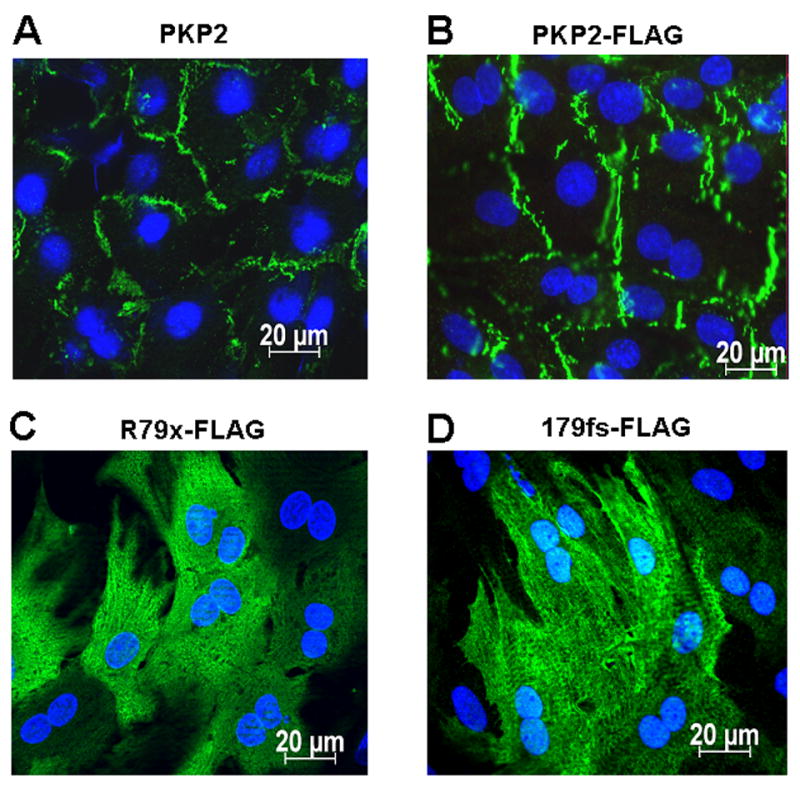
Panel A: Localization of endogenous PKP2. Panels B, C, D: Immunolocalization of exogenously-expressed FLAG-tagged wtPKP2, R79x and 179fs, respectively, using an antibody to the FLAG epitope. Notice intracellular localization of R79x and 179fs.
Immunochemical analysis of endogenous PKP2, DP and Cx43 in NRVMs expressing wtPKP2-FLAG, R79x-FLAG or 179fs-FLAG
Figure 3 shows immunolocalization of endogenous PKP2 (green) and the exogenous FLAG-tagged proteins (red), in cells expressing either wtPKP2-FLAG (panels A-C), R79x-FLAG (panels D-F) or 179fs-FLAG (panels G-I). The images show that, while unable to properly localize to sites of cell apposition, the mutants did not cause an apparent redistribution of endogenous PKP2. Similarly, the apparent localization of endogenous DP (Figure 4) or Cx43 (Figure 5) was not different in cells expressing mutant constructs (panels D-F and G-I) when compared to cells expressing the exogenous wild-type protein (panels A-C). Yet, we did observe an apparent reduction in the amount of Cx43-immunoreactive signal obtained from cells expressing the R79x construct when compared to those expressing wtPKP2 or 179fs (compare figure 5D-F with 5A-C, or G-I).
Figure 3. Immunolocalization of endogenous plakophilin2 (PKP2) following expression of FLAG-tagged PKP2 proteins.

Panels A, D, G: Detection of exogenous, FLAG -tagged wtPKP2, R79x and 179fs, respectively, using an anti-FLAG antibody. Panels B, E and H,: endogenous PKP2. Panels C, F and I, merged images.
Figure 4. Immunolocalization of endogenous desmoplakin (DP) following expression of FLAG-tagged PKP2 proteins.
