

Abstract
ADAM12 has recently emerged as a Candidate Cancer Gene in a comprehensive genetic analysis of human breast cancers. Three somatic mutations in ADAM12 were observed at significant frequencies in breast cancers: D301H, G479E, and L792F. The first two of these mutations involve highly conserved residues in ADAM12 and our computational sequence analysis confirms that they may be cancer-related. We show that the corresponding mutations in mouse ADAM12 inhibit the proteolytic processing and activation of ADAM12 in NIH3T3, COS-7, CHO-K1 cells and in MCF-7 breast cancer cells. The D/H and G/E ADAM12 mutants exert a dominant-negative effect on the processing of the wild-type ADAM12. Immunofluorescence analysis and cell surface biotinylation experiments demonstrate that the D/H and G/E mutants are retained inside the cell and are not transported to the cell surface. Consequently, the D/H and G/E mutants, unlike the wild-type ADAM12, are not capable of shedding Delta-like l, a ligand for Notch receptor, at the cell surface, or of stimulating cell migration. Our results suggest that the breast cancer-associated mutations interfere with the intracellular trafficking of ADAM12 and result in loss of the functional ADAM12 at the cell surface.
Keywords: proteolytic processing, cell surface, endoplasmic reticulum, intracellular trafficking
Introduction
Metalloprotease Disintegrin 12 (ADAM12) is highly expressed in cancer of the breast (1,2), prostate (3), liver (4), stomach (5), colon (3), bladder (6), and in glioblastoma (7). ADAM12 appears to be selectively up-regulated in cancer cells, and its expression is nearly undetectable in normal breast, prostate, colon and liver epithelium (1-6). In mouse models of breast, prostate and colon cancer, ADAM12 is found in a subpopulation of stromal cells adjacent to epithelial tumor cells (2,3). Although this localization suggests that ADAM12 may play a role in stromal-tumor crosstalk, the molecular mechanism of ADAM12 action during cancer progression is not known.
Studies utilizing mouse models of breast and prostate cancers suggested that ADAM12 promotes tumor progression. Forced expression of ADAM12 in breast tumors of MMTV-PyMT mice (carrying the polyomavirus middle T oncogene under mouse mammary tumor virus promoter and developing multifocal mammary adenocarcinomas) accelerated tumor progression (2). Knocking out ADAM12 expression in prostate tumors of W10 mice (expressing SV40 large T-antigen under control of probasin promoter and developing prostate carcinomas) resulted in smaller and better differentiated tumors (3).
Recently, ADAM12 has been identified as one of the Candidate Cancer Genes in a comprehensive mutational analysis of human breast cancers (8). Three mutations were found to be associated with breast cancer: D301H in the metalloprotease domain, G479E in the disintegrin domain, and L792F in the cytoplasmic tail (Fig. 1a). The metalloprotease domain of ADAM12 is catalytically active and cleaves several protein substrates, including insulin growth factor-binding protein (IGFBP)-3 and IGFBP-5 (9), epidermal growth factor and betacellulin (10), placental leucine aminopeptidase (11), and Delta-like 1, a ligand for Notch receptors (12). The disintegrin domain of ADAM12, in co-operation with the following cysteine-rich domain, was reported to interact with integrin receptors, syndecans, and with TGF-β type II receptor (13-16). The cytoplasmic domain has the ability to bind to several signaling, cytoskeletal, and adaptor proteins (17-21).
Figure 1.

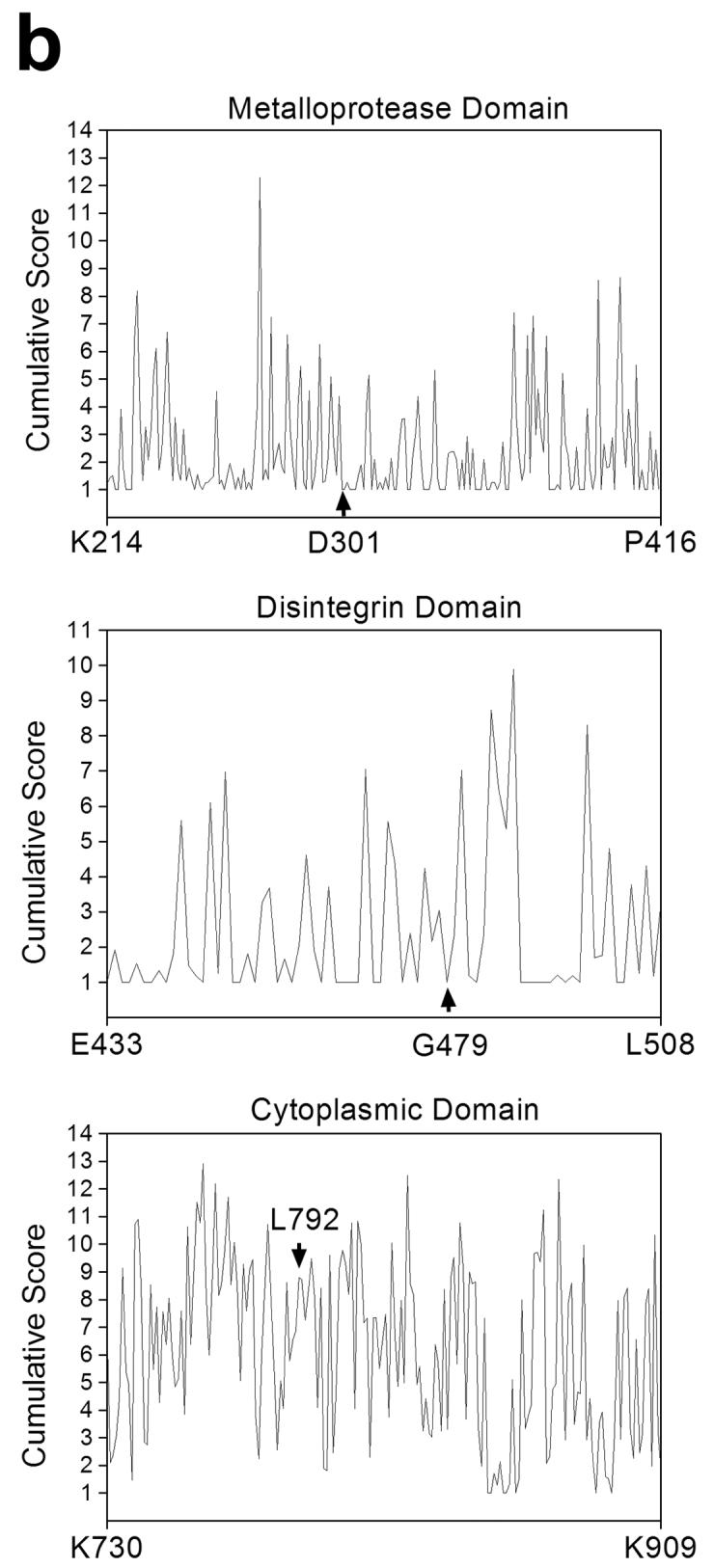
(a) Domain organization of ADAM12 and localization of the three mutations identified in a large-scale screen of breast cancer genome (8). Alignments of amino acid sequences of human, mouse, bovine and xenopus ADAM12 surrounding the mutated sites are shown below the diagram, the mutated amino acids are bold and underlined. (b) Cumulative SIFT (Sorting Intolerant From Tolerant) scores for individual positions in the metalloprotease domain (residues 214-416), the disintegrin domain (residues 433-508), and the cytoplasmic domain (residues 730-909) of human ADAM12. At each position, SIFT scores for individual amino acid substitutions were calculated using the software available at http://blocks.fhcrc.org/sift/SIFT.html, the scores for all possible substitutions at a particular position were then added up. Positions of D301, G479, and L792 are indicated by arrows. Notice that the cumulative score at D301 and G479 is 1, the minimal cumulative score value possible, indicating that no substitutions are allowed at these positions.
