

Abstract
The death receptor pathway is coupled to the mitochondria apoptosis pathway. However, mitochondrial participation, which is stimulated by Bid but suppressed by Bcl-2/Bcl-xL, is required in certain cells (Type II), but not in others (Type I). While these differences were originally characterized in the lymphoid cell lines, the typical Type II cells are represented by hepatocytes in vivo. The molecular mechanisms that distinguish Type II from Type I cells and the regulation are not fully understood. Fas can be sequestered by the HGF receptor c-Met and high doses of HGF can promote cell death by freeing Fas from c-Met complex. We thus reasoned that treatment of the Type II cells with high doses of HGF could enhance Fas-mediated apoptosis and spare the mitochondria amplification. Indeed, such treatment led to increased apoptosis in Type II lymphoid cells, which could not be blocked by Bcl-xL. Moreover, significant hepatocyte apoptosis was induced by this scheme in the absence of Bid with increased dissociation of Fas from c-Met. These findings indicate that high doses of HGF could be used to promote apoptosis in Type II cells bypassing the requirement for mitochondria activation.
Two major apoptosis pathways are present in the mammalian cells. The death receptor pathway is initiated at the cell surface via the death receptor, Fas, TNF-R1 and TRAIL-receptors (Ashkenazi and Dixit, 1998). The mitochondria pathway is initiated intracellularly at the level of mitochondria and regulated by the Bcl-2 family proteins (Green and Kroemer, 2004). The death receptor Fas can be activated by its natural ligand, FasL, or by agonistic antibodies. Upon ligation Fas receptors recruit the adapter molecule, FADD, which recruits the initiator caspase, caspase-8. The complex of Fas, FADD and caspase-8 is called the death inducing signaling complex (DISC), which is responsible for the activation of caspase-8 and the downstream effector caspases, committing the cell to apoptosis.
The death receptor pathway can be coupled to the mitochondria pathway mainly via the activation of the pro-death Bcl-2 family protein, Bid, which is cleaved by caspase-8 (Gross et al., 1999; Yin et al., 1999; Zhao et al., 2001). Early studies in the lymphoid cell lines indicated that cells with similar sensitivity to anti-Fas-induced apoptosis could differ in the dependence on the mitochondria pathway. Thus effective killing does not require mitochondrial involvement in Type I cells, such as SKW6.4 and H9, but does in Type II cells, such as Jurkat and CEM (Scaffidi et al., 1998). Consequently over-expression of the anti-death Bcl-2 or Bcl-xL could block Fas-mediated apoptosis in Type II cells, but not in Type I cells (Scaffidi et al., 1998; Sun et al., 2002).
Such a characterization is not an artifact of in vitro cell lines. It has been demonstrated that Fas-mediated apoptosis in primary hepatocytes requires the mitochondrial participation. Thus over-expression of Bcl-2 or Bcl-xL in hepatocytes suppresses apoptosis (Lacronique et al., 1996; de la Coste et al., 1999) and deletion of Bcl-xL in the liver leads to spontaneous apoptosis (Takehara et al., 2004). The deletion of Bid also renders hepatocytes resistant to Fas-mediated apoptosis (Yin et al., 1999; Li et al., 2002). These data clearly indicate that hepatocytes are in vivo representatives of the Type II cells.
Mitochondria activation, as characterized by the release of cytochrome c and membrane depolarization, occurs in both Type I and Type II cells, which can be equally blocked by Bcl-2 (Scaffidi et al., 1998). This indicates that the two types of cells do not differ in the mitochondria activation, which is intact in both. But other fundamental differences make the mitochondria pathway more important in Type II cells, but not in Type I cells. One such difference could be that the DISC assembly is rapid and potent in Type I cells, but weak and delayed in Type II cells (Scaffidi et al., 1998). This leads to a strong caspase-8 activation in the former, but weak caspase-8 activation in the latter (Scaffidi et al., 1998). As such, the downstream caspase-3 activation is also ineffective in Type II cells, such that it could not overcome the inhibitory effects of XIAP, which binds to it (Li et al., 2002; Sun et al., 2002). The mitochondria pathway is required to reverse XIAP inhibition via the release of Smac, which binds to XIAP and liberates the suppressed caspase-3 (Du et al., 2000; Li et al., 2002; Sun et al., 2002).
It is not fully clear as to why DISC assembly upon Fas engagement is different in the two types of cells. One possibility is the cellular location where the DISC is assembled could affect the quality of the DISC (Lee et al., 2006). Another hypothesis is that the level or status of Fas available for the ligation by either the agonistic antibody or the natural ligand, FasL, could be different. The available Fas may be less in the Type II cells, either at the absolute number per cell, or at the relative number due to functional defect. Recently it has been shown that c-Met, the receptor for hepatocyte growth factor (HGF), can bind to and sequester Fas in several types of epithelial cells, including hepatocytes as well as endothelial cells, which prevents the bound Fas from being activated by its ligand or the agonistic antibodies (Wang et al., 2002; Smyth and Brady, 2005). Disruption of c-Met/Fas interactions, such as by high doses of HGF, could increase the sensitivity of these cells to Fas-mediated apoptosis (Wang et al., 2002; Smyth and Brady, 2005). Based on these findings, we postulated that disruption of such an interaction in the Type II cells may increase the amount of Fas available for ligation and thus enhance the activation of caspase-8, leading to stronger downstream caspase-3 activation and bypassing the requirement for the mitochondria pathway.
The current study examined this hypothesis and found that co-treatment of the Type II lymphoid cells with the anti-Fas antibody and a high dose of HGF did enhance apoptosis, which could not be blocked by the over-expression of Bcl-xL. Furthermore, high doses of HGF could sensitize bid-deficient hepatocytes to anti-Fas treatment in both in vivo and in vitro models with increased dissociation of Fas from c-Met. As the result, the combined HGF and anti-Fas caused significant liver injury in bid-deficient mice, which would be otherwise resistant to anti-Fas alone. These findings thus suggest that disruption of c-Met/Fas interaction could facilitate the conversion of the Type II response to the Type I response by enhancing Fas signaling and eliminating the requirement for the mitochondrial participation.