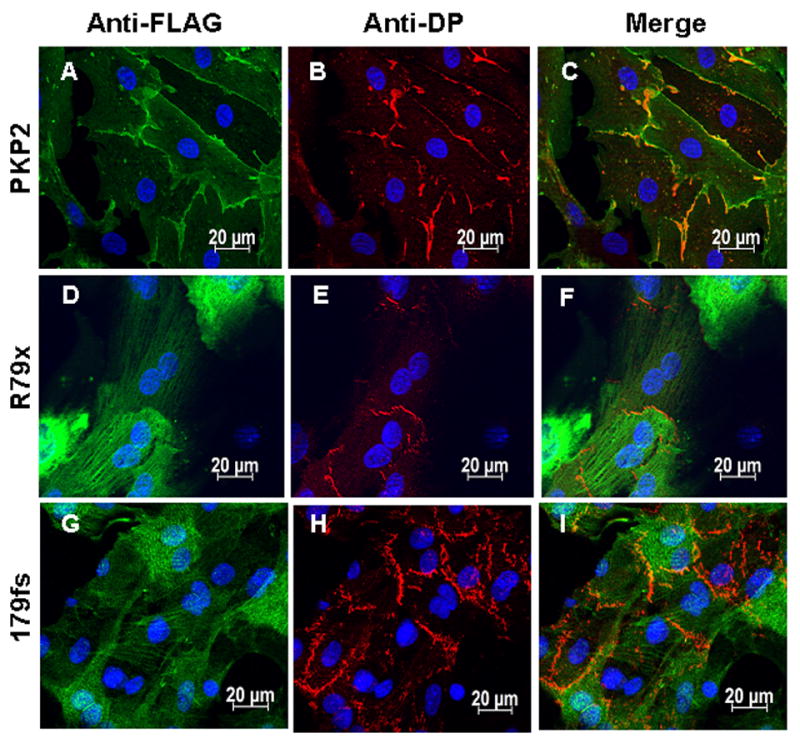
Panels A-C: Immunodetection of FLAG-tagged wtPKP2, endogenous DP and merged image, respectively. Panels D-F and G-I follow the same order, but the FLAG-tagged protein was either R79x (D and F) or 179fs (G and I).
Figure 5. Immunolocalization of endogenous Cx43 following expression of FLAG-tagged PKP2 proteins.

Panels A-C: Immunodetection of wtPKP2-FLAG, endogenous Cx43 and merged image, respectively. Panels D-F and G-I follow the same order, but the FLAG-tagged protein was either R79x (D and F) or 179fs (G and I).
Epitope masking, and/or the absence of gap junction plaques, can lead to apparent decreases in Cx43 immunofluorescent signals without changes in protein content. Here, we used a triton solubility assay to explore the effect of PKP2 mutants on Cx43 expression and distribution. A similar assay explored the abundance and distribution of DP. The blot presented in Figure 6A was probed with an anti-DP antibody, whereas the blot in panel B was tested for Cx43-immunoreactive proteins. As expected, DP was largely triton-insoluble.11 Expression of either wtPKP2 or the mutants did not modify DP segregation into the triton-insoluble fraction, suggesting that the truncated PKP2 proteins do not cause significant remodeling of DP. However, we did observe a reduction in total Cx43 in cells expressing R79x (see Figure 6B). The latter occurred at the expense of the low-mobility Cx43 band (often associated with phosphorylated protein, segregating into the insoluble fraction12). Yet, expression of either 179fs or wtPKP2 did not modify Cx43 content or distribution into the subcellular fractions.
Figure 6.

Panel A: Triton X-dependent segregation of DP in NRVM’s expressing PKP2 mutants R79x or 179fs. Fractions: T (total), I (insoluble) or S (soluble). Panel B: Similar experiment; blot probed for Cx43. In average of three experiments, ratio of low/high mobility band density was 0.96, 0.50 and 0.96 for samples obtained from T, S or I fractions of PKP2-FLAG-expressing cells. Numbers were similar for 179fs-expressing cells (0.97, 0.56 and 1.34) but lower in the T and I fractions of cells expressing R79x (0.51, 0.53 and 0.7). Cx43 band density in samples labeled “T” obtained from cells expressing R79x-FLAG was 37%, when compared to cells expressing wtPKP2-FLAG (band density adjusted for the corresponding actin control). ß-actin was used as loading control. Panel C: Dye transfer between NRVM cell pairs. Plot of averaged data (+/- SEM). Fluorescence intensity in each cell (relative to maximum in cell 1) was measured as a function of time after patch break. Graph shows rise in dye intensity in both control and R79X donor cells (■,●) and recipient cells (□,○) as a function of time.