In the present study, we investigated the effects of cancer-associated mutations on ADAM12 function. We show that two mutations that are classified as cancer-causing by a bioinformatic approach block the generation of the mature, active form of ADAM12, lead to retention of ADAM12 in the endoplasmic reticulum, and result in loss of ADAM12 function at the cell surface.
Materials and Methods
Expression constructs
Full-length mouse ADAM12 cDNA was cloned into pcDNA3.1 vector or into pBABEpuro retroviral expression vector. In pcDNA3.1 vector, the stop codon was either placed after c-myc and 6xHis tags (constructs used in experiments shown in Figs 2 and 3) or immediately after the ADAM12 sequence (this construct was used in co-expression experiments shown in Fig. 3). The retroviral expression constructs did not contain any tags. The D299H and G477E mutants were generated by site-directed mutagenesis using QuickChange kit (Stratagene). The entire lengths of ADAM12 inserts were then sequenced to eliminate the possibility that errors might have been introduced during the mutagenesis. The expression construct of mouse Dll1 was described earlier (12).
Figure 2.

Proteolytic processing of the wild-type and mutant forms of ADAM12 in different cell types. COS-7 (a), CHO-K1 (b) or MCF-7 cells (c) were transiently transfected with the wild-type mouse ADAM12 (wt), the D/H mutant (D299H), the G/E mutant (G477E), or with empty vector (Vec). (d) NIH3T3 cells were transiently transfected with ADAM12 plasmid constructs (left panel), were infected with ADAM12 retroviruses (middle panel), or were selected with puromycin for stable expression of ADAM12 proteins after retroviral infection. Cell extracts (a, b, c) or glycoprotein-enriched fractions of cell extracts obtained after passing through concanavalin A sepharose columns (d) were analyzed by Western blotting using antibodies specific for the cytoplasmic tail of the recombinant proteins. Arrows show the position of the full-length, nascent ADAM12, asterisks indicate the mature, processed, catalytically active form of ADAM12.
Figure 3.

The effect of D/H and G/E mutations on the proteolytic processing of the wild-type ADAM12. COS-7 cells were transfected with the wild-type ADAM12 (wt, lanes 1-3) or with empty vector (Vec, lanes 4-6). Cells were co-transfected with the D/H (lanes 2 and 4) or G/E (lanes 3 and 5) ADAM12 mutant containing C-terminal c-myc and 6xHis tags. Alternatively, cells were co-transfected with empty vector (lanes 1 and 6). (a) The expression of all ADAM12 proteins was examined by immunoblotting with anti-ADAM12 antibody (left); the D/H and G/E mutants were visualized by immunoblotting with anti-c-myc tag antibody (right). The position of the full-length wild-type ADAM12 is indicated by arrow, the full-length ADAM12 mutants are indicated by arrowhead, the processed form of ADAM12 is designated by asterisk. Notice that the proteolytic processing of the wild-type ADAM12 is diminished in cells co-expressing the D/H or G/E ADAM12 mutants. (b) The D/H and G/E mutants were immunoprecipitated using anti-c-myc tag antibody, the immunoprecipitates were analyzed by immunoblotting with anti-ADAM12 antibody (left) or anti-c-myc tag antibody (right). Notice the presence of the full-length wild-type ADAM12 in the immunoprecipitates obtained from cells that were co-transfected with the wild-type and mutant ADAM12.
Cells
COS-7, NIH3T3, CHO-K1, and MCF-7 cells were obtained from American Tissue Culture Collection. The retroviral packaging cell line Phoenix Eco was provided by Dr. Garry P. Nolan (Stanford University). COS-7, NIH3T3, and Phoenix Eco cells were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% FBS. CHO-K1 cells were grown in F12K nutrient mixture with 10% FBS. MCF-7 cells were maintained in DMEM with 10% FBS and 1 μg/ml insulin. COS-7, NIH3T3, CHO-K1, and MCF-7 cells were transfected with ADAM12-pcDNA3.1 plasmids using Fugene 6 transfection reagent, cells were lyzed 48 h after transfection. Phoenix Eco cells were transfected with ADAM12-pBABEpuro vectors using calcium phosphate precipitation method, viral supernatants were harvested 48 h later and used to infect NIH3T3 cells. Cells were either collected 48 h post-infection or were selected for 10 days with 2 μg/ml of puromycin.
