

Abstract
Liver X receptors (LXRs) are important regulators of cholesterol and lipid metabolism. LXR agonists have been shown to limit the cellular cholesterol content by inducing reverse cholesterol transport, increasing bile acid production, and inhibiting intestinal cholesterol absorption. Most of them, however, also increase lipogenesis via sterol regulatory element-binding protein-1c (SREBP1c) and carbohydrate response element-binding protein activation resulting in hypertriglyceridemia and liver steatosis. We report on the antiatherogenic properties of the steroidal liver X receptor agonist N,N-dimethyl-3β-hydroxy-cholenamide (DMHCA) in apolipoprotein E (apoE)-deficient mice. Long-term administration of DMHCA (11 weeks) significantly reduced lesion formation in male and female apoE-null mice. Notably, DMHCA neither increased hepatic triglyceride (TG) levels in male nor female apoE-deficient mice. ATP binding cassette transporter A1 and G1 and cholesterol 7α-hydroxylase mRNA abundances were increased, whereas SREBP1c mRNA expression was unchanged in liver, and even decreased in macrophages and intestine. Short-term treatment revealed even higher changes on mRNA regulation. Our data provide evidence that DMHCA is a strong candidate as therapeutic agent for the treatment or prevention of atherosclerosis, circumventing the negative side effects of other LXR agonists.
Keywords: atherogenesis, liver X receptor, cholesterol catabolism, ABC transporter, gene expression
Nuclear liver X receptors (LXRs) are involved in the control of cholesterol and lipid metabolism. LXRα (NR1H3) and LXRβ (NR1H2) are sterol sensors that bind oxysterols to act as a transcriptional switch for the coordinated regulation of genes involved in cellular cholesterol homeostasis, cholesterol transport, catabolism, and absorption (1). In peripheral cells such as macrophages, LXRs are likely to coordinate a physiological response to cholesterol loading by regulating the transcription of several genes involved in cholesterol efflux and catabolism, including ATP-binding cassette (ABC)A1 and G1 (2–6). In the intestine, LXR ligands were shown to reduce dietary cholesterol absorption by ABCA1, ABCG1, and ABCG5/G8 activation (3, 7, 8). In the liver, LXRs apparently regulate cholesterol, fatty acid, and triglyceride (TG) metabolism. This latter effect is partly mediated by sterol regulatory element-binding protein-1c (SREBP1c) activation (9, 10) and the upregulation of its downstream target genes [e.g., lipoprotein lipase (11), fatty acid synthase (FAS) (12), and stearoyl-CoA desaturase 1 (13)]. Additionally, carbohydrate response element-binding protein (ChREBP) is activated by LXR (14) and enhances hepatic fatty acid synthesis. In rodents but not in humans, LXRs enable bile acid synthesis by activation of cholesterol 7α-hydroxylase (CYP7A1) (15). Recent publications in atherosclerotic mouse models have shown that LXR activation by GW3965 and T0901317, two synthetic nonsteroidal LXR ligands, results in decreased atherosclerosis (16, 17). Unfortunately, the concomitant induction of lipogenic genes leads to hypertriglyceridemia and liver steatosis, which is an undesirable effect of most LXR agonists.
N,N-dimethyl-3β-hydroxy-cholenamide (DMHCA) has been identified as a potent synthetic steroidal LXR activator in vitro and in vivo (18). It induces ABCA1 expression in macrophages and liver, but has only negligible effects on hepatic SREBP1c activation (approximately one-tenth of that observed for either T0901317 or GW3965) (18). These properties give DMHCA a novel and selective in vivo profile.
In the present study, we have evaluated the effects of different doses and feeding periods of DMHCA in normal chow and Western type diet (WTD) on lipid parameters and gene regulation in wild-type mice. Furthermore, we determined whether DMHCA administration to apolipoprotein E (apoE)-deficient mice had an impact on atherosclerotic plaque formation. We found that DMHCA reduced atherosclerosis in male and female apoE-null mice without the development of hepatic steatosis and hypertriglyceridemia. These observations suggest that DMHCA may represent a promising therapeutic agent for intervention in atherosclerosis.
MATERIALS AND METHODS
Animals and diets
Animal experiments were performed in accordance with the standards established by the Austrian Federal Ministry of Science and Research, Division of Genetic Engineering and Animal Experiments (Vienna, Austria). C57Bl/6 (Himberg, Austria) and apoE-deficient mice on a C57Bl/6 background (Charles River WIGA GesmbH, Sulzfeld, Germany) were maintained in a clean environment on a regular light-dark cycle (14 h light, 10 h dark). Before the initiation of the corresponding diets mice were kept on a standard laboratory chow diet. Male C57Bl/6 mice were fed chow diet or Western type diet (WTD; TD88137 mod. containing 21% fat and 0.2% cholesterol, ssniff, Soest, Germany and Harlan Teklad, Madison, WI) ± DMHCA or T0901317 (80 mg/kg body weight/day) (Cayman Chemicals, Ann Arbor, MI) for 4 or 15 days. To study atherosclerotic lesion formation, male and female apoE-deficient mice were fed ad libitum the WTD ± DMHCA (8 mg/kg body weight/day) for 11 weeks. To elucidate gene regulation by DMHCA in apoE-null mice in short time experiments, male mice were fed chow diet ± DMHCA (80 mg/kg body weight/day) for 4 days or WTD ± DMHCA (8 mg/kg body weight/day) for 15 days. Diets were supplemented with the respective LXR ligand at a level sufficient to provide the appropriate mg/kg food dose on consumption of a 5 g diet by a 25 g mouse per day. Body weight and food intake were monitored regularly. DMHCA was synthesized as described (19) and/or was obtained from Dr. E. M. Quinet (18). Purity was checked by thin layer chromatography and 1H-NMR.