Dye transfer in R79x-expressing cells
Dye transfer experiments allowed us to assess intercellular communication in R79x-expressing cell pairs. Figure 6C shows a plot of fluorescence intensity recorded from either the patched (“cell 1”) or the partner (“cell 2”) cell, as a function of time after patch break. Filled and open squares represent data from cells expressing wtPKP2-FLAG; filled and open circles indicate data from R79x-FLAG-expressing cells. Our results show that dye transfer was unaffected by R79x expression, despite the decrease in Cx43 protein observed by immunochemistry.
Co-immunoprecipitation and pull-down experiments
Previous studies have shown that both DP and Cx43 are found in a PKP2 immunoprecipitate.7,13 Here, co-immunoprecipitation experiments determined whether the association of PKP2 with either DP13 or Cx437 was disrupted by the mutations. NRVMs expressing either wtPKP2-FLAG, R79x-FLAG or 179fs-FLAG were lysed, lysates exposed to agarose beads coated with anti-FLAG antibody, and complexes precipitated by centrifugation. A Western blot confirmed that FLAG-tagged proteins were precipitated by the anti-FLAG coated beads (Figure 7A). Separate Western blots assessed the presence of either DP (Figure 7B) or Cx43 (Figure 7C) in the precipitate. Spurious bands were observed in the immunoblots for DP or Cx43, likely consequent to non-specific reactivity of the secondary antibody with a component of the precipitate (see, e.g., bands noted by thick arrow on panel 7C). Most importantly, wtPKP2 and, to a lesser extent, 179fs retained the ability to associate with both DP and Cx43. Yet, neither DP nor Cx43 were present in the precipitate of cells expressing R79x. Notice that DP bands were more intensely defined than those for Cx43. The latter may be consequent to the fact that the DP-PKP2 interaction is likely direct, and of a higher affinity than the PKP2-Cx43 interaction.
Figure 7. PKP2a mutant R79x does not co-immunoprecipitate with DP or with Cx43.
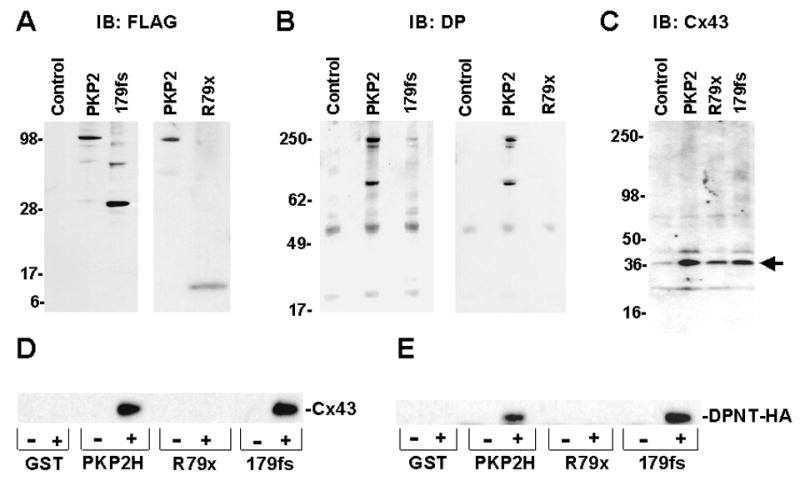
Constructs wtPKP2-FLAG, R79x-FLAG and 179fs-FLAG were used. Precipitates (using FLAG antibodies) were immunoblotted (IB) for FLAG (panel A), DP (panel B) or Cx43 (panel C). Lane labeled “control” refers to cells not infected with FLAG-tagged proteins. Thick arrow to right of panel C shows spurious band detected non-specifically by the secondary antibody. Panel D: GST-PKP2 was presented to rat heart lysate. Precipitate was probed for Cx43. Panel E: GST-PKP2 constructs were used to precipitate DPNT-HA from HEK cells. Both DPNT and Cx43 were able to interact with either PKP2H or 179fs, but not with R79x.
Failure of R79x-FLAG to co-precipitate DP, or Cx43 may be consequent to the low level of R79x-FLAG expression. To exclude this possibility, we conducted “pull-down” experiments where the bait proteins, i.e., the head domain of PKP2 (PKP2H), R79x or 179fs, were GST-fused, produced in bacteria, purified and bound to glutathione beads. Care was taken to ensure that the concentrations of GST-fused proteins were the same for all constructs. GST-fused proteins were exposed to adult rat heart lysates (see also7) and then probed for Cx43. The results were similar to those from co-precipitation experiments: Cx43 was recovered after exposure of the lysate to either PKP2H or 179fs; yet, this protein was not found associated with GST-R79x (See Figure 7D).