Cell surface biotinylation, immunoprecipitation, and Western blotting
Cells were washed with DPBS, incubated for 30 min at room temperature with 0.5 mg/ml N-hydroxysulfosuccinimidobiotin (sulfo-NHS-biotin; Pierce), and then washed with growth medium and DPBS. Cellular proteins were extracted with extraction buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1% Triton X-100, 1% sodium deoxycholate, 0.1% SDS, 1 mM 4-(2-aminoethyl)-benzene-sulfonylfluoride hydrochloride (AEBSF), 5 μg/ml aprotinin, 5 μg/ml leupeptin, 5 μg/ml pepstatin A, 10 mM 1,10-phenanthroline; 0.5 ml extraction buffer/well in a 6-well plate). Cell extracts were centrifuged at 21,000g for 15 min, supernatants were incubated for 1 h at 4°C with streptavidin sepharose (Pierce; 1 ml cell extract/50 μl of resin). The resin was washed three times with extraction buffer, eluted with SDS sample buffer; samples were then resolved by SDS-PAGE and transferred to a nitrocellulose membrane. ADAM12 enrichment on concanavalin A sepharose columns and immunoprecipitation were performed as described (12). Primary antibodies were: rabbit anti-ADAM12 cytoplasmic peptide (1:1000), rabbit anti-Dll1 (Santa Cruz Biotechnology, H-265, 0.2 μg/ml), or mouse anti-c-myc tag (Cell Signaling, 9E10, 1 μg/ml); secondary antibodies were horseradish peroxidase (HRP)-conjugated anti-rabbit or anti-mouse IgG.
Immunofluorescence
Cells plated on coverslips placed in a 6-well plate were transfected with wild-type or mutant forms of ADAM12. Two days later, cells were fixed with 3.7% paraformaldehyde in DPBS for 20 min and permeabilized with 0.1% Triton X-100 in DPBS for 5 min. The coverslips were incubated with anti-ADAM12 rabbit antibody (1:400 dilution) and mouse anti-KDEL antibody (1:200 dilution, Stressgen), followed by incubation with Alexa 488-conjugated anti-rabbit IgG antibody, rhodamine Red-X-conjugated anti-mouse IgG antibody, and DAPI. After washing in DPBS, coverslips were mounted on glass slides and examined by Axiovert 200 inverted fluorescent microscope.
Cell migration
The scratch motility assay was used to evaluate two-dimensional cell migration. NIH3T3 cells stably expressing the wild-type or mutant forms of ADAM12 were grown to confluence in 6-well plates. Several scratches were then made on cell monolayers using a sterile 200-μl pipette tip. Cells were rinsed three times with DPBS and placed in growth medium. The same regions of each scratch were analyzed after 0 h, 4 h, and 8 h with Axiovert 200 inverted microscope, using phase contrast optics at 10x magnification. The distance between the edges of each wounded cell monolayer was measured at 15-20 different points using AxioVision software. Experiments were repeated three times.
Results
Out of the three mutations in ADAM12 identified in a large-scale analysis of breast cancer genome, two mutations involve amino acids that are conserved between different species (D301 and G479), and one mutation involves an amino acid that is not conserved (L792; Fig. 1a). We first used two computational web-based approaches to determine a potential significance of mutating these sites. SIFT algorithm (Sorting Intolerant From Tolerant, available at http://blocks.fhcrc.org/sift/SIFT.html) predicts whether an amino acid substitution affects protein function based on sequence homology and the physical properties of amino acids (22-24). SIFT calculates a score for each possible amino acid substitution at a particular position in a protein. Scores range from 0 to 1, where 0 is damaging and 1 is neutral. The minimum cumulative score (for all possible substitutions, including no change) at any position is 1. Fig. 1b shows the results of SIFT analysis for the metalloprotease, disintegrin, and cytoplasmic domains of human ADAM12. The cumulative scores at D301 and G479 are 1, meaning that no changes are allowed and all mutations are predicted to be deleterious. In contrasts, the cumulative score at L792 is close to 9, which indicates that many substitutions are tolerated at this site. The score for the L792F mutation is 0.71, suggesting that this change should not have a significant effect on ADAM12 function.
The second algorithm, CanPredict (available at http://www.cgl.ucsf.edu/Research/genentech/canpredict/), combines the information from SIFT, the Pfam-based LogR.E-value, and the Gene Ontology Similarity Score (GOSS) metrics to distinguish cancer-associated missense mutations from common polymorphisms (25,26). The Pfam-based LogR.E-values for the D301E, G479E, and L792E mutations were 1.15, 1.21, and -0.08, respectively (as stated in ref. 27, LogR.E-value scores > 0.5 are likely to indicate deleterious changes); the GOSS score for all three mutations was 5.31. Based on the scores obtained from SIFT, Pfam-based LogR.E-value, and the GOSS metrics, the D301H and G479E mutations were predicted by the CanPredict algorithm to be likely cancer-causing, whereas the L792F mutation was classified as likely non-cancer. Similar predictions were obtained for the corresponding mutations in mouse ADAM12: D299H, G477E, and S788F. The effects of the D299H and G477E mutations on the intracellular trafficking and processing of mouse ADAM12 were then examined in the remaining part of this study.
When COS-7, CHO-K1, MCF-7, or NIH3T3 cells were transiently transfected with a plasmid encoding the wild-type ADAM12, two forms of ADAM12 were readily detected in the immunoblots: the nascent, full-length form of ~120 kDa and the mature, processed form of ~90 kDa that lacks the pro-domain and represents the catalytically active enzyme (Fig. 2a-d). In contrast, transfections with plasmids encoding the D299H and G477E ADAM12 mutants (designated D/H and G/E, respectively) produced exclusively the 120-kDa nascent form of ADAM12 and the 90-kDa processed form was not detected (Fig. 2a-d). To exclude the possibility that the difference in processing of the wild-type ADAM12 and the D/H and G/E mutants was caused by high levels of protein expression in plasmid-transfected cells, we used a retroviral expression system that resulted in more moderate levels of ADAM12 expression. NIH3T3 cells were infected with ecotrophic viruses containing the wild-type, D/H, or G/E ADAM12 genes. Two days after infection, 50-70% cells stained positively for ADAM12, and the expression level of ADAM12 in individual cells was significantly lower than in plasmidtransfected cells (result not shown). Importantly, Western blot analysis of infected cells showed that, similarly to the transfected cells, the D/H and G/E mutants were not processed (Fig. 2d, middle panel). Furthermore, the lack of processing of the D/H and G/E mutants was observed after stable expression in NIH3T3 cells, when steady state levels of protein expression were achieved (Fig. 2d, right panel). These results demonstrate that the D/H and G/E mutations block the proteolytic processing and the generation of the active, 90-kDa form of ADAM12.