Preparation of histological sections and lesion analysis
For analyses of atherosclerotic lesions at the aortic root, the upper two-thirds of the heart were fixed in 4% formaldehyde, embedded in tissue medium (Tissue-Tek O.C.T, Sanova Pharma GesmbH Diagnostik, Vienna, Austria), and frozen at −20°C. After approximately 600 μm, 8 μm cryosections of the aortic root were cut and lipid-rich regions were stained with Oil Red O (Sigma, Vienna, Austria) and counterstained with hemotoxylin (Richard-Allen Scientific, Kalamazoo, MI). Macrophages were detected with monoclonal rat anti-mouse antibody MOMA-2 (Acris, Hiddenhausen, Germany). Tissue specimens were fixed with acetone for 10 min at room temperature (RT). The slides were ultraviolet blocked and the sections were incubated with MOMA-2 antibody (1:600) for 30 min at RT. Subsequently, the sections were incubated with a biotinylated polyclonal rabbit anti-rat IgG (1:75, Dako Österreich GmbH, Vienna, Austria) for another 30 min at RT. The substrate chromogen AEC (Dako) was applied for 10 min at RT. The sections were counterstained with hemotoxylin. Images were taken with ScanScope T3 whole slide scanner (Aperio Technologies, Bristol, UK). Mean lesion area at the tricuspid valves was analyzed using Adobe Photoshop.
For en face analysis, apoE-deficient mice were euthanized using 300 μl of Nembutal (diluted 1:5) and the heart was perfused for 10 min with PBS and for 15 min with 4% formaldehyde. Then the aorta was dissected, opened longitudinally from the heart to the iliac arteries, and stained with Oil Red O. Images were analyzed with Adobe Photoshop and a software algorithm which had been programmed with Interactive Data Language (ITT Visual Information Solutions, Boulder, CO). Briefly, the amount of plaques was calculated by the ratio Aplaque/Atotal after segmentation steps of the images with background and needle separation. The extent of lesion area is expressed as the percentage of the total area of aorta section covered by the lesion. Statistical analysis of the plaque area was performed using the statistic program SPSS 14.0, applying a Student's t-test.
Cell fractionation and immunoblotting
Nuclear extraction (20) and protein quantitation were performed as described (21). Aliquots (100 μg of protein) were separated by SDS-PAGE and blotted onto nitrocellulose membranes. For immunoblot analysis, SREBP1 antibody (Santa Cruz Biotechnology, Heidelberg, Germany) (1:1,000) was visualized with horseradish peroxidase-conjugated goat anti-rabbit IgG (1:2,000, Dako Österreich GmbH, Vienna, Austria) using the Enhanced Chemiluminescence (ECL) Western Blotting Detection System Kit (Amersham Biosciences, GE Healthcare Europe GmbH, Vienna, Austria). Gels were calibrated with the Sigma marker wide range (Sigma, Vienna, Austria). Membranes were exposed to ECL Hyperfilm (Amersham Biosciences).
Plasma and hepatic lipid parameters
Blood was collected by retro orbital bleeding and EDTA-plasma was prepared within 20 min. Plasma TG (DiaSys, Holzheim, Germany), total cholesterol (TC) (Greiner Diagnostics AG, Langenthal, Switzerland), HDL cholesterol (Technoclone GmbH, Vienna, Austria), alanine aminotransferase (ALT) (Roche Diagnostics, Mannheim, Germany) and aspartate aminotransferase (AST) (Thermo Electron Corporation, Louisville, CO) concentrations were measured enzymatically. To determine hepatic lipid contents, total lipids were extracted from livers and lipid parameters were determined using above mentioned kits. For analysis of hepatic TG-associated fatty acids lipids from livers were extracted and separated by TLC. The TG band was scraped, methylated, and analyzed by gas-liquid chromatography using heptadecanoic acid as internal standard (22).
Lipoprotein profile
Plasma samples of seven overnight fasted female apoE-deficient mice fed WTD plus DMHCA for 11 weeks were pooled and compared with those of seven apoE-deficient mice fed WTD (control). Lipoproteins were isolated by fast protein liquid chromatography (FPLC) on a Pharmacia FPLC system (Pfizer Pharma, Karlsruhe, Germany) equipped with a Superose 6 column (Amersham Biosciences, Piscataway, NJ). Two hundred μl pooled plasma samples were diluted, subjected to FPLC analysis and lipoproteins were eluted with 10 mM Tris-HCl, 1 mM EDTA, 0.9% NaCl, and 0.02% NaN3 (pH 7.4). Fractions of 0.5 ml each were collected and TG and TC concentrations were assayed enzymatically using above mentioned kit. To enhance sensitivity, reaction buffers were supplemented by the addition of sodium 3,5-dichloro-2-hydroxy-benzenesulfonate (Sigma-Aldrich, Vienna, Austria).