Materials and Methods
Chemicals and antibodies
All medium, serum and regular supplements were from Invitrogen-GIBCO (Carlsbad, CA). Recombinant human HGF protein was obtained from SnowBrand Pharmaceuticals (Osaka, Japan). Collagenase H was from Boehringer Ingelheim (Ridgefield, CT). Hoechst 33342 was obtained from Invitrogen-Molecular Probe (Carlsbad, CA). Caspase substrates were obtained from Biomol, Plymouth Meeting, PA. All other chemicals were from Sigma (St. Louis, MO).
The CH11 clone of anti-human Fas antibody (Upstate Biotechnology, Charlottesville, VA) was used for apoptosis induction in the human cell lines. The Jo2 clone of anti-mouse Fas antibody (BD PharMingen, San Diego, CA) was used for apoptosis induction in mouse hepatocytes. The following antibodies were used in immunoblot or immunoprecipitation assay: anti-β-actin (Sigma, AC15) (1:5,000); anti-human caspase-3 (Stressgen, Victoria, Canada, AAP-113) (1:1,000); anti-caspase-8 (Stressgen, AAP-108) (1:1,000); anti-caspase-7 (Cell Signaling, Danvers, MA) (1:1,000); anti-Bcl-2 (6C8) (BD Bioscience, San Diego, CA) (1:1,000); anti-Bcl-xL (Transduction Laboratories, San Jose, CA, B61220) (1:1,000); anti-14-3-3ε (Santa Cruz, CA, T-16, sc-1020) (1:1,000); anti-c-Met (Santa Cruz, B6) (1:1,000), anti-Fas (Santa Cruz, FL335) (1:500) and anti-HGF antibody (Yang et al., 2001).
Cell lines and cell culture
The Type I cell lines, SKW6.4 (Neo and Bcl-2 transfected) were kindly provided by Dr. Andreas Strasser (Walter and Eliza Hall Institute, Australia) and Dr. Marcus E. Peter (University of Chicago) (Scaffidi et al., 1998). The Type II cells, Jurkat and its derivates Jurkat-Neo, Jurkat-Bcl-2 and Jurkat-Bcl-xL, and CEM and its derivate CEM-Bcl-2 were kindly provided by Dr. Gerald M. Cohen (University of Leicester) (Sun et al., 2002). Caspase-8 deficient Jurkat cells (JB-6), and caspase-8 reconstituted JB6 (BC-22) were obtained form Dr. Shigekazu Nagata (Osaka University) (Kawahara et al., 1998).
These cell lines were maintained in RPMI 1640 with 10% fetal calf serum and standard supplements. G418 (500 μg/ml) was included in the medium for cell lines containing a neomycin-based vector. To induce apoptosis, an anti-human Fas antibody (CH11, 100 ng/ml) was added to the culture.
Mice and animal procedures
Mice deficient in bid were established by gene targeting technique (Yin et al., 1999). These mice have been bred to C57BL/6J background. All mice used in the experiments were male littermates ~25–30 g in weight. They were maintained in a specific pathogen-free facility in compliance with National Institutes of Health and University of Pittsburgh guidelines.
For induction of liver injury, mice were intravenously given an anti-Fas antibodies (Jo-2, BD PharMingen) at the dose of 0.25 μg/g of body weight. Each group contained three to five mice. Animals were then sacrificed at the designated time points. Blood was collected for the measurement of the activity of alanine aminotransferase (ALT) and asparate aminotransferase (AST). Livers were harvested and cytosols were prepared for the biochemical analysis as previously described (Zhao et al., 2001). A portion of the liver was fixed in 10% balanced formalin for paraffin embedding and TUNEL staining.
To deliver the naked DNA to the liver in vivo, the control pcDNA3 vector and pcDNA3-hHGF (kindly provided by Dr. Youhua Liu, University of Pittsburgh) were administrated into mice by a hydrodynamic-based gene delivery technique (Yang et al., 2001). Briefly, a large volume (1.6 ml) of DNA solution containing 1 μg DNA/g of mouse body weight was rapidly injected into the tail vein in 5–10 sec. Gene expression could be detected in 16–24 h and mice were then subjected to anti-Fas treatment.
Induction of apoptosis in primary hepatocytes
Murine hepatocytes were isolated through a reverse perfusion-based method as previously described (Zhao et al., 2003). The cells were plated on Falcon “Primaria” 24-well plates (Falcon 3847) at a density of 8 ×104 cells/well and cultured in William’s medium E supplemented with 10% fetal bovine serum and all other standard supplements. To induce apoptosis, the Jo-2 anti-Fas antibody (0.5 μg/ml) was given together with cycloheximide (CHX) (10 μg/ml) as previously described (Zhao et al., 2003).
Analysis of cell death
Cell viability was determined by either trypan blue dye exclusion or the MTT assay as previously described (Zhao et al., 2003). Caspase activities were measured using 25 μg of proteins and 20 μM of fluorescent substrates (Ac-DEVD-AFC and AC-IETD-AFC for caspase-3, and -8, respectively) (Zhao et al., 2003). The fluorescence signals were detected by a fluorescence microplate reader (GENios, Tecan) at excitation and emission wavelengths of 400 and 510 nm, respectively.
Immunoblot and immunoprecipitation assays
For immunoblot assay, cell lines were lysed with RIPA buffer supplemented with a cocktail of protease inhibitors (Roche, Palo Alto, CA). The liver cytosol was prepared, separated by 12.5% SDS-PAGE and transferred to a polyvinylidene difluoride membrane (PVDF) for immunoblot assay as previously describe (Zhao et al., 2001).
Immunoprecipitation assay with anti-Fas antibody was performed as previously described (Wang et al., 2002). Briefly, total cell lysates containing approximately 500 μg of protein was incubated with 10 μg of the anti-Fas antibody (Santa Cruz, FL335) in a volume of 1 ml for 1 h. The mixture was further incubated with the IgG-conjugated agarose beads overnight a 4°C on a nutator. The beads were pelleted and washed before the precipitated proteins were eluted in the boiled SDS-PAGE sample buffer and separated by 8% SDS-PAGE. The proteins were then transferred to the PVDF membranes and blotted with an anti-c-Met antibody.