The D/H and G/E mutations in ADAM12 observed in breast cancers were monoallelic (8). This suggests that another copy of the ADAM12 gene should be intact and should produce the wild-type ADAM12 protein. To examine whether the presence of the mutated forms of ADAM12 affects the processing of the wild-type form, we co-expressed both the wild-type and the mutant forms in COS-7 cells. The D/H and G/E forms contained C-terminal c-myc and 6xHis tags and migrated in SDS-PAGE gels with slightly lower mobilities than the wild-type ADAM12 that did not contain any tags. As shown in Fig. 3a, the presence of either the D/H or G/E mutant drastically diminished the amount of the 90-kDa active form of the wild-type ADAM12 in cotransfected cells, suggesting that the D/H and G/E mutants exerted a dominant-negative effect on the processing of the wild-type ADAM12. One possible mechanism of such dominant-negative effect could be an association between the wild-type and mutant forms of ADAM12. Coimmunoprecipitation experiments using anti-c-myc tag antibody revealed that the wild-type, untagged ADAM12 did indeed associated with c-myc-tagged D/H and G/E mutants (Fig. 3b).
The processing of ADAM12 is mediated by furin-like proteases and it takes place in the Golgi, after the protein leaves the endoplasmic reticulum (ER). The lack of processing of the D/H and G/E mutants suggested that these mutants might be exported from the ER less efficiently than the wild-type ADAM12. Indeed, immunofluorescence analysis of transfected NIH3T3 cells showed that while a sizeable amount of the wild-type ADAM12 was present in post-ER compartments (Fig. 4a-c), the D/H or G/E mutants were retained in the ER (Fig. 4d-i). Moreover, we observed that cells expressing either the wild-type ADAM12 or the D/H or G/E mutant frequently showed stronger ER-staining than the neighboring, non-transfected cells (for example, see Fig. 4b and 4h). Since the main proteins recognized by the antibody to stain the ER, are molecular chaperones BiP and Grp94, this result suggests that the D/H and G/E mutants might be partially misfolded, leading to the up-regulation of chaperone expression and the inability to leave the ER.
Figure 4.

The effect of D/H and G/E mutations on the intracellular localization of ADAM12. NIH3T3 cells were transfected with the wild-type (wt; a-c), D/H (d-f) or G/E (g-i) mutant forms of ADAM12. Cells were co-stained with anti-ADAM12 antibody (green), anti-KDEL antibody, an endoplasmic reticulum (ER) marker (red), and DAPI (blue). Notice that while the wild-type ADAM12 is detected both in the ER and in post-ER compartments, the D/H and G/E mutants are retained within the ER.
We next determined the effect of D/H and G/E mutations on the cell surface expression of ADAM12. NIH3T3 cells were subjected to surface biotinylation using sulfo-NHS-biotin, a membrane impermeable protein biotinylation reagent, followed by binding of the biotinylated proteins to streptavidin sepharose and detection of ADAM12 by Western blotting. In the wild-type ADAM12-expressing cells, the 90-kDa mature form was biotinylated and the nascent 120-kDa form was not (Fig. 5a). In mutant-expressing cells, no biotinylation of ADAM12 was detected, which is consistent with their lack of proteolytic processing and retention in the ER. Finally, we performed two functional tests for ADAM12 at the cell surface. First, we examined the proteolytic processing of Delta-like 1 (Dll1), a ligand for Notch receptor and a recently identified cell surface substrate for ADAM12 (12). NIH3T3 cells stably expressing the wild-type, D/H, or G/E mutant forms of ADAM12 or control NIH3T3 cells were transfected with mouse Delta-like 1 (Dll1). In NIH3T3 cells expressing the wild-type ADAM12, the extent of Dll1 processing was significantly higher than in control cells. In contrast, in cells expressing either the D/H or the G/E mutant, there was no increase in Dll1 processing (Fig. 5b). Second, we compared the effect of overexpression of the wild-type ADAM12 and ADAM12 mutants on cell migration. Our recent studies indicate that ADAM12 increases two-dimensional migration of several cell types, including NIH3T3 fibroblasts (Syta and Zolkiewska, manuscript in preparation), and the mechanism by which ADAM12 stimulates cell migration is currently under investigation in our laboratory. Here, NIH3T3 cells stably expressing the wild-type ADAM12 showed higher mobility than control cells or cells expressing the D/H or G/E mutants (Fig. 5c). These distinct effects on cell migration were not caused by different expression levels of the recombinant proteins, as both the wild-type ADAM12 and the two mutant forms were expressed at similar levels (Fig. 2d). Rather, these results suggested that proper processing of ADAM12 and its cell surface localization were required for increased cell migration. Collectively, the results in Fig. 5 demonstrated that the D/H and G/E mutations blocked the expression of a functionally active ADAM12 at the cell surface.
Figure 5.

The D/H and G/E mutations result in loss of the functional ADAM12 at the cell surface. (a) NIH3T3 cells infected with the wild-type (wt), D/H, or G/E mutant forms of mouse ADAM12 or with control (Vec) retroviruses were cell surface biotinylated. The biotinylated proteins were isolated on streptavidin sepharose and subjected to SDS-PAGE and Western blotting with anti-ADAM12 antibody. Notice that the main biotinylated form of ADAM12 corresponds to the 90-kDa processed form (asterisk) and is detected in the wild-type ADAM12-expressing cells, but not in the D/H or G/E mutant-expressing cells. (b) NIH3T3 cells stably expressing the wild-type, D/H, or G/E mutant forms of ADAM12 or control NIH3T3 cells (Vec) were transfected with mouse Delta-like 1 (Dll1). Cell extracts were subjected to Western blotting with an antibody recognizing the C-terminal part of Dll1. Notice that while the full length (FL) Dll1 is processed by the wild-type ADAM12 and the amount of the C-terminal fragment (CTF) of Dll1 is increased, the D/H and G/E mutants do not process Dll1. (c) Two-dimensional migration of control NIH3T3 cells (Vec) or NIH3T3 cells stably expressing the wild-type, D/H, or G/E mutant forms of ADAM12.