Cell culture
Thioglycollate-elicited peritoneal macrophages (MPM) were isolated with 10 ml PBS from mice 3 days after peritoneal injection of 3 ml 3% thioglycollate medium. MPM were centrifuged, washed with PBS, and cultured in 75 cm2 flasks in DMEM (Gibco, Invitrogen, Lofer, Austria) supplemented with 10% FCS, 1% L-glutamine, and 1% streptomycin/penicillin under standard cell culture conditions (37°C, 5% CO2) for 2 h. Thereafter, nonadherent cells were removed and MPM were cultured in the same medium overnight. Macrophage differentiation to foam cells (from C57Bl/6 mice) was performed by incubation of MPM with 50 μg/ml aggregated LDL for 24 h in the absence or presence of 2.5 μM DMHCA.
RNA isolation and quantitative real-time PCR
Total RNA from mouse tissues was isolated using the Trizol procedure according to the manufacturer's protocol (Invitrogen, Lofer, Austria). Total RNA from cells was isolated using RNeasy Mini Kit (Qiagen, Vienna, Austria). Quantitative gene expression analysis was performed on a LightCycler 480 (Roche Diagnostics, Mannheim, Germany) using the Quantifast™ SYBR®GREEN PCR Kit (Qiagen). For RNA quantification, 1–2 μg of total RNA were reverse transcribed according to the manufacturer's instructions using random hexamer primers (Finnzymes, Espoo, Finland). In general, 6 ng template cDNA was used for each real time PCR. PCR primers used for real-time PCR (see supplementary information) were designed with primer designer version 2.0.
Melting curve analysis was performed to ensure that a single PCR product was amplified and no primer dimers were generated. PCR efficiency of each transcript was determined using four dilutions (1:5, 1:25, 1:125, 1:625) in triplicate of a pool of all available cDNAs of one experiment. To confirm accuracy and reproducibility a minimum of three samples per condition were measured in triplicate. Crossing points were determined by Second Derivate Maximum Method and PCR efficiencies were calculated from the slope, according to the established equation E = 10[-1/slope]. Data calculations and determination of statistical parameters were performed with the public domain program relative expression software tool REST (http://www.gene-quantification.com/download.html) using a pair-wise fixed reallocation randomization test (23). Data are displayed as expression ratios normalized to a reference gene. Initially, amplifications of murine cyclophilin A were performed as internal controls for variations in mRNA amounts. In the liver, cyclophilin A expression varied between mouse genotypes and therefore hypoxanthine guanine phosphoribosyl transferase (HPRT) was used as reference gene. In macrophages, aorta, and ileum data are displayed as expression ratios normalized to cyclophilin A as reference gene.
Statistics
Statistical analyses in experiments except real-time PCR analyses (see above) were performed using the Student's t-test. Data are expressed as mean ± SD. * P < 0.05; ** P ≤ 0.01; *** P ≤ 0.001.
RESULTS
DMHCA increases ABCA1 mRNA levels in macrophages and foam cells
MPM isolated from C57Bl/6 mice and foam cells were incubated in the absence or presence of 2.5 μM DMHCA for 24 h. As expected, DMHCA was found to stimulate ABCA1 mRNA expression in MPM and in aggregated LDL-laden foam cells in vitro by 7.0- and 4.8-fold, respectively, compared with untreated macrophages and foam cells (Fig. 1).