Histology and histochemistry assay
Liver tissues were fixed in 10% neutral buffered formalin, processed and stained by the histology core laboratory at the Department of Pathology. Paraffin-embedded blocks were cut at 5 μm in thickness and the sections were stained with hematoxylin and eosin (H–E). For terminal deoxynucleotidyl transferase mediated dUTP-biotin nick end labeling (TUNLE) assay, a commercial kit (ApopTag kit, Chemicon International, Temecula, CA) was used according to the manufacturer’s instruction. All images were taken with a digital camera (SPOT, Diagnostic Instruments, Sterling Height, MI) under a light microscope (Nikon Eclipse TE200, Melville, CA).
Results
Fas-mediated apoptosis in Type II lymphoid cells could be protected by anti-death Bcl-2 family proteins
Previous work has indicated that Jurkat is a Type II cell line (Scaffidi et al., 1998). Four different Jurkat cell lines were examined here. While stimulation of Jurkat cells expressing the neomycin vector (J6-Neo) with an agonistic antibody (CH11) induced significant cell death, Jurkat cells lacking caspase-8 (JB6) were completely resistant to this killing (Fig. 1A,B), reaffirming the notion that Fas-induced cell death depends on the DISC and caspase-8 (Ashkenazi and Dixit, 1998). Jurkat cells over-expressing the anti-death Bcl-2 family proteins, Bcl-xL or Bcl-2, were also very well protected from anti-Fas-induced apoptosis (Fig. 1A,B). The cleavage of the pro-form of caspase-3 and caspase-7 were both reduced in these cells, compared to J6-Neo cells (Fig. 1C). The activation of caspase and the protection of Bcl-2/Bcl-xL indicated the apoptotic nature of the cell death.
Fig. 1.

Anti-Fas antibody-induced apoptosis is dependent on caspase-8 and can be blocked by Bcl-2 or Bcl-xL in Type II cells. A: Immunoblot analysis using indicated antibodies on four Type II Jurket-derived cell lines, J6-Neo (stably transfected with the neomycin vector only), J6-Bcl-xL (stably transfected with Bcl-xL), J6-Bcl-2(stably transfected with Bcl-2)and JB-6(caspase-8-deficient). B: Four Type II Jurkat cell line sand two Type I cell lines, SKW-Neo (stably transfected with the neomycin vector only) and SKW-Bcl-2 (stably transfected with Bcl-2) were treated with an anti-human Fas antibody (CH11, 100 ng/ml) for 12 h. Cell death was assessed by trypan blue staining (mean ± s.d.). C: Immunoblot analysis using indicated antibodies on four Type II Jurket-derived cell lines following anti-Fas treatment (CH11, 100 ng/ml) for 8 h. A stronger caspase activation was associated with a more prominent reduction of the pro-form of the caspases due to cleavage. 14-3-3ε served as the loading control. D,E: The Type I cell lines, SKW-Neo and SKW-Bcl-2, and the Type II cell lines, CEM-Neo (stably transfected with the neomycin vector only) and CEM-Bcl-2 (stably transfected with Bcl-2)were treated with anti-Fas as in C. Immunoblot analysis was conducted using the indicated antibodies(D). A stronger caspase-3 activation was associated with a more prominent cleavage product. Caspase-3 activity was measured using Ac-DEVD-AFC as the substrate (E). The values (mean ± s.d.) were expressed as the percentage of the CEM or SKW cells expressing the vector, respectively.
The protection conferred by the anti-death Bcl-2 proteins in the Jurkat cells could be confirmed in another Type II cell line, CEM (Fig. 1D,E). Over-expression of Bcl-2 in CEM cells completely blocked caspase-3 activation, suggesting its dependency on the mitochondria pathway. On the other hand, caspase activation in the Type I cells, SKW, expressing Bcl-2 was not significantly affected. These results were consistent with previous studies and indicate that mitochondria activation is important in Type II, but not in Type I cells for Fas-mediated apoptosis (Scaffidi et al., 1998).
High doses of HGF overcome the protection of Bcl-xL in Fas-mediated apoptosis in Type II lymphoid cells
It seems that the requirement for mitochondria activation in Type II cells could be related to the weak and delayed assembly of DISC in these cells (Scaffidi et al., 1998). Recent studies in different types of cells indicate that a significant amount of Fas could be in complex with the HGF receptor c-Met at the cell surface (Wang et al., 2002; Smyth and Brady, 2005). A competitive binding of HGF with c-Met can cause the disruption of Fas-c-Met complex and release Fas from this complex. Resistance to Fas-mediated killing could be due to the sequestering of Fas by c-Met and co-treatment of cells with HGF can enhance the sensitivity of cells to Fas-mediated killing due to increased availability of free Fas (Wang et al., 2002). We reasoned that such an activity of HGF in Type II cells could lead to a stronger DISC assembly and bypass the requirement for the mitochondria pathway, so that the inhibition of the later would not be able to block anti-Fas induced apoptosis.
Indeed, HGF promoted a dose-dependent increase of anti-Fas induced apoptosis in Jurkat cells expressing Bcl-xL (Fig. 2A). Consistently, caspase-8 activity was raised by HGF co-treatment, leading to the enhancement of effector caspase-3 activity (Fig. 2B,C). These results indicated that the mitochondria activation, which was suppressed by Bcl-xL or Bcl-2, could be bypassed by the application of HGF in Type II cells, which rendered the killing much like that in Type I cells.
Fig. 2.

Dose-dependent promotion of Fas-induced apoptosis by HGF in Jurkat cells expressing Bcl-xL orBcl-2. A: Jurkat cells were treated with the CH11anti-Fas antibody (100ng/ml) for 12h in the absence or presence of human HGF(0.5 μg/ml or as indicated). Cell death was assessed by trypan blue staining (mean ± s.d.). B,C: Jurkat-Neo (white column) and Jurkat-Bcl-xL (black column) were incubated with medium (Ctl) or anti-Fas (100 ng/ml) with or without HGF (0.5 μg/ml) for 8 h. Note the cleavage of caspase-3 was reduced in the presence of Bcl-2, but enhanced in the presence of HGF. Caspase-8 (B) and -3 (C) activities were determined and presented as fold increase over non-treated controls (mean ± s.d.).