Attempts to pull-down full-length DP using GST-fusion proteins were ineffective, likely because of difficulty in extracting DP from the adult heart lysate. As an alternative, we tested the ability of recombinant GST-PKP2 constructs to pull down “DPNT-HA,” that is, a protein sequence containing the first 584 amino acids of DP13 concatenated to an HA epitope tag. DPNT-HA was expressed in HEK cells, and lysates presented to the GST-bait proteins. Results are shown in Figure 7E. The symbols “+” and “-” indicate whether GST fusion proteins were exposed (+), or not (-), to the HEK lysate. Clearly, an HA-immunoreactive protein bound to both PKP2H and 179fs, but not to R79x or to control beads (GST alone). Overall, the results show that the first 79 amino acids of PKP2 lack the necessary sequences for interaction with either DP or Cx43, whereas these interactions are present for mutant 179fs.
PKP2 mutant expression and cell function
Previous studies have suggested that ARVC-relevant mutations may induce apoptosis, thus participating in the phenotype of the disease.14,15,16 As an initial assessment of this hypothesis, we tested the presence or absence of the apoptosis-related protein, HSP90, in the lysate of NRVMs expressing either wtPKP2-FLAG or its mutants. As shown in Figure 8, there was a drastic loss of HSP90 expression in cells infected with R79x-FLAG, but not with the other constructs. The latter suggests that, though R79x lacks the ability to interact with two known molecular partners, it is not an innocuous mutation. Rather, it may actively modify the cellular and molecular phenotype of the cells in which it is expressed.
Figure 8. Western blot for HSP90 obtained from cells expressing FLAG-tagged PKP2, R79x or 179fs.

Cell lysates were separated by triton solubility as described in methods and immunoprobed for HSP90. Notice absence of immunodetectable HSP90 signal in samples collected from cells expressing R79x. ß-actin was used as loading control.
Discussion
A number of PKP2 mutations have been associated with ARVC. However, a correlate as to the effects of disease-associated mutations on cell/molecular function is still lacking. We have shown that loss of PKP2 expression leads to Cx43 remodeling and decreased intercellular coupling.7 Here, we show that mutants R79x and 179fs failed to localize to the cell membrane, but did not alter the localization of endogenous PKP2, DP or Cx43. The well-known interaction of PKP2 with DP was preserved for mutant 179fs, but not for the shorter, R79x construct. A similar result was observed when testing the PKP2-Cx43 interaction. Additional experiments demonstrated that R79x expression caused a reduction in the total amount of Cx43, at the expense of the low mobility (“phosphorylated”) band. Finally, expression of R79x brought about a reduction in expression of HSP90, a protein related to preservation of myocyte survival.17 Altogether, these results suggest that R79x, though present in amounts significantly lower than wild type PKP2, actively affects myocyte function, in a manner consistent with the loss of myocardial integrity characteristic of ARVC.
Expression levels were markedly different between constructs. As such, we considered whether to use different MOIs, so that protein levels were comparable (or at least detectable), or to match MOIs, at the expense of having markedly different (or undetectable) protein levels. We favored the former, since our study aimed at characterizing R79x expression on cellular/molecular phenotype. Even at higher MOIs, R79x level was lower than that observed when either control, or 179fs constructs were used. Whether the low R79x levels were consequent to low protein production, or an increase in degradation, remains to be determined. Yet, R79x expression caused drastic changes in the abundance of at least two proteins of fundamental importance to myocyte biology: Cx43 and HSP90. Our results suggest that the ARVC phenotype in these patients may be consequent not only to PKP2 haploinsufficiency, but to the presence of the R79x fragment. Yet, though our results reflect the phenotype of the mutant in an exogenous system, experiments beyond the present study would be required to address the actual levels of R79x protein in an ARVC afflicted heart.