Fig. 1.
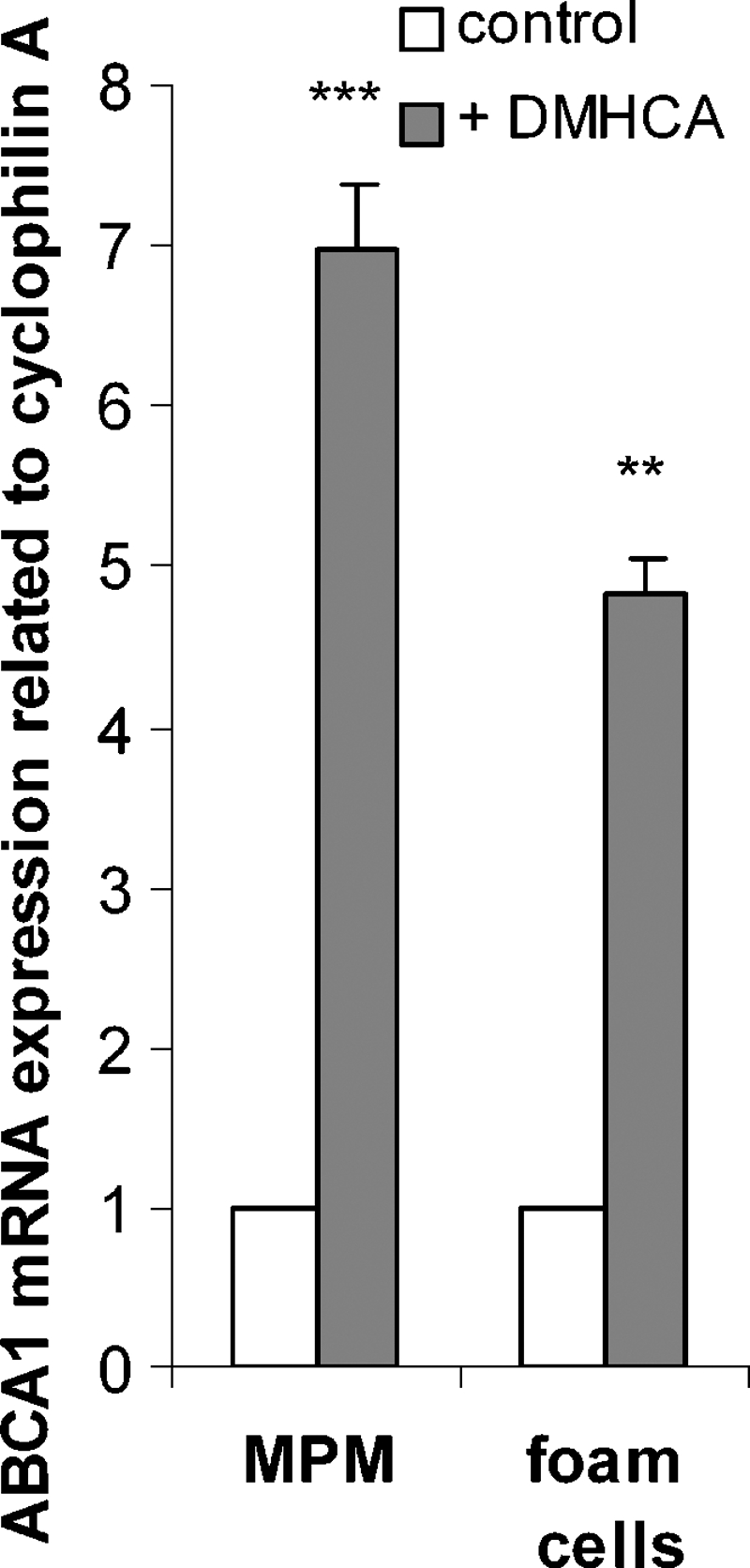
N,N-dimethyl-3β-hydroxy-cholenamide (DMHCA) induces ATP-binding cassette (ABC)A1 mRNA expression in macrophages and foam cells. Peritoneal macrophages (MPM) from C57Bl/6 mice were isolated and incubated ± 50 μg/ml aggregated LDL for 24 h in the absence or presence of 2.5 μM DMHCA. ABCA1 mRNA expression ratio in MPM and foam cells plus DMHCA including cyclophilin A normalization was calculated by pairwise fixed reallocation test relative to control MPM and foam cells (arbitrarily set to 1). Data are expressed as mean ± SD. ** P ≤ 0.01; *** P ≤ 0.001.
DMHCA does not induce liver damage or hypertriglyceridemia in wild-type mice
To study possible effects of a short-term treatment, C57Bl/6 mice were fed chow diet containing a high dose of either DMHCA or T0901317 (80 mg/kg body weight/day) for 4 days. Although all mice ate comparable amounts of food and showed similar body weights, liver weights were significantly increased in T0901317-fed animals (1.54 ± 0.24 g) when compared with controls (1.12 ± 0.12 g) (Fig. 2A). Macroscopically, administration of T0901317 resulted in a yellow-brown change in color of the liver and moderate to severe steatosis (Fig. 2A). This effect was also confirmed by measuring the TG concentration, which was significantly increased by 2.4-fold when compared with controls (Fig. 2, inset). In addition, plasma alanine aminotransferase (ALT) concentration was increased (241 ± 95 U/l) indicating liver damage following treatment with T0901317 (Fig. 2, inset). Furthermore, T0901317 treatment significantly enhanced SREBP1c mRNA levels by 4.4-fold (Fig. 2B). There were no differences in the content of the precursor SREBP1 protein between the different diets, however, T0901317 markedly increased mature, transcriptionally active hepatic nuclear SREBP1 (nSREBP1) protein by 2.9-fold (Fig. 2C). In contrast, liver weight remained unchanged (1.15 ± 0.04 g) and the liver color appeared normal in DMHCA-fed mice (Fig. 2A). Oil Red O staining of liver specimens from DMHCA-treated animals revealed minimal steatosis (Fig. 2A) with slightly increased hepatic TG levels (24.5 ± 4.8 mg/g) when compared with controls (19.7 ± 1.6 mg/g) (Fig. 2, inset). In accordance, DMHCA treatment resulted in only 1.6-fold increased SREBP1c mRNA (Fig. 2B) and there were no changes in nSREBP1 protein levels (Fig. 2C). Plasma ALT levels were not significantly altered (Fig. 2, inset). These data clearly demonstrate that T0901317 had a profound effect on hepatic fat content with a drastic increase in plasma ALT levels after 4 days of feeding, while DMHCA only mildly increased hepatic TG concentrations with no detectable liver damage.
Fig. 2.

Liver analyses of C57Bl/6 mice fed chow diet ± T0901317 or DMHCA (80 mg/kg body weight/day) for 4 days. A: Livers were weighed and cryo-sections were stained with Oil Red O and hemotoxylin. Original magnification: 100× (top row), 600× (bottom row). Real-time PCR analysis of sterol regulatory element-binding protein-1c (SREBP1c) mRNA expression (B) and Western blot analysis of precursor (p) and mature (n) SREBP1 expression (C). Data represent the mean values ± SD. * P < 0.05; ** P ≤ 0.01; *** P ≤ 0.001.