Discussion
ADAMs and ADAMTSs (ADAMs with thrombospondin motifs) emerge as important regulators of cancer progression (28,29). Human breast cancers are often associated with elevated levels of ADAM9, 12, 15, 17, and 28 (1,2,30-33). Transgenic expression of ADAM12 in breast tumors in mice promotes tumor growth (2) and overexpression of ADAM17 in human breast cancer cells increases cell proliferation and invasion (33). ADAMTS1 is up-regulated in breast tumors that develop bone metastases (34). Elevated levels of ADAMTS8 in breast cancers, together with low levels of ADAMTS15, are associated with a poor clinical prognosis (35). ADAMTS18 has been recently identified as a tumor suppressor gene that is frequently inactivated epigenetically in breast carcinomas (36). Furthermore, matrix metalloproteases (MMPs), a protease family related to ADAMs, have a well established role in breast cancer progression (37).
A genome-wide analysis of breast and colorectal cancers identified 122 genes that are frequently mutated in breast cancers and 69 genes mutated with high frequencies in colorectal cancers (8). The list of genes mutated in colorectal cancers included five ADAM-related genes, ADAM29, ADAMTS15, ADAMTS18, ADAMTSL3 (ADAMTS-like 3/punctin-2), and MMP2. ADAMTSL3, which had the highest cancer mutation prevalence (CaMP) score among these metalloproteases, has been recently found to be significantly reduced in colon cancer compared to normal colon (38). It was pointed out that if cancer-associated mutations lead to the loss of function of ADAMTSL3, then they would additionally decrease the level of ADAMTSL3 in cancer cells and this is how they may contribute towards development and progression the colorectal cancer (38).
In contrast, among the 122 genes found to be mutated with high frequencies in breast cancers, there was only one ADAM, namely ADAM12 (8). The only other gene coding for an extracellular protease was TMPRSS6 (transmembrane protease, serine 6), which is not related to ADAMs. Furthermore, only 14 genes had a higher cancer mutation prevalence score than ADAM12. High mutation frequency in the ADAM12 gene, together with a strongly up-regulated expression of ADAM12 in breast cancer (1) suggests that ADAM12 may play an important role in breast cancer progression.
Our computational, web-based analyses indicated that two out the three reported mutations in ADAM12, D301H and G479E, are most likely cancer-associated. Introduction of the corresponding mutations in mouse ADAM12 resulted in lack of the proteolytic processing of ADAM12, retention of the protein in the ER, and loss of the functional ADAM12 at the cell surface. We used two independent methods to assay ADAM12 at the cell surface, proteolytic processing of Dll1 and measurement of cell migration. The ability of ADAM12 to cleave Dll1, the cell surface localization of this reaction, and the effect of ADAM12-mediated cleavage of Dll1 on Notch signaling have been recently described (12). The role of ADAM12 in cell migration may be more complex. It was reported that ADAM12 inhibited integrin α4β1-, but not integrin α5β1-mediated migration of CHO cells on fibronectin (39). Several other reports showed that different ADAMs can either inhibit or stimulate migration of various cell types (40,41). As shown in Fig. 5c, introduction of the wild-type ADAM12 into NIH3T3 cells expressing endogenous levels of integrin receptors resulted in enhanced cell migration, and the D/H and G/E ADAM12 mutants were significantly less potent in this assay. While the mechanism of ADAM12-stimulated cell migration is currently under investigation in our laboratory, the lower ability of the D/H and G/E mutants to enhance cell migration is consistent with their impaired intracellular processing and transport to the cell surface.
All three mutations in ADAM12 found in breast cancers, including D/H and G/E, were monoallelic (8). If loss of the functional ADAM12 at the cell surface is going to be relevant to cancer progression, the D/H and G/E mutations should either lead to ADAM12 haploinsufficiency or should exert a dominant-negative effect on the wild-type ADAM12 protein that is produced in the presence of a single copy of the wild-type ADAM12 gene. Our co-expression experiments show that the D/H or the G/E mutants associate with the wild-type ADAM12 and interfere with its proteolytic processing and activation, suggesting that they indeed might exert a dominant-negative effect. Although self-association properties of ADAM12 or other ADAMs have not been extensively studied, at least two different ADAMs, ADAM1 and ADAM2, were previously shown to form heterodimers (42,43). Thus, it possible that ADAMs in general, or ADAM12 in particular, have a tendency to form multimers. It is also conceivable that the associative properties of ADAM12 are changed upon introduction of the D/H or G/E mutation, a possibility that we are currently investigating.
Loss-of-function of cancer-associated D/H and G/E ADAM12 mutations is unexpected, as it seems to contradict the in vivo studies in which overexpression of ADAM12 in breast tumors of MMTV-PyMT mice accelerated tumor progression (2). It has to be pointed out that the ADAM12 construct overexpressed in MMTV-PyMT mice lacked the cytoplasmic tail and thus certain unidentified tumor-suppressing functions of the cytoplasmic domain of ADAM12 might have been overlooked. On the other hand, knocking out the ADAM12 gene in W10 mice resulted in reduced prostate tumor size and better differentiated prostate tumors (3), which is consistent with an oncogenic function of ADAM12. Although the effect of deleting ADAM12 expression in mouse models of breast cancer is not known yet, one might predict that the development of mammary tumors will be delayed in the ADAM12-/- background.
Our findings linking breast-cancer associated mutations in ADAM12 to the lack of expression of the functional protein at the cell surface and its retention in the ER may be explained in several ways. First, it is possible that ADAM12 has a dual effect on breast cancer progression. This effect would be similar to the effect exerted, for example, by transforming growth factor β (TGF-β). TGF-β has the tumor growth-inhibiting activity at the early stage of tumor development and tumor-promoting activity during later stages of tumor progression and invasion (44,45). If this is the case for ADAM12, breast cancer-associated mutations in ADAM12, by interfering with production of the functional ADAM12, would compromise the tumor growth-inhibiting function of ADAM12 and would effectively promote tumor development. Interestingly, ADAM12 has been recently shown to interact with TGF-β type II receptor (TβRII), to stabilize the receptor, and to enhance TGF-β signaling (16). Thus, the interaction with TβRII may represent a tumor-suppressing aspect of ADAM12. Furthermore, we have recently demonstrated that ADAM12 sheds Dll1, a ligand for Notch receptor, from the cell surface and modulates the Notch signaling (12). Dll1, similar to ADAM12, is highly expressed in breast tumors (46). Although Notch signaling stimulates mammary tumorigenesis (47,48), the Notch pathway has also been reported to have tumor-suppressive functions (49,50). In consequence, the effect of Dll1 shedding on breast cancer progression may be complex and it may also depend on tumor stage.