Expression of R79x correlated with decreased abundance of Cx43. While the mechanism responsible for this effect remains unclear, we speculate that PKP2 integrity may relate to transcriptional control of relevant membrane proteins. Indeed, PKP2 is known to associate with beta-catenin,13 which in turn can regulate Cx43 expression,18,19 Moreover, small amounts of PKP2 can be normally detected in the nucleus. Early termination may free the N-terminal fragment from its interactions with desmosomal proteins and facilitate nuclear translocation, thus disrupting transcriptional control.
R79x expression caused a decrease in the amount of Cx43. Yet, despite this reduction, dye transfer was normal in R79x-containing cells. This is not particularly surprising, since Cx43 was still present at sites of cell apposition, and previous studies have shown that Cx43 amount does not necessarily correlate with functional coupling (e.g., conduction velocity in heterozygote Cx43-null mice is not different from that of control mice20). From that standpoint, expression of R79x may not be causative of poor electrical coupling. Yet, in afflicted patients, two factors converge: PKP2 haploinsufficiency, and R79x expression. Previous studies have shown that loss of PKP2 expression leads to Cx43 internalization and decrease in intercellular coupling.7 Haploinsufficiency and R79x expression may cause separate effects on Cx43 protein levels and distribution, adding (non-linearly) to disrupt gap junction-associated cellular functions, including electrical coupling and electrical stability between myocytes. Moreover, while dye transfer is an established method for assessing coupling, it provides only a coarse view of connexin function that does not assess small changes in channel properties or permselectivity. In addition, some connexin functions are not necessarily linked to expression of functional channels (e.g., control of cell proliferation21,22,23 or apoptosis24). Whether these connexin-related functions are modified by expression of PKP2 mutations requires further investigation.
Previous studies have shown increased apoptotic activity associated with ARVC.14,15,25 Our studies do not provide a direct link between expression of R79x and apoptosis. Yet, our data showing that R79x expression drastically reduced HSP90 levels is intriguing. HSP90 plays a cardioprotective role. It is essential for myoblast survival, and its inhibition causes depletion of HSP90-dependent protein kinases ErbB2, Fyn and AKT, and induction of apoptosis.17,26,27 Moreover, Rodriguez-Sinovas et al27 have demonstrated that Cx43 co-immunoprecipitate HSP90 and that this interaction is important for Cx43 translocation to the inner mitochondrial membrane. We propose that reduced expression of both molecules (HSP90 and Cx43), caused by expression of R79x, leads to facilitated activation of myocyte apoptotic pathways, with consequent loss of viable myocardium as time progresses. Yet, our argument is only speculative and our data should be interpreted with caution before a more direct link between R79x expression and apoptosis can be demonstrated.
Our results showed that the ability of PKP2 to associate to both Cx43 and DP remained present for the 179fs construct, but was absent in the case of R79x. The latter suggests that amino acids 79-178 in PKP2 are necessary for the interactions (either direct or indirect) that maintain these three proteins within the same molecular complex. It is important to note that we chose to study the actual 179fs mutation, to better reproduce the molecular environment present in the afflicted patients. It is therefore possible, though unlikely, that the additional non-sense sequence resulting from the frameshift (11 amino acids) rather than the PKP2 sequence that is absent in the mutant R79x, allowed for interaction with the partner proteins. Yet, whether through one sequence, the other, or both, mutant 179fs was still capable of interacting with Cx43 and DP. These differences in molecular behavior for both constructs and yet, their commonality as disease-causing mutations suggests that multiple cellular pathways are affected by PKP2 mutations. This is reminiscent of what has been observed in other arrhythmogenic diseases (long QT syndrome, or Brugada), where the position of the mutation within the sequence of the affected genes varies significantly and yet, there is convergence in terms of the basic features of the disease. It will be interesting to determine whether, for ARVC, variations in clinical phenotype correlate with the severity of the molecular and cellular changes observed, as more ARVC-relevant PKP2 mutations are studied.
In summary, we have presented the first characterization of these ARVC-relevant PKP2 mutations at the cellular/molecular level. To address all aspects of cell function, or all molecular interactions, would be unrealistic. We focused on two mutations, and two known PKP2 molecular partners (Cx43 and DP). Our data show important differences regarding protein interactions, protein localization and likely, cell function. These initial observations will be expanded by additional studies with other mutations so that we can begin to discern the molecular mechanisms leading to the main clinical phenotype characteristic of this disease.
Acknowledgments
We thank April Lazarus for generating the viruses and Li Gao and Christine Burrer for assistance with experiments.