To check whether a treatment longer than 4 days or WTD affects lipid parameters, male C57Bl/6 mice were fed chow or WTD ± 80 mg DMHCA/kg body weight/day for 15 days. Plasma TG concentrations were not significantly altered by DMHCA on both diets suggesting that a high dose of DMHCA fed for more than 2 weeks does not induce hypertriglyceridemia in wild-type animals (Table 1). Interestingly, plasma TC levels were significantly reduced by 32% in mice fed chow diet plus DMHCA but remained unchanged in mice fed WTD plus DMHCA when compared with controls. Hepatic TC concentrations were markedly decreased on both diets by DMHCA (54% and 56%, respectively). In contrast, hepatic TG levels were not changed by DMHCA in WTD, but were significantly increased by 2.4-fold by DMHCA in chow diet. Plasma AST levels were slightly but not significantly increased on chow diet plus DMHCA and unaltered by DMHCA in WTD-fed animals (Table 1). Oil Red O staining of liver sections from DMHCA-treated mice revealed macroscopically no differences in lipid accumulation upon the absence or presence of DMHCA in both diets, while T0901317 treatment resulted in severe steatosis with enlarged lipid droplets on both diets (see supplementary Figure I). Because we found slightly increased TG levels in DMHCA-treated livers on chow diet, we checked the hepatic fatty acid composition in the TG fraction, which was changed upon high DMHCA concentrations in the chow diet. Oleate (C18:1), palmitate (C16:0), linoleate (C18:2), and stearate (C16:1) were significantly increased (Fig. 3A). No alterations were observed in mice fed WTD plus DMHCA compared with WTD alone (Fig. 3B).
TABLE 1.
Plasma and hepatic triglyceride and total cholesterol concentrations of mice fed N,N-dimethyl-3β-hydroxy-cholenamide (DMHCA) for 15 days
| Plasma
|
Liver
|
||||
|---|---|---|---|---|---|
| TG (mg/dl) | TC (mg/dl) | AST (IU/l) | TG (mg/g tissue) | TC (mg/g tissue) | |
| Chow | 62.3 ± 10.3 | 127 ± 16.4 | 30.1 ± 13.0 | 14.0 ± 6.4 | 2.16 ± 0.40 |
| Chow + DMHCA | 52.6 ± 8.2 | 86.8 ± 15.5b | 47.7 ± 13.2 | 33.8 ± 14.9a | 1.00 ± 0.45b |
| WTD | 45.6 ± 8.1 | 208 ± 65.9 | 26.7 ± 8.7 | 32.1 ± 12.8 | 4.28 ± 0.55 |
| WTD + DMHCA | 54.9 ± 10.8 | 217 ± 50.6 | 21.4 ± 7.3 | 35.1 ± 11.1 | 1.88 ± 0.44b |
AST, aspartate aminotransferase; DMHCA, N,N-dimethyl-3β-hydroxy-cholenamide. Plasma and hepatic triglyceride (TG) and total cholesterol (TC) concentrations of male C57Bl/6 mice fed chow or Western type diet (WTD) ± N,N-dimethyl-3β-hydroxy-cholenamide (DMHCA) (80 mg/kg body weight/day) for 15 days. Data are expressed as mean ± SD values of 6-8 mice aged 8–12 weeks.
P < 0.05.
P ≤ 0.001.
Fig. 3.

Effect of DMHCA on hepatic fatty acid composition. C57Bl/6 mice were fed chow diet (A) or Western type diet (WTD) (B) (containing 80 mg DMHCA/kg body weight/day) for 15 days. Fatty acid composition in the livers was determined by gas chromatography after methylation using heptadecanoic acid as internal standard. Data represent the mean values ± SD; n = 8. * P < 0.05; ** P ≤ 0.01.
Effect of DMHCA on mRNA expression of LXR target genes in wild-type mice
Target gene expression levels in livers of C57Bl/6 mice fed chow or WTD ± T0901317 or DMHCA (80 mg/kg body weight/day) for 15 days were analyzed by real time PCR. In chow diet, treatment with DMHCA led to a significant 2.5-fold increase of CYP7A1 mRNA expression (Fig. 4A). Additionally, the expression levels of ABCA1, ABCG1, ABCG5, and ABCG8 were significantly increased by 1.9-, 2.1-, 3.5- and 2.8-fold, respectively. Hepatic SREBP1c, fatty acid synthase (FAS) and ChREBP mRNA quantities were unaltered upon DMHCA treatment. T0901317 in chow diet resulted in more pronounced increase of CYP7A1, ABCA1, ABCG1, and ABCG5/G8 mRNA expression compared with controls, but also enhanced SREBP1c, FAS, and ChREBP mRNA by 4.7-, 3.4-, and 1.3-fold, respectively (Fig. 4A). Similar real-time PCR results were obtained when mRNA levels were determined from livers of mice fed WTD plus the LXR agonists. Both ligands significantly increased CYP7A1 and ABCG8 mRNA, only T0901317 resulted in an increase in ABCG1 and ABCG5 mRNA, while ABCA1 mRNA was unaltered by T0901317 and DMHCA treatment. While T0901317 significantly increased SREBP1c and FAS by 3.4- and 47.3-fold, respectively, DMHCA did not alter SREBP1c mRNA expression. While FAS mRNA was significantly increased by 2.7-fold, ChREBP was significantly decreased upon DMHCA treatment by 47%. We also analyzed ABCA1, ABCG1, ABCG5, and ABCG8 mRNA levels in ileum of T0901317 and DMHCA in chow diet and found these ABC transporters significantly increased by both ligands; again the upregulation was higher by T0901317 treatment (see supplementary Figure II).