HGF by itself had no toxicity at different doses (Fig. 2A and data not shown) and the enhancement of Fas-mediated apoptosis required the presence of caspase-8 (Fig. 3A). Etoposide, a DNA damaging agent, typically induces apoptosis via the mitochondria pathway independently of caspase-8 (Fig. 3B). While over-expression of Bcl-2 could effectively reduce etoposide-induced apoptosis, this protection could not be reversed by HGF treatment (Fig. 3B). These findings thus lend additional support to the notion that high doses of HGF was likely working at the receptor level to facilitate Fas activation, and therefore caspase-8 activation and cell death.
Fig. 3.
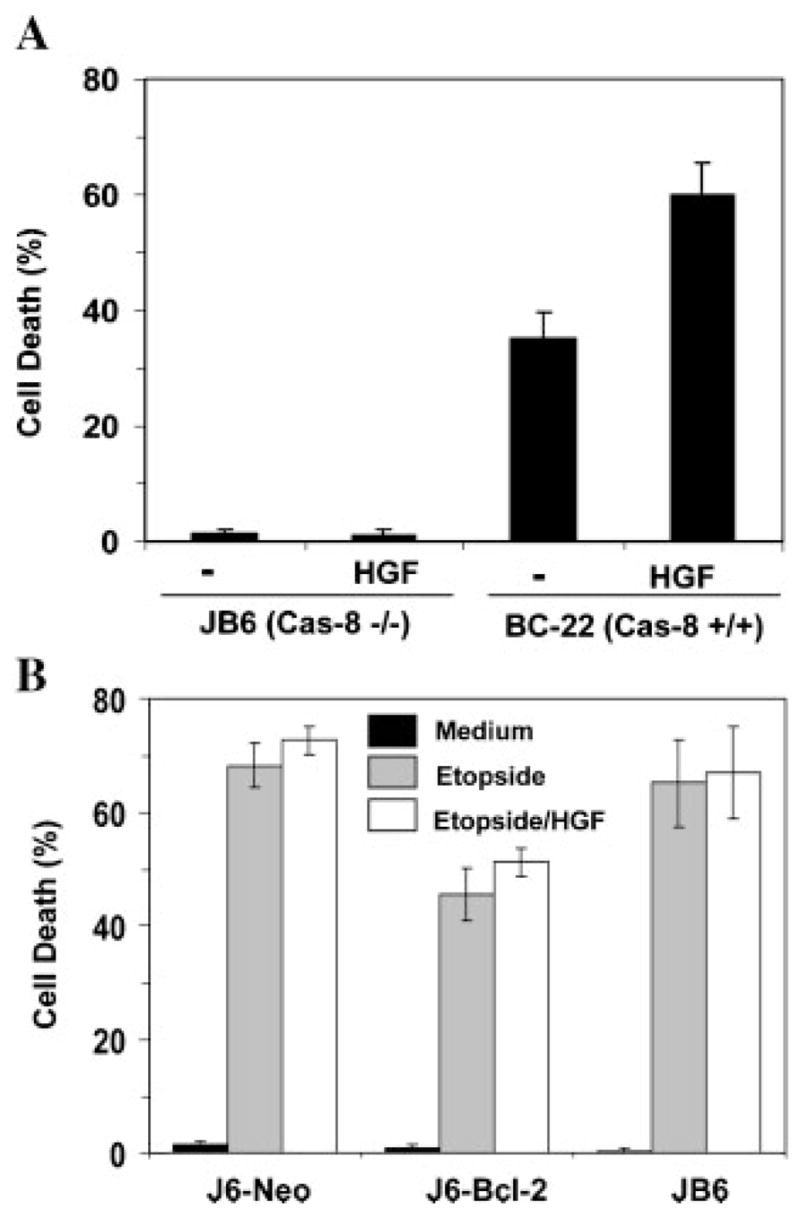
HGF enhances anti-Fas-induced apoptosis likely at the level of caspase-8 activation. A: Jurkat cells deficient in caspase-8 (JB6) and its caspase-8 reconstituted subline (BC-22) were treated with CH11 anti-Fas antibody (100 ng/ml) with or without HGF (0.5 μg/ml) for 12 h. Cell death was assessed by trypan blue staining (mean ± s.d.). B: Different Jurkat cell lines as indicated were treated with medium (black column), etoposide (20 μM, gray column) or etoposide plus 0.5 μg/ml of HGF (white column) for 12 h and analyzed for cell death by the trypan blue assay (mean ± s.d.).
High doses of HGF restore the sensitivity of bid-deficient hepatocytes to Fas-induced apoptosis and promote anti-Fas induced bid-independent liver injury
The lymphoid cell lines are generally quite sensitive to anti-Fas-induced apoptosis without HGF despite that mitochondria amplification is required for the Type II cells. This would suggest that a sufficiently large amount of Fas receptors in these cells are free from the c-Met complex, which is supported by the observation that c-Met expression in Jurkat cells was low (data not shown and Smyth and Brady, 2005). Application of HGF could further increase the amount of free Fas, overcoming the mitochondria requirement in the Type II cells. To seek further evidence that this mechanism could be functional, we examined whether HGF could enhance Fas-mediated killing in a primary Type II cell, the hepatocyte, where c-Met expression was abundant.
Primary hepatocytes in culture were resistant to anti-Fas killing, unless a sensitizing agent, such as the protein synthesis inhibitor cycloheximide, is present (Ni et al., 1994). Furthermore, previous work had shown that Fas-mediated apoptosis in hepatocytes largely depended on the pro-death Bcl-2 family protein, Bid, which mediated the mitochondria activation (Yin et al., 1999; Zhao et al., 2001, 2003; Li et al., 2002). Hepatocytes deficient in Bid were resistant to anti-Fas induced killing (Fig. 4) (Zhao et al., 2003), much like the Bcl-xL/Bcl-2-overexpressing Jurkat cells. However, addition of a high dose of HGF enhanced the sensitivity of the bid-deficient hepatocytes to anti-Fas (Fig. 4), indicating a decrease in mitochondria requirement. The killing of the wild type cells by the anti-Fas antibody was potent due to the participation of the mitochondria pathway and was thus not obviously further enhanced by the co-treatment of HGF.