Supported by NIH grants HL39707, HL087226 and GM57691, and by a Predoctoral Fellowship from the American Heart Association (EO).
Footnotes
Disclosures None.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Thiene G, Corrado D, Basso C. Arrhythmogenic right ventricular cardiomyopathy/dysplasia. Orphanet J Rare Dis. 2007;14:45. doi: 10.1186/1750-1172-2-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Antoniades L, Tsatsopoulou A, Anastasakis A, et al. Arrhythmogenic right ventricular cardiomyopathy caused by deletions in plakophilin-2 and plakoglobin (Naxos disease) in families from Greece and Cyprus: genotype-phenotype relations, diagnostic features and prognosis. Eur Heart J. 2006;27:2208–16. doi: 10.1093/eurheartj/ehl184. [DOI] [PubMed] [Google Scholar]
- 3.Kannankeril PJ, Bhuiyan ZA, Darbar D, et al. Arrhythmogenic right ventricular cardiomyopathy due to a novel plakophilin 2 mutation: wide spectrum of disease in mutation carriers within a family. Heart Rhythm. 2006;3:939–44. doi: 10.1016/j.hrthm.2006.04.028. [DOI] [PubMed] [Google Scholar]
- 4.van Tintelen JP, Entius MM, Bhuiyan ZA, et al. Plakophilin-2 mutations are the major determinant of familial arrhythmogenic right ventricular dysplasia/cardiomyopathy. Circulation. 2006;113:1650–8. doi: 10.1161/CIRCULATIONAHA.105.609719. [DOI] [PubMed] [Google Scholar]
- 5.Dalal D, James C, Devanagondi R, et al. Penetrance of mutations in plakophilin-2 among families with arrhythmogenic right ventricular dysplasia/cardiomyopathy. J Am Coll Cardiol. 2006;48:1416–24. doi: 10.1016/j.jacc.2006.06.045. [DOI] [PubMed] [Google Scholar]
- 6.Gerull B, Heuser A, Wichter T, et al. Mutations in the desmosomal protein plakophilin-2 are common in arrhythmogenic right ventricular cardiomyopathy. Nat Genet. 2004;36:1162–4. doi: 10.1038/ng1461. [DOI] [PubMed] [Google Scholar]
- 7.Oxford EM, Musa H, Maass K, et al. Connexin43 remodeling caused by inhibition of plakophilin-2 expression in cardiac cells. Circ Res. 2007;101:703–11. doi: 10.1161/CIRCRESAHA.107.154252. [DOI] [PubMed] [Google Scholar]
- 8.Kaplan SR, Gard JJ, Protonotarios N, et al. Remodeling of myocyte gap junctions in arrhythmogenic right ventricular cardiomyopathy due to a deletion in plakoglobin (Naxos disease) Heart Rhythm. 2004;1:3–11. doi: 10.1016/j.hrthm.2004.01.001. [DOI] [PubMed] [Google Scholar]
- 9.Kaplan SR, Gard JJ, Carvajal-Huerta L, Ruiz-Cabezas JC, Thiene G, Saffitz JE. Structural and molecular pathology of the heart in Carvajal syndrome. Cardiovasc Pathol. 2004;13:26–32. doi: 10.1016/S1054-8807(03)00107-8. [DOI] [PubMed] [Google Scholar]
- 10.Hofmann I, Casella M, Schnölzer M, et al. Identification of the junctional plaque protein plakophilin 3 in cytoplasmic particles containing RNA-binding proteins and the recruitment of plakophilins 1 and 3 to stress granules. Mol Biol Cell. 2006;17:1388–98. doi: 10.1091/mbc.E05-08-0708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grossmann KS, Grund C, Huelsken J, et al. Requirement of plakophilin 2 for heart morphogenesis and cardiac junction formation. J Cell Biol. 2004;167:149–60. doi: 10.1083/jcb.200402096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Musil LS, Goodenough DA. Biochemical analysis of connexin43 intracellular transport, phosphorylation, and assembly into gap junctional plaques. J Cell Biol. 1991;115:1357–74. doi: 10.1083/jcb.115.5.1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen X, Bonne S, Hatzfeld M, et al. Protein binding and functional characterization of plakophilin 2. Evidence for its diverse roles in desmosomes and beta -catenin signaling. J Biol Chem. 2002;277:10512–22. doi: 10.1074/jbc.M108765200. [DOI] [PubMed] [Google Scholar]