Second, it is possible that the main tumor-promoting activity of ADAM12 takes place in the ER. The wild-type ADAM12 is found both at the cell surface and the ER, and the D/H and G/E mutants localize predominantly to the ER. The loss of ADAM12 at the cell surface as a result of cancer-associated mutations is obviously very different from the loss caused by gene knockout, where ADAM12 protein is never expressed. If the presence of ADAM12 in the ER promotes tumor growth, then the retention of the D/H and G/E mutants in the ER may augment this effect. One possible consequence of accumulation of ADAM12 in the ER might be induction of the ER stress and the unfolded protein response (UPR). The UPR has been observed in certain tumors, where it seems to play a protective role during tumor development and to alter the sensitivity of tumor cells to chemotherapeutic agents (51,52). Future studies on the molecular and cellular consequences of mutating ADAM12 should shed more light on the role of ADAM12 in tumorigenesis and should help develop therapeutic interventions in breast cancer based on the functionality of ADAM12.
Acknowledgments
This work was supported by NIH grants GM065528 to AZ. This is contribution 08-63-J from the Kansas Agricultural Experiment Station.
Abbreviations
- ADAM
protein containing a disintegrin and metalloprotease
- Dll1
Delta-like 1
- DMEM
Dulbecco's modified Eagle's medium
- DPBS
Dulbecco's phosphate-buffered saline
- FBS
fetal bovine serum
- AEBSF
4-(2-aminoethyl)benzenesulfonyl fluoride hydrochloride
- sulfo-NHS
N-hydroxysulfosuccinimidobiotin
- DAPI
4',6-diamidino-2-phenylindole
Footnotes
Novelty: This is the first time examination of the effect of breast cancer-associated mutations in ADAM12 on the intracellular processing and localization of the protein.
Impact: These studies may help develop therapeutic interventions in breast cancer based on functionality of ADAM12 and/or related signaling pathways.
References
- 1.Iba K, Albrechtsen R, Gilpin BJ, Loechel F, Wewer UM. Cysteine-rich domain of human ADAM 12 (meltrin α) supports tumor cell adhesion. Am J Pathol. 1999;154:1489–501. doi: 10.1016/s0002-9440(10)65403-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kveiborg M, Frohlich C, Albrechtsen R, Tischler V, Dietrich N, Holck P, Kronqvist P, Rank F, Mercurio AM, Wewer UM. A role for ADAM12 in breast tumor progression and stromal cell apoptosis. Cancer Res. 2005;65:4754–61. doi: 10.1158/0008-5472.CAN-05-0262. [DOI] [PubMed] [Google Scholar]
- 3.Peduto L, Reuter VE, Sehara-Fujisawa A, Shaffer DR, Scher HI, Blobel CP. ADAM12 is highly expressed in carcinoma-associated stroma and is required for mouse prostate tumor progression. Oncogene. 2006;25:5462–6. doi: 10.1038/sj.onc.1209536. [DOI] [PubMed] [Google Scholar]
- 4.Le Pabic H, Bonnier D, Wewer UM, Coutand A, Musso O, Baffet G, Clement B, Theret N. ADAM12 in human liver cancers: TGF-β-regulated expression in stellate cells is associated with matrix remodeling. Hepatology. 2003;37:1056–66. doi: 10.1053/jhep.2003.50205. [DOI] [PubMed] [Google Scholar]
- 5.Carl-McGrath S, Lendeckel U, Ebert M, Roessner A, Rocken C. The disintegrinmetalloproteinases ADAM9, ADAM12, and ADAM15 are upregulated in gastric cancer. Int J Oncol. 2005;26:17–24. [PubMed] [Google Scholar]
- 6.Frohlich C, Albrechtsen R, Dyrskjot L, Rudkjaer L, Orntoft TF, Wewer UM. Molecular profiling of ADAM12 in human bladder cancer. Clin Cancer Res. 2006;12:7359–68. doi: 10.1158/1078-0432.CCR-06-1066. [DOI] [PubMed] [Google Scholar]
- 7.Kodama T, Ikeda E, Okada A, Ohtsuka T, Shimoda M, Shiomi T, Yoshida K, Nakada M, Ohuchi E, Okada Y. ADAM12 is selectively overexpressed in human glioblastomas and is associated with glioblastoma cell proliferation and shedding of heparin-binding epidermal growth factor. Am J Pathol. 2004;165:1743–53. doi: 10.1016/S0002-9440(10)63429-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sjöblom T, Jones S, Wood LD, Parsons DW, Lin J, Barber TD, Mandelker D, Leary RJ, Ptak J, Silliman N, Szabo S, Buckhaults P, et al. The consensus coding sequences of human breast and colorectal cancers. Science. 2006;314:268–74. doi: 10.1126/science.1133427. [DOI] [PubMed] [Google Scholar]
- 9.Loechel F, Fox JW, Murphy G, Albrechtsen R, Wewer UM. ADAM 12-S cleaves IGFBP-3 and IGFBP-5 and is inhibited by TIMP-3. Biochem Biophys Res Commun. 2000;278:511–5. doi: 10.1006/bbrc.2000.3835. [DOI] [PubMed] [Google Scholar]
- 10.Horiuchi K, Le Gall S, Schulte M, Yamaguchi T, Reiss K, Murphy G, Toyama Y, Hartmann D, Saftig P, Blobel CP. Substrate selectivity of epidermal growth factor-receptor ligand sheddases and their regulation by phorbol esters and calcium influx. Mol Biol Cell. 2007;18:176–88. doi: 10.1091/mbc.E06-01-0014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ito N, Nomura S, Iwase A, Ito T, Kikkawa F, Tsujimoto M, Ishiura S, Mizutani S. ADAMs, a disintegrin and metalloproteinases, mediate shedding of oxytocinase. Biochem Biophys Res Commun. 2004;314:1008–13. doi: 10.1016/j.bbrc.2003.12.183. [DOI] [PubMed] [Google Scholar]