Fig. 4.
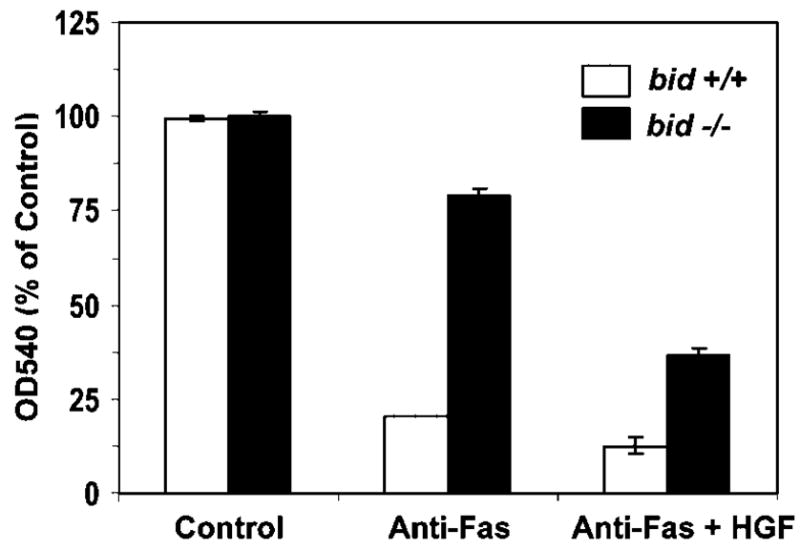
HGF enhances anti-Fas-induced apoptosis in bid-deficient hepatocytes. Wild-type (white column) and bid-deficient (black column) hepatocytes were treated with medium, anti-mouse Fas antibody (Jo-2, 0.5 μg/ml) plus CHX (10 ng/ml) in the absence or the presence of human HGF (0.5 μg/ml) as indicated for 12 h. Viability was measured by the MTT assay and expressed as the relative change of OD540 readings over the non-treated control group (mean ± s.d.).
To determine whether this strategy of enhancing the apoptotic response of Type II cells has any pathophysiological significance in vivo, we injected a plasmid encoding the human HGF gene into the bid-deficient mice via the hydrodynamics method, which instituted a rapid infusion of a large volume (1.6 ml) of solution through the tail vein, resulting in the retention of the injected plasmid in the liver (Yang et al., 2001). This led to a rapid over-expression of functional HGF protein in the liver in 16–24 h (Fig. 5A) (Yang et al., 2001). Immunoprecipitation of Fas could bring down c-Met as a complex and the amount of c-Met in the complex was significantly reduced in the presence of HGF (Fig. 5B), suggesting an increased availability of free Fas. The similar pattern to what had been reported in earlier publications using the same approach (Wang et al., 2002; Smyth and Brady, 2005) rendered a firm support to this notion, despite that reciprocal immunoprecipitation using anti-c-Met to pull down Fas was unfortunately not practical due to the same migration size of Fas as the precipitating antibodies (~55 kDa) on SDS-PAGE, which prevented its definite recognition by immunoblot (Wang et al., 2002; Smyth and Brady, 2005). In addition, the disruptive effect of HGF was independent of Bid expression in the liver.
Fig. 5.
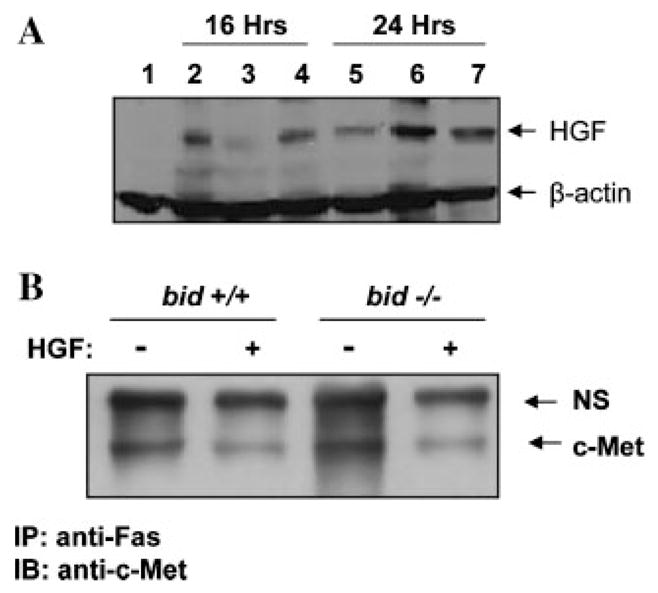
Expression of HGF in the liver disrupts Fas-c-Met complex. A: Mice were intravenously given either saline (lane 1) or pc-DNA-hHGF (1 μg/g of body weight, lanes 2–7) for 16 or 24 h as indicated. Liver samples were then prepared and subjected to immunoblot assay using the anti-HGF and anti-β-actin antibodies. Each lane represented one liver sample. B: Wild type or bid-deficient mice were intravenously given vector control or the vector encoding human HGF for 24 h. Subsequently, the mice were injected with the Jo-2 anti-Fas antibody (0.25 μg/g). Liver samples were prepared 4 h later and subjected to immunoprecipitation with an anti-Fas antibody (Santa Cruz, FL335), followed by immunoblot with an anti-c-Met antibody. NS: a non-specific product cross-reactive with the anti-c-Met antibody served as the loading control.