Fig. 4.

Liver analyses of C57Bl/6 mice fed chow diet or WTD ± T0901317 or DMHCA (80 mg/kg body weight/day) for 15 days. Real-time PCR expression ratios upon liver X receptors (LXR) agonist treatment in chow (A) and WTD (B) fed mice including hypoxanthine guanine phosphoribosyl transferase (HPRT) normalization were calculated by pairwise fixed reallocation test. Controls fed chow or WTD without LXR ligand were arbitrarily set to 1. Data are expressed as mean values ± SD; n = 8 performed in duplicate. * P < 0.05; ** P ≤ 0.01; *** P ≤ 0.001.
Effect of DMHCA on plasma lipid parameters in apoE-deficient mice
Because we observed beneficial effects of DMHCA in wild-type mice, we further investigated the effect of DMHCA on body weight and lipid parameters in apoE-deficient mice during a long-term treatment. For that purpose, animals were fed WTD with or without DMHCA (8 mg/kg body weight/day) for 11 weeks. Although the food intake was similar in the different groups, male and female mice fed WTD plus DMHCA showed slightly decreased body weight gain when compared with mice fed WTD without DMHCA, which reached significance in female mice after 8 weeks of treatment (Fig. 5A, B). Plasma TC and TG levels were determined biweekly in the fed state. While no significant differences were observed in plasma TC (Fig. 5C) and TG (Fig. 5E) concentrations in male mice, female mice showed a significant decrease in TC (Fig. 5D) and TG (Fig. 5F) levels. Similar changes in plasma lipid levels were observed when mice were fasted overnight before blood was taken after 11 weeks of feeding WTD plus DMHCA (Table 2). We found reduced plasma TG levels in male (140 ± 10 mg/dl) and female (163 ± 9 mg/dl) apoE-deficient mice compared with mice fed WTD alone (197 ± 26 and 220 ± 5 mg/dl, respectively), whereas TC concentrations were significantly decreased only in male (799 ± 65 mg/dl vs. 1131 ± 52 mg/dl) but not in female animals (587 ± 33 mg/dl vs. 638 ± 16 mg/dl, respectively). No significant changes were found in HDL cholesterol levels. Determination of plasma AST or ALT levels did not give reliable results due to lipemic samples. Yet we determined bilirubin concentrations and found that they were comparable upon all diets. Therefore, long-time treatment did not lead to liver damage in apoE-deficient mice. Additionally, hepatic TG concentrations were unaltered in male (15.6 ± 0.1 mg/g) and female (12.7 ± 0.7 mg/g) DMHCA-treated animals compared with control males (16.6 ± 2.8 mg/g) and females (13.2 ± 0.3 mg/g) (Table 2). Upon FPLC separation of plasma lipoproteins from fasted female animals, the TG content of the VLDL fraction was 55% decreased after DMHCA treatment (Fig. 6A), and the TC content was decreased by 29% (Fig. 6B).
Fig. 5.

Body weight and plasma triglyceride (TG) and total cholesterol (TC) concentrations of male and female apolipoprotein E (apoE)-deficient mice. Male (n = 5) and female (n = 7) apoE-deficient mice were fed WTD ± DMHCA (8 mg/kg body weight/day) for 11 weeks. Body weight (A, B), plasma TC (C, D), and TG concentrations (E, F) in the fed state were determined at 0, 2, 4, 6, 8, 10, and 11 weeks. Data are expressed as mean values ± SD. * P < 0.05; ** P ≤ 0.01; *** P ≤ 0.001.
TABLE 2.
Plasma and hepatic TG and TC concentrations of overnight fasted apoE-deficient mice fed DMHCA for 11 weeks
| Plasma (mg/dl)
|
Liver (mg/g)
|
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Male apoE-ko | TG | TC | HDL-C | Bilirubin | TG | TC | ||||||
| WTD | 197 ± 26 | 1131 ± 52 | 13 ± 0.41 | 2.7 ± 1.4 | 16.6 ± 2.8 | 9.5 ± 2.5 | ||||||
| WTD+DMHCA | 140 ± 10a | 799 ± 65a | 21 ± 3.0 | 2.4 ± 1.2 | 15.6 ± 0.1 | 7.6 ± 1.5 | ||||||
| Female apoE-ko | ||||||||||||
| WTD | 220 ± 5 | 638 ± 16 | 33 ± 0.71 | 2.6 ± 1.1 | 13.2 ± 0.3 | 9.3 ± 1.1 | ||||||
| WTD+DMHCA | 163 ± 9b | 587 ± 33 | 34 ± 0.78 | 2.2 ± 1.4 | 12.7 ± 0.7 | 8.1 ± 0.4 | ||||||
Male and female apoE-deficient mice were fed WTD ± DMHCA (8 mg/kg body weight/day) for 11 weeks. Data are expressed as mean ± SD values of four animals.
P < 0.05.
P ≤ 0.01.
Fig. 6.

Effect of DMHCA on distribution of triglycerides and cholesterol in plasma lipoprotein fractions of apoE-deficient mice. Lipoprotein profile of plasma pools of five fasted female apoE-deficient mice fed WTD ± DMHCA (8 mg/kg body weight/day) for 11 weeks. Plasma lipoproteins were separated by fast protein liquid chromatography. TG (A) and TC (B) concentrations in each fraction were measured enzymatically.