- 12.Dyczynska E, Sun D, Yi H, Sehara-Fujisawa A, Blobel CP, Zolkiewska A. Proteolytic processing of Delta-like 1 by ADAM proteases. J Biol Chem. 2007;282:436–44. doi: 10.1074/jbc.M605451200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Iba K, Albrechtsen R, Gilpin B, Frohlich C, Loechel F, Zolkiewska A, Ishiguro K, Kojima T, Liu W, Langford JK, Sanderson RD, Brakebusch C, Fassler R, Wewer UM. The cysteine-rich domain of human ADAM 12 supports cell adhesion through syndecans and triggers signaling events that lead to β1 integrin-dependent cell spreading. J Cell Biol. 2000;149:1143–56. doi: 10.1083/jcb.149.5.1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Eto K, Puzon-McLaughlin W, Sheppard D, Sehara-Fujisawa A, Zhang XP, Takada Y. RGD-independent binding of integrin α9β1 to the ADAM-12 and -15 disintegrin domains mediates cell-cell interaction. J Biol Chem. 2000;275:34922–30. doi: 10.1074/jbc.M001953200. [DOI] [PubMed] [Google Scholar]
- 15.Zhao Z, Gruszczynska-Biegala J, Cheuvront T, Yi H, von der MH, von der MK, Kaufman SJ, Zolkiewska A. Interaction of the disintegrin and cysteine-rich domains of ADAM12 with integrin α7β1. Exp Cell Res. 2004;298:28–37. doi: 10.1016/j.yexcr.2004.04.005. [DOI] [PubMed] [Google Scholar]
- 16.Atfi A, Dumont E, Colland F, Bonnier D, L'Helgoualc'h A, Prunier C, Ferrand N, Clement B, Wewer UM, Theret N. The disintegrin and metalloproteinase ADAM12 contributes to TGF-β signaling through interaction with the type II receptor. J Cell Biol. 2007;178:201–8. doi: 10.1083/jcb.200612046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kang Q, Cao Y, Zolkiewska A. Metalloprotease-disintegrin ADAM 12 binds to the SH3 domain of Src and activates Src tyrosine kinase in C2C12 cells. Biochem J. 2000;352(Pt 3):883–92. [PMC free article] [PubMed] [Google Scholar]
- 18.Kang Q, Cao Y, Zolkiewska A. Direct interaction between the cytoplasmic tail of ADAM 12 and the Src homology 3 domain of p85α activates phosphatidylinositol 3-kinase in C2C12 cells. J Biol Chem. 2001;276:24466–72. doi: 10.1074/jbc.M101162200. [DOI] [PubMed] [Google Scholar]
- 19.Galliano MF, Huet C, Frygelius J, Polgren A, Wewer UM, Engvall E. Binding of ADAM12, a marker of skeletal muscle regeneration, to the muscle-specific actin-binding protein, α-actinin-2, is required for myoblast fusion. J Biol Chem. 2000;275:13933–9. doi: 10.1074/jbc.275.18.13933. [DOI] [PubMed] [Google Scholar]
- 20.Cao Y, Kang Q, Zolkiewska A. Metalloprotease-disintegrin ADAM 12 interacts with αactinin-1. Biochem J. 2001;357:353–61. doi: 10.1042/0264-6021:3570353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Abram CL, Seals DF, Pass I, Salinsky D, Maurer L, Roth TM, Courtneidge SA. The adaptor protein fish associates with members of the ADAMs family and localizes to podosomes of Src-transformed cells. J Biol Chem. 2003;278:16844–51. doi: 10.1074/jbc.M300267200. [DOI] [PubMed] [Google Scholar]
- 22.Ng PC, Henikoff S. Predicting deleterious amino acid substitutions. Genome Res. 2001;11:863–74. doi: 10.1101/gr.176601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ng PC, Henikoff S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31:3812–4. doi: 10.1093/nar/gkg509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ng PC, Henikoff S. Predicting the effects of amino acid substitutions on protein function. Annu Rev Genomics Hum Genet. 2006;7:61–80. doi: 10.1146/annurev.genom.7.080505.115630. [DOI] [PubMed] [Google Scholar]
- 25.Kaminker JS, Zhang Y, Waugh A, Haverty PM, Peters B, Sebisanovic D, Stinson J, Forrest WF, Bazan JF, Seshagiri S, Zhang Z. Distinguishing cancer-associated missense mutations from common polymorphisms. Cancer Res. 2007;67:465–73. doi: 10.1158/0008-5472.CAN-06-1736. [DOI] [PubMed] [Google Scholar]
- 26.Kaminker JS, Zhang Y, Watanabe C, Zhang Z. CanPredict: a computational tool for predicting cancer-associated missense mutations. Nucleic Acids Res. 2007;35:W595–W598. doi: 10.1093/nar/gkm405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Clifford RJ, Edmonson MN, Nguyen C, Buetow KH. Large-scale analysis of non-synonymous coding region single nucleotide polymorphisms. Bioinformatics. 2004;20:1006–14. doi: 10.1093/bioinformatics/bth029. [DOI] [PubMed] [Google Scholar]
- 28.Rocks N, Paulissen G, El Hour M, Quesada F, Crahay C, Gueders M, Foidart JM, Noel A, Cataldo D. Emerging roles of ADAM and ADAMTS metalloproteinases in cancer. Biochimie. doi: 10.1016/j.biochi.2007.08.008. in press. [DOI] [PubMed] [Google Scholar]
- 29.Mochizuki S, Okada Y. ADAMs in cancer cell proliferation and progression. Cancer Sci. 2007;98:621–8. doi: 10.1111/j.1349-7006.2007.00434.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kuefer R, Day KC, Kleer CG, Sabel MS, Hofer MD, Varambally S, Zorn CS, Chinnaiyan AM, Rubin MA, Day ML. ADAM15 disintegrin is associated with aggressive prostate and breast cancer disease. Neoplasia. 2006;8:319–29. doi: 10.1593/neo.05682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lendeckel U, Kohl J, Arndt M, Carl-McGrath S, Donat H, Rocken C. Increased expression of ADAM family members in human breast cancer and breast cancer cell lines. J Cancer Res Clin Oncol. 2005;131:41–8. doi: 10.1007/s00432-004-0619-y. [DOI] [PubMed] [Google Scholar]
- 32.Mitsui Y, Mochizuki S, Kodama T, Shimoda M, Ohtsuka T, Shiomi T, Chijiiwa M, Ikeda T, Kitajima M, Okada Y. ADAM28 is overexpressed in human breast carcinomas: implications for carcinoma cell proliferation through cleavage of insulin-like growth factor binding protein-3. Cancer Res. 2006;66:9913–20. doi: 10.1158/0008-5472.CAN-06-0377. [DOI] [PubMed] [Google Scholar]