Most importantly, a subsequent treatment of the mice with the agonistic anti-Fas antibody induced a much stronger activation of caspase-8 and caspase-3 in the bid-deficient livers (Fig. 6A–C). Thus functionally HGF treatment led to a stronger Fas signaling, which overcame the deficiency of mitochondria activation. Consequently, a significantly enhanced liver injury was observed in anti-Fas-stimulated bid-deficient mice. There were severe hepatic apoptosis in multiple parts of livers, based on the TUNEL staining (Fig. 6D), and the serum levels of the liver enzymes, AST and ALT, were significantly raised in these mice (Fig. 6E).
Fig. 6.

HGF promotes anti-Fas-induced liver injury in bid-deficient mice. A–D: Wild type and bid-deficient mice were pre-treated with the vector or pcDNA-hHGF for 24 h. Mice were then given the Jo-2 anti-Fas antibody (0.25 μg/g) as indicated and sacrificed 4 h (wild type mice) or 24 h (bid-deficient mice) later. Liver samples were prepared and subjected to immunoblot assay with the anti-caspase-3 antibody (A), procaspase-3 was shown to the determination of caspase-8 (B) and caspase-3 (C) activities (mean ± s.d.), and to TUNEL staining (D). E: bid-deficient mice were treated with saline (white column), anti-Fas plus the vector (gray column) or anti-Fas plus pcDNA-hHGF (black column). Twenty-four hours after the anti-Fas injection, mice were sacrificed and the sera were analyzed for the activity of AST and ALT (mean ± s.d.). [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
The findings in primary cultured hepatocytes and in the in vivo liver injury model were well correlated with those in the lymphoid cell lines. Together they indicate that an enhanced Fas-mediated killing could be achieved in Type II cells without the need for mitochondria activation if a high level of HGF is present, which likely increases the availability of Fas to the agonistic antibody.
Discussion
Fas is physiologically activated by its natural ligand, FasL, in vivo. Experimentally, Fas can also be potently activated by the agonistic antibodies, such as the CH11 clone of anti-human Fas antibody and the Jo2 clone of anti-mouse Fas antibody. The availability of Fas receptor, either physically or functionally, and its ability to recruit FADD and caspase-8 seems to be the main determining factor in the regulation of the quantity and quality of the DISC and therefore the activation of caspase-8. It is assumed that the level of caspase-8 activity in turn affects whether the mitochondrial pathway is required for the effective transmission of death signal to the downstream effector caspases, such as caspase-3 (Scaffidi et al., 1998). Low levels of caspase-8 activity will not generate sufficient amounts of activated caspase-3 to overcome the inhibitory effects of XIAP, which, however, could be reversed by the mitochondria-released Smac (Du et al., 2000; Li et al., 2002; Sun et al., 2002). This seems to be the case for Type II cells, which require the mitochondrial participation for an effective Fas-mediated killing.
We hypothesized that by increasing the Fas availability on the cell surface of the Type II cells we could enhance Fas-mediated caspase-8 activation and promote cell death without a significant contribution of the mitochondria pathway. There are plenty of examples that death signals could promote apoptosis by increasing the availability of Fas molecules on the cell surface. This could be achieved by the upregulation of Fas expression at the transcription level (Ashkenazi and Dixit, 1998), the promotion of the transportation of existing Fas molecules from the intracellular location to the cell surface (Bennett et al., 1998; Sodeman et al., 2000), and the release of Fas from the complex with c-Met on the cell surface (Wang et al., 2002; Smyth and Brady, 2005).
Previous studies have shown that the natural ligand of c-Met, HGF, although non-toxic by itself, could disrupt c-Met-Fas interaction, thereby increasing the amount of free Fas and enhancing anti-Fas-mediated cell death (Wang et al., 2002). Indeed, when treating the Type II cells with an anti-Fas antibody in the presence of HGF, cell death was significantly increased and the need for mitochondria activation was dramatically reduced. The finding is made not only in the lymphoid cell lines, but also in primary hepatocytes and in an animal model of liver injury. The significance of HGF treatment is more noticeable in bid-deficient hepatocytes, which expressed a high level of c-Met in association with Fas (Fig. 5). The level of c-Met in Jurkat cells is much lower than in hepatocytes and endothelial cells (Smyth and Brady, 2005 and data not shown), which could contribute to their generally higher sensitivity to Fas-mediated killing. However, it is interesting to note that HGF treatment could still enhance the killing in Jurkat cells so that the mitochondria amplification could be spared. Thus the balance of the various components in controlling Fas-mediated killing could be quite delicate and a small shift could change the outcome.
The ability of HGF to enhance the killing independently from mitochondria activation is determined in Bcl-xL or Bcl-2 over-expressed cells, or in bid-deficient cells, in which mitochondria activation is suppressed or lacking, respectively. However, the enhancing effect of HGF seems to require the presence of caspase-8 and is not observed for apoptosis induced by chemicals that directly activate the mitochondria pathway. Moreover, the amount of c-Met that could be immunoprecipitated with Fas was reduced in the presence of HGF. All the evidence supports the notion that HGF works at the Fas receptor level, that Fas signaling strength can be an important contributing factor in determining Type II versus Type I response and that Fas-c-Met interaction can serve as a regulatory mechanism. On the other hand, we certainly could not exclude any additional mechanisms by which the conversion of Type II cells to Type I cells by HGF could occur. These mechanisms might include a direct impact on the intracellular location where the DISC is assembled, which could also affect the quality of the DISC (Lee et al., 2006).
Genetic evidence supports the regulation of c-Met on Fas-mediated apoptosis in vivo. Transgenic mice over-expressing the extracellular domain of c-Met (with no receptor tyrosine kinase activity associated with the intracellular domain) were resistant to anti-Fas-induced liver injury (Wang et al., 2002), most likely due to the increased sequestration of Fas in the complex. On the other hand, conditional knockout of c-Met gene in the liver rendered the mice hypersensitive to anti-Fas antibody-induced hepatocyte apoptosis and liver injury (Huh et al., 2004). HGF, when used at a low dose or when applied several days before the death stimulation, can be associated with a protective effect by initiating c-Met-mediated delayed transcriptional response, which could lead to the upregulation of certain protective molecules, such as Bcl-xL or Mcl-1, via the PI3-kinase/Akt pathway (Kosai et al., 1998; Suzuki et al., 2000; Schulze-Bergkamen et al., 2004; Khai et al., 2006). However, when applied at a higher dose and at the same time of Fas stimulation, the disruption of c-Met/Fas complex could constitute an independent pro-death effect that could overcome the slow-acting transcription-dependent protective effect. The toxic effect of HGF had been well noted in several earlier reports (Conner et al., 1999; Matteucci et al., 2003; Rasola et al., 2004). Furthermore, transgenic mice over-expressing dHGF chronically in the liver (under the albumin promoter) were paradoxically more sensitive to Fas-mediated apoptosis (R. Zarnegar, unpublished observations). This study thus took the advantage of this unique action of HGF and examined the issue of whether enhanced Fas signaling in the Type II cells could allow the bypass of the mitochondria requirement by the use of high doses of HGF to disrupt the c-Met-Fas interaction.