DMHCA reduces plaque formation in apoE-deficient mice
To examine the impact of DMHCA on atherogenesis in apoE-deficient mice, atherosclerotic lesions were evaluated by aortic valve section and en face analyses after 11 weeks on WTD in the absence or presence of DMHCA (Fig. 7). Mice receiving 8 mg DMHCA/kg body weight/day showed a decrease in average lesion area compared with controls by both en face and aortic valve section analysis. Immunohistochemical staining of macrophages in the aortic valves with a macrophage-specific anti-MOMA-2 antibody and Oil Red O staining of lipids demonstrated that lipids colocalize with macrophages and are more abundant in control apoE-deficient mice (Fig. 7A). Quantification of Oil Red O-stained aortic valve sections revealed that treatment with DMHCA resulted in a significant 45.9% and 48.4% decrease in lesion area in male (Fig. 7B) and female (Fig. 7C) apoE-deficient mice when compared with controls. To further document positive effects of DMHCA on atherosclerosis, Oil Red O-stained lesions in en face preparations of aortas were quantified. Treatment with DMHCA led to a significant reduction in lesion area of male and female apoE-deficient mice compared with controls (27.7% and 29.5%, respectively) (Fig. 7D, E).
Fig. 7.

En face and aortic root section analysis of atherosclerosis in male apoE-deficient mice fed WTD ± DMHCA (8 mg/kg body weight/day) for 11 weeks. A: Representative staining of aorta and aortic valves with Oil Red O and MOMA-2 antibody. Lesions in aortic valves were analyzed in male (n = 6) (B) and female (n = 6) (C) apoE-deficient mice. Box plot graphics of en face analysis of male (n = 6) (D) and female (n = 7) (E) apoE-deficient mice. * P < 0.05; *** P ≤ 0.001.
Effect of DMHCA administration on LXR target gene expression in apoE-deficient mice
To elucidate the long-term effect of DMHCA administration on gene expression levels in apoE-deficient mice after 11 weeks on WTD with or without DMHCA, gene expression levels in liver, MPM, and small intestine were analyzed by real time PCR. In the liver, ABCG1 and CYP7A1 mRNA expression levels were significantly increased by 2.3- and 2.8-fold, respectively, whereas ABCA1, ABCG5, and ABCG8 mRNAs remained unchanged (Fig. 8A). Importantly, hepatic SREBP1c, FAS, and ChREBP mRNA expressions were not upregulated upon DMHCA treatment. These results together with unchanged hepatic TG concentrations corroborate our data that chronic DMHCA administration does not induce liver steatosis.
Fig. 8.

Regulation of LXR target mRNA levels after chronic administration of DMHCA. ApoE-deficient mice were fed WTD ± DMHCA (8 mg/kg body weight/day) for 11 weeks. Real-time PCR expression ratios in liver (A), peritoneal macrophages (MPM) (B), and ileum (C) including HPRT or cyclophilin A normalization were calculated by pairwise fixed reallocation test. Controls fed WTD were arbitrarily set to 1. Data are expressed as mean values ± SD; n = 3 performed in triplicate. * P < 0.05; ** P ≤ 0.01; *** P ≤ 0.001.
In MPM, mRNA expression of SREBP1c was slightly but significantly reduced by 13%, whereas LDLR and acetyl-CoA carboxylase (ACC) were markedly decreased by 34% and 24%, respectively (Fig. 8B). No significant differences were observed in ABCA1 and ABCG1 mRNA levels (Fig. 8B). In the ileum, ABCA1, ABCG1, and ABCG5 mRNAs remained unchanged upon DMHCA administration (Fig. 8C), while SREBP1c was significantly decreased by 53%.
Finally, we determined the effects of short-term DMHCA administration on target gene expression in apoE-deficient mice. Mice were fed normal chow diet (in the absence or presence of 80 mg DMHCA/kg body weight/day) for 4 days or WTD (in the absence or presence of 8 mg DMHCA/kg body weight/day) for 15 days. In the liver, treatment with DMHCA resulted in a significant induction of CYP7A1 mRNA expression on both diets, while ABCG5 and ABCG8 were increased by DMHCA only in the chow diet (1.4- and 1.3-fold, respectively). ABCA1, LDLR, and HMGCR mRNA were not altered upon feeding both diets. SREBP1c decreased upon DMHCA feeding with the chow diet by 52% (Fig. 9A, E). In addition, we checked whether DMHCA had an influence on gene expression in the aortae. Administration of DMHCA to chow and WTD increased ABCA1 mRNA levels in the aortae by 1.4- and 1.7-fold, respectively, while ABCG1 was significantly elevated by DMHCA only in chow diet by 1.9-fold (Fig. 9B, F). Additionally, ABCA1 mRNA was found to be increased by DMHCA upon chow and WTD feeding (1.7- and 3.1-fold, respectively) (Fig. 9C, G). In the ileum, ABCA1, ABCG1, and ABCG5 were all increased upon DMHCA administration in the chow diet (2.7-, 1.9- and 1.3-fold, respectively) (Fig. 9D). While ABCA1 mRNA quantity was significantly 1.7-fold increased in mice fed WTD supplemented with DMHCA, no changes were observed in ABCG1 and ABCG5 mRNA levels (Fig. 9H). To summarize, all effects on mRNA expression levels were similar in apoE-deficient mice after short-time feeding of both diets with the high dose of DMHCA in chow diet resulting in higher and more significant changes.