- 33.McGowan PM, Ryan BM, Hill AD, McDermott E, O'Higgins N, Duffy MJ. ADAM-17 expression in breast cancer correlates with variables of tumor progression. Clin Cancer Res. 2007;13:2335–43. doi: 10.1158/1078-0432.CCR-06-2092. [DOI] [PubMed] [Google Scholar]
- 34.Minn AJ, Kang Y, Serganova I, Gupta GP, Giri DD, Doubrovin M, Ponomarev V, Gerald WL, Blasberg R, Massague J. Distinct organ-specific metastatic potential of individual breast cancer cells and primary tumors. J Clin Invest. 2005;115:44–55. doi: 10.1172/JCI22320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Porter S, Span PN, Sweep FC, Tjan-Heijnen VC, Pennington CJ, Pedersen TX, Johnsen M, Lund LR, Romer J, Edwards DR. ADAMTS8 and ADAMTS15 expression predicts survival in human breast carcinoma. Int J Cancer. 2006;118:1241–7. doi: 10.1002/ijc.21476. [DOI] [PubMed] [Google Scholar]
- 36.Jin H, Wang X, Ying J, Wong AHY, Li H, Lee KY, Srivastava G, Chan ATC, Yeo W, Ma BBY, Putti TC, Lung ML, Shen ZY, Xu LY, Langford C, Tao Q. Epigenetic identification of ADAMTS18 as a novel 16q23.1 tumor suppressor frequently silenced in esophageal, nasopharyngeal and multiple other carcinomas. Oncogene. 2007 doi: 10.1038/sj.onc.1210559. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Folgueras AR, Pendás AM, Sánchez LM, López-Otín C. Matrix metalloproteinases in cancer: from new functions to improved inhibition strategies. Int J Dev Biol. 2004;48:411–24. doi: 10.1387/ijdb.041811af. [DOI] [PubMed] [Google Scholar]
- 38.Koo BH, Hurskainen T, Mielke K, Aung PP, Casey G, Autio-Harmainen H, Apte SS. ADAMTSL3/punctin-2, a gene frequently mutated in colorectal tumors, is widely expressed in normal and malignant epithelial cells, vascular endothelial cells and other cell types, and its mRNA is reduced in colon cancer. Int J Cancer. 2007;121:1710–6. doi: 10.1002/ijc.22882. [DOI] [PubMed] [Google Scholar]
- 39.Huang J, Bridges LC, White JM. Selective modulation of integrin-mediated cell migration by distinct ADAM family members. Mol Biol Cell. 2005;16:4982–91. doi: 10.1091/mbc.E05-03-0258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Alfandari D, Cousin H, Gaultier A, Smith K, White JM, Darribere T, DeSimone DW. Xenopus ADAM 13 is a metalloprotease required for cranial neural crest-cell migration. Curr Biol. 2001;11:918–30. doi: 10.1016/s0960-9822(01)00263-9. [DOI] [PubMed] [Google Scholar]
- 41.Arribas J, Bech-Serra JJ, Santiago-Josefat B. ADAMs, cell migration and cancer. Cancer Metastasis Rev. 2006;25:57–68. doi: 10.1007/s10555-006-7889-6. [DOI] [PubMed] [Google Scholar]
- 42.Blobel CP, Wolfsberg TG, Turck CW, Myles DG, Primakoff P, White JM. A potential fusion peptide and an integrin ligand domain in a protein active in sperm-egg fusion. Nature. 1992;356:248–52. doi: 10.1038/356248a0. [DOI] [PubMed] [Google Scholar]
- 43.Cho C, Ge H, Branciforte D, Primakoff P, Myles DG. Analysis of mouse fertilin in wild-type and fertilin β-/- sperm: Evidence for C-terminal modification, α/β dimerization, and lack of essential role of fertilin α in sperm-egg fusion. Developmental Biology. 2000;222:289–95. doi: 10.1006/dbio.2000.9703. [DOI] [PubMed] [Google Scholar]
- 44.Bierie B, Moses HL. Tumour microenvironment: TGFβ: the molecular Jekyll and Hyde of cancer. Nat Rev Cancer. 2006;6:506–20. doi: 10.1038/nrc1926. [DOI] [PubMed] [Google Scholar]
- 45.Siegel PM, Massague J. Cytostatic and apoptotic actions of TGF-β in homeostasis and cancer. Nat Rev Cancer. 2003;3:807–21. doi: 10.1038/nrc1208. [DOI] [PubMed] [Google Scholar]
- 46.Reedijk M, Odorcic S, Chang L, Zhang H, Miller N, McCready DR, Lockwood G, Egan SE. High-level coexpression of JAG1 and NOTCH1 is observed in human breast cancer and is associated with poor overall survival. Cancer Res. 2005;65:8530–7. doi: 10.1158/0008-5472.CAN-05-1069. [DOI] [PubMed] [Google Scholar]
- 47.Stylianou S, Clarke RB, Brennan K. Aberrant activation of notch signaling in human breast cancer. Cancer Res. 2006;66:1517–25. doi: 10.1158/0008-5472.CAN-05-3054. [DOI] [PubMed] [Google Scholar]
- 48.Leong KG, Karsan A. Recent insights into the role of Notch signaling in tumorigenesis. Blood. 2006;107:2223–33. doi: 10.1182/blood-2005-08-3329. [DOI] [PubMed] [Google Scholar]
- 49.Niimi H, Pardali K, Vanlandewijck M, Heldin CH, Moustakas A. Notch signaling is necessary for epithelial growth arrest by TGF-β. J Cell Biol. 2007;176:695–707. doi: 10.1083/jcb.200612129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Parr C, Watkins G, Jiang WG. The possible correlation of Notch-1 and Notch-2 with clinical outcome and tumour clinicopathological parameters in human breast cancer. Int J Mol Med. 2004;14:779–86. doi: 10.3892/ijmm.14.5.779. [DOI] [PubMed] [Google Scholar]
- 51.Ma Y, Hendershot LM. The role of the unfolded protein response in tumour development: friend or foe? Nat Rev Cancer. 2004;4:966–77. doi: 10.1038/nrc1505. [DOI] [PubMed] [Google Scholar]
- 52.Zhao L, Ackerman SL. Endoplasmic reticulum stress in health and disease. Curr Opin Cell Biol. 2006;18:444–52. doi: 10.1016/j.ceb.2006.06.005. [DOI] [PubMed] [Google Scholar]