In conclusion, our study demonstrates a plausible mechanism by which Type I and Type II response could be regulated. Furthermore, it also points out a potential use of high doses of HGF in promoting apoptosis in cells otherwise resistant to Fas-mediated killing due to mutations in the mitochondria pathway. The latter may be explored for cancer therapy, such as for the leukemia/lymphoma of the Type II category.
Acknowledgments
We would like to thank Dr. Andreas Strasser (Walter and Eliza Hall Institute) and Dr. Marcus Peter (University of Chicago) for the SKW6.4 (Neo and Bcl-2 transfected). Jurkat and its derivates Jurkat-Neo, Jurkat-Bcl-2 and Jurkat-Bcl-xL, and CEM and its derivate CEM-Bcl-2 were kindly provided by Dr. Gerald M. Cohen (University of Leicester). We are also indebted to Dr. Shigekazu Nagata (Osaka University) for the caspase-8 deficient Jurkat cells (JB-6) and caspase-8 reconstituted JB6 cells (BC-22). pcDNA3 vector and pcDNA3-hHGF were kindly provided by Dr. Youhua Liu (University of Pittsburgh). The authors are in part supported by NIH grants (CA84817 and CA111456 for X-MY, CA35373 for GKM, and CA95782 and ES006109 for RZ) and the research is in part supported by Rangos Fund for Enhancement of Pathology Research.
Literature Cited
- Ashkenazi A, Dixit VM. Death receptors: Signaling and modulation. Science. 1998;281:1305–1308. doi: 10.1126/science.281.5381.1305. [DOI] [PubMed] [Google Scholar]
- Bennett M, Macdonald K, Chan SW, Luzio JP, Simari R, Weissberg P. Cell surface trafficking of Fas: A rapid mechanism of p53-mediated apoptosis. Science. 1998;282:290–293. doi: 10.1126/science.282.5387.290. [DOI] [PubMed] [Google Scholar]
- Conner EA, Teramoto T, Wirth PJ, Kiss A, Garfield S, Thorgeirsson SS. HGF-mediated apoptosis via p53/bax-independent pathway activating JNK1. Carcinogenesis. 1999;20:583–590. doi: 10.1093/carcin/20.4.583. [DOI] [PubMed] [Google Scholar]
- de la Coste A, Fabre M, McDonell N, Porteu A, Gilgenkrantz H, Perret C, Kahn A, Mignon A. Differential protective effects of Bcl-xL and Bcl-2 on apoptotic liver injury in transgenic mice. Am J Physiol Gastrointest Liver Physiol. 1999;277:G702–G708. doi: 10.1152/ajpgi.1999.277.3.G702. [DOI] [PubMed] [Google Scholar]
- Du C, Fang M, Li Y, Li L, Wang X. Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell. 2000;102:33–42. doi: 10.1016/s0092-8674(00)00008-8. [DOI] [PubMed] [Google Scholar]
- Green DR, Kroemer G. The pathophysiology of mitochondrial cell death. Science. 2004;305:626–629. doi: 10.1126/science.1099320. [DOI] [PubMed] [Google Scholar]
- Gross A, Yin XM, Wang K, Wei MC, Jockel J, Milliman C, Erdjument-Bromage H, Tempst P, Korsmeyer SJ. Caspase cleaved BID targets mitochondria and is required for cytochrome c release, while BCL-XL prevents this release but not tumor necrosis factor-R1/Fas death. J Biol Chem. 1999;274:1156–1163. doi: 10.1074/jbc.274.2.1156. [DOI] [PubMed] [Google Scholar]
- Huh C-G, Factor VM, Sanchez A, Uchida K, Conner EA, Thorgeirsson SS. Hepatocyte growth factor/c-met signaling pathway is required for efficient liver regeneration and repair. PNAS. 2004;101:4477–4482. doi: 10.1073/pnas.0306068101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawahara A, Ohsawa Y, Matsumura H, Uchiyama Y, Nagata S. Caspase-independent cell killing by fas-associated protein with death domain. J Cell Biol. 1998;143:1353–1360. doi: 10.1083/jcb.143.5.1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khai NC, Takahashi T, Ushikoshi H, Nagano S, Yuge K, Esaki M, Kawai T, Goto K, Murofushi Y, Fujiwara T, Fujiwara H, Kosai K-I. In vivo hepatic HB-EGF gene transduction inhibits Fas-induced liver injury and induces liver regeneration in mice: A comparative study to HGF. J Hepatol. 2006;44:1046–1054. doi: 10.1016/j.jhep.2005.10.027. [DOI] [PubMed] [Google Scholar]
- Kosai K, Matsumoto K, Nagata S, Tsujimoto Y, Nakamura T. Abrogation of Fas-induced fulminant hepatic failure in mice by hepatocyte growth factor. Biochem Biophys Res Commun. 1998;244:683–690. doi: 10.1006/bbrc.1998.8293. [DOI] [PubMed] [Google Scholar]
- Lacronique V, Mignon A, Fabre M, Viollet B, Rouquet N, Molina T, Porteu A, Henrion A, Bouscary D, Varlet P, Joulin V, Kahn A. Bcl-2 protects from lethal hepatic apoptosis induced by an anti-Fas antibody in mice. Nat Med. 1996;2:80–86. doi: 10.1038/nm0196-80. [DOI] [PubMed] [Google Scholar]