- 14.Valente M, Calabrese F, Thiene G, et al. In vivo evidence of apoptosis in arrhythmogenic right ventricular cardiomyopathy. Am J Pathol. 1998;152:479–84. [PMC free article] [PubMed] [Google Scholar]
- 15.Nagata M, Hiroe M, Ishiyama S, et al. Apoptotic cell death in arrhythmogenic right ventricular cardiomyopathy: a comparative study with idiopathic sustained ventricular tachycardia. Jpn Heart J. 2000;41:733–41. doi: 10.1536/jhj.41.733. [DOI] [PubMed] [Google Scholar]
- 16.Yamaji K, Fujimoto S, Ikeda Y, et al. Apoptotic myocardial cell death in the setting of arrhythmogenic right ventricular cardiomyopathy. Acta Cardiol. 2005;60:465–70. doi: 10.2143/AC.60.5.2004965. [DOI] [PubMed] [Google Scholar]
- 17.Yun BG, Matts RL. Differential effects of Hsp90 inhibition on protein kinases regulating signal transduction pathways required for myoblast differentiation. Exp Cell Res. 2005;307:212–23. doi: 10.1016/j.yexcr.2005.03.003. [DOI] [PubMed] [Google Scholar]
- 18.Ai Z, Fischer A, Spray DC, et al. Wnt-1 regulation of connexin43 in cardiac myocytes. J Clin Invest. 2000;105:161–71. doi: 10.1172/JCI7798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Czyz J, Guan K, Zeng Q, et al. Loss of beta 1 integrin function results in upregulation of connexin expression in embryonic stem cell-derived cardiomyocytes. Int J Dev Biol. 2005;49:33–41. doi: 10.1387/ijdb.041835jc. [DOI] [PubMed] [Google Scholar]
- 20.Morley GE, Vaidya D, Samie FH, et al. Characterization of conduction in the ventricles of normal and heterozygous Cx43 knockout mice using optical mapping. J Cardiovasc Electrophysiol. 1999;10:1361–75. doi: 10.1111/j.1540-8167.1999.tb00192.x. [DOI] [PubMed] [Google Scholar]
- 21.Zhang YW, Nakayama K, Nakayama K, et al. A novel route for connexin 43 to inhibit cell proliferation: negative regulation of S-phase kinase-associated protein (Skp 2) Cancer Res. 2003;63:1623–30. [PubMed] [Google Scholar]
- 22.Zhang YW, Kaneda M, Morita I. The gap junction-independent tumor-suppressing effect of connexin 43. J Biol Chem. 2003;278:44852–6. doi: 10.1074/jbc.M305072200. [DOI] [PubMed] [Google Scholar]
- 23.Kardami E, Dang X, Iacobas DA, et al. The role of connexins in controlling cell growth and gene expression. Prog Biophys Mol Biol. 2007;94:245–64. doi: 10.1016/j.pbiomolbio.2007.03.009. [DOI] [PubMed] [Google Scholar]
- 24.Giardina SF, Mikami M, Goubaeva F, et al. Connexin 43 confers resistance to hydrogen peroxide-mediated apoptosis. Biochem Biophys Res Commun. 2007;362:747–52. doi: 10.1016/j.bbrc.2007.08.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mallat Z, Tedgui A, Fontaliran F, et al. Evidence of apoptosis in arrhythmogenic right ventricular dysplasia. N Engl J Med. 1996;335:1190–6. doi: 10.1056/NEJM199610173351604. [DOI] [PubMed] [Google Scholar]
- 26.Griffin TM, Valdez TV, Mestril R. Radicicol activates heat shock protein expression and cardioprotection in neonatal rat cardiomyocytes. Am J Physiol Heart Circ Physiol. 2004;287:H1081–8. doi: 10.1152/ajpheart.00921.2003. [DOI] [PubMed] [Google Scholar]
- 27.Rodriguez-Sinovas A, Boengler K, Cabestrero A, et al. Translocation of connexin 43 to the inner mitochondrial membrane of cardiomyocytes through the heat shock protein 90-dependent TOM pathway and its importance for cardioprotection. Circ Res. 2006;99:93–101. doi: 10.1161/01.RES.0000230315.56904.de. [DOI] [PubMed] [Google Scholar]