- Lee KH, Feig C, Tchikov V, Schickel R, Hallas C, Schutze S, Peter ME, Chan AC. The role of receptor internalization in CD95 signaling. EMBO J. 2006;25:1009–1023. doi: 10.1038/sj.emboj.7601016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Zhao Y, He X, Kim T-H, Kuharsky DK, Rabinowich H, Chen J, Du C, Yin X-M. Relief of extrinsic pathway inhibition by the Bid-dependent mitochondrial release of Smac in Fas-mediated hepatocyte apoptosis. J Biol Chem. 2002;277:26912–26920. doi: 10.1074/jbc.M200726200. [DOI] [PubMed] [Google Scholar]
- Matteucci E, Modora S, Simone M, Desiderio MA. Hepatocyte growth factor induces apoptosis through the extrinsic pathway in hepatoma cells: Favouring role of hypoxia-inducible factor-1 deficiency. Oncogene. 2003;22:4062–4073. doi: 10.1038/sj.onc.1206519. [DOI] [PubMed] [Google Scholar]
- Ni R, Tomita Y, Matsuda K, Ichihara A, Ishimura K, Ogasawara J, Nagata S. Fas-mediated apoptosis in primary cultured mouse hepatocytes. Exp Cell Res. 1994;215:332–337. doi: 10.1006/excr.1994.1349. [DOI] [PubMed] [Google Scholar]
- Rasola A, Anguissola S, Ferrero N, Gramaglia D, Maffe A, Maggiora P, Comoglio PM, Di Renzo MF. Hepatocyte growth factor sensitizes human ovarian carcinoma cell lines to paclitaxel and cisplatin. Cancer Res. 2004;64:1744–1750. doi: 10.1158/0008-5472.can-03-2383. [DOI] [PubMed] [Google Scholar]
- Scaffidi C, Fulda S, Srinivasan A, Friesen C, Li F, Tomaselli KJ, Debatin KM, Krammer PH, Peter ME. Two CD95 (APO-1/Fas) signaling pathways. EMBO J. 1998;17:1675–1687. doi: 10.1093/emboj/17.6.1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulze-Bergkamen H, Brenner D, Krueger A, Suess D, Fas SC, Frey CR, Dax A, Zink D, Buchler P, Muller M, Krammer PH. Hepatocyte growth factor induces Mcl-1 in primary human hepatocytes and inhibits CD95-mediated apoptosis via Akt. Hepatology. 2004;39:645–654. doi: 10.1002/hep.20138. [DOI] [PubMed] [Google Scholar]
- Smyth LA, Brady HJ. cMet and Fas receptor interaction inhibits death-inducing signaling complex formation in endothelial cells. Hypertension. 2005;46:100–106. doi: 10.1161/01.HYP.0000167991.82153.16. [DOI] [PubMed] [Google Scholar]
- Sodeman T, Bronk SF, Roberts PJ, Miyoshi H, Gores GJ. Bile salts mediate hepatocyte apoptosis by increasing cell surface trafficking of Fas. Am J Physiol Gastrointest Liver Physiol. 2000;278:G992–G999. doi: 10.1152/ajpgi.2000.278.6.G992. [DOI] [PubMed] [Google Scholar]
- Sun XM, Bratton SB, Butterworth M, MacFarlane M, Cohen GM. Bcl-2 and Bcl-xL inhibit CD95-mediated apoptosis by preventing mitochondrial release of Smac/DIABLO and subsequent inactivation of X-linked inhibitor-of-apoptosis protein. J Biol Chem. 2002;277:11345–11351. doi: 10.1074/jbc.M109893200. [DOI] [PubMed] [Google Scholar]
- Suzuki A, Hayashida M, Kawano H, Sugimoto K, Nakano T, Shiraki K. Hepatocyte growth factor promotes cell survival from fas-mediated cell death in hepatocellular carcinoma cells via Akt activation and Fas-death-inducing signaling complex suppression. Hepatology. 2000;32:796–802. doi: 10.1053/jhep.2000.17738. [DOI] [PubMed] [Google Scholar]
- Takehara T, Tatsumi T, Suzuki T, Rucker EB, Hennighausen L, Jinushi M, Miyagi T, Kanazawa Y, Hayashi N. Hepatocyte-specific disruption of Bcl-xL leads to continuous hepatocyte apoptosis and liver fibrotic responses. Gastroenterology. 2004;127:1189–1197. doi: 10.1053/j.gastro.2004.07.019. [DOI] [PubMed] [Google Scholar]
- Wang X, DeFrances MC, Dai Y, Pediaditakis P, Johnson C, Bell A, Michalopoulos GK, Zarnegar R. A mechanism of cell survival: Sequestration of Fas by the HGF receptor Met. Mol Cell. 2002;9:411–421. doi: 10.1016/s1097-2765(02)00439-2. [DOI] [PubMed] [Google Scholar]
- Yang J, Chen S, Huang L, Michalopoulos GK, Liu Y. Sustained expression of naked plasmid DNA encoding hepatocyte growth factor in mice promotes liver and overall body growth. Hepatology. 2001;33:848–859. doi: 10.1053/jhep.2001.23438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin XM, Wang K, Gross A, Zhao Y, Zinkel S, Klocke B, Roth KA, Korsmeyer SJ. Bid-deficient mice are resistant to Fas-induced hepatocellular apoptosis. Nature. 1999;400:886–891. doi: 10.1038/23730. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Li S, Childs EE, Kuharsky DK, Yin X-M. Activation of Pro-death Bcl-2 family proteins and mitochondria apoptosis pathway in tumor necrosis factor-alpha-induced liver injury. J Biol Chem. 2001;276:27432–27440. doi: 10.1074/jbc.M102465200. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Ding WX, Qian T, Watkins S, Lemasters JJ, Yin XM. Bid activates multiple mitochondrial apoptotic mechanisms in primary hepatocytes after death receptor engagement. Gastroenterology. 2003;125:854–867. doi: 10.1016/s0016-5085(03)01066-7. [DOI] [PubMed] [Google Scholar]