Fig. 9.

Regulation of LXR target mRNA levels in apoE-deficient mice by short-term administration of DMHCA. Male mice were fed chow diet ± DMHCA (80 mg/kg body weight/day) for 4 days (A–D) or WTD ± DMHCA (8 mg/kg body weight/day) for 15 days (E–H). Real-time PCR expression ratios in liver (A, E), aorta (B, F), MPM (C, G), and ileum (D, H) including HPRT or cyclophilin A normalization were calculated by pairwise fixed reallocation test. Controls fed diets without DMHCA were arbitrarily set to 1. Data are expressed as mean values from three samples performed in triplicate ± SD. * P < 0.05; ** P ≤ 0.01; *** P ≤ 0.001.
DMHCA does not induce liver steatosis or hypertriglyceridemia in apoE-deficient mice
To examine whether DMHCA treatment affects plasma and hepatic lipid parameters in apoE-deficient animals in a short-term treatment, male mice were fed chow ± 80 mg DMHCA/kg body weight/day for 4 days. In addition, we wanted to check whether WTD fed for a shorter time (15 days) than 11 weeks ± 8 mg DMHCA/kg body weight/day has an effect on lipid parameters. We found that plasma TG and TC concentrations were not significantly changed upon DMHCA treatment on both diets, confirming our data that DMHCA does not induce hypertriglyceridemia in apoE-null mice (Table 3). Following treatment with DMHCA, there were no changes in liver TC content. Liver TG levels were significantly decreased in mice fed a chow diet plus DMHCA (Fig. 10A), and they were not changed in mice fed a WTD plus DMHCA (Fig. 10B). Plasma AST levels were similar in all animals. Furthermore, the hepatic fatty acid composition in the TG fraction especially oleate (C18:1), palmitate (C16:0), and linoleate (C18:2) were significantly decreased (Fig. 10A). In contrast, no alterations in the hepatic fatty acid composition was observed in mice fed WTD plus DMHCA compared with WTD alone (Fig. 10B). However, when mice were fed WTD and high concentration of DMHCA (80 mg/kg body weight/day) for 4 days, this also resulted in a drastic decrease of all measured fatty acids, while no changes in fatty acid composition were observed when mice were fed chow diet plus 8 mg DMHCA/kg body weight/day (data not shown). These data indicate that 8 mg/kg/day of DMHCA, which is sufficient to reduce atherosclerosis in apoE-deficient mice when fed for 11 weeks, does not affect hepatic fatty acid composition after 15 days, while 80 mg/kg/day significantly reduces fatty acid concentrations in the liver even after a 4 day treatment.
TABLE 3.
Plasma and hepatic TG and TC concentrations of overnight fasted apoE-deficient mice after shorttime administration of DMHCA
| Plasma
|
Liver
|
||||
|---|---|---|---|---|---|
| TG (mg/dl) | TC (mg/dl) | AST (U/l) | TG (mg/g) | TC (mg/g) | |
| Chow | 103 ± 35 | 567 ± 111 | 30.9 ± 11.1 | 39.3 ± 8.2 | 2.5 ± 0.43 |
| Chow+DMHCA (4d, 80 mg) | 121 ± 21 | 531 ± 19 | 29.4 ± 14.3 | 18.1 ± 6.1a | 2.0 ± 0.45 |
| WTD | 187 ± 67 | 1,110 ± 285 | 38.1 ± 20.5 | 32.6 ± 6.4 | 7.5 ± 2.2 |
| WTD+DMHCA (15d, 8 mg) | 205 ± 61 | 1,038 ± 203 | 29.2 ± 15.6 | 34.6 ± 10.1 | 5.5 ± 0.64 |
Male apoE-deficient mice were fed chow diet ± DMHCA (80 mg/kg body weight/day) for 4 days or WTD ± DMHCA (8 mg/kg body weight/day) for 15 days. Data are expressed as mean ± SD values of six animals.
P ≤ 0.01.
Fig. 10.

Effect of DMHCA on hepatic fatty acid composition in apoE-deficient mice. Male apoE-deficient mice were fed chow diet ± DMHCA (80 mg/kg body weight/day) for 4 days (A) or WTD ± DMHCA (8 mg/kg body weight/day) for 15 days (B). Fatty acid composition in the livers was determined by gas chromatography after methylation using heptadecanoic acid as internal standard. Data represent the mean values ± SD; n = 8. * P < 0.05; ** P ≤ 0.01; ***P ≤ 0.001.
DISCUSSION
Several studies have shown that LXRs are important regulators of cholesterol and lipid metabolism (8, 24). Additionally, antiatherogenic effects of LXR agonists are well documented (16, 17). Observations in mouse models of atherosclerosis revealed that the nonsteroidal LXR ligands T0901317 (17) and GW3965 (16) reduced the development of atherosclerosis in LDLR- or apoE-deficient mice. Despite their ability to reduce atherosclerosis, LXR ligands are known to exhibit less-favorable effects on overall lipid metabolism including increase of plasma and hepatic TG concentrations (9). The molecular mechanism responsible for LXR-mediated hepatic lipogenesis has been largely attributed to the drastic increase in the expression of SREBP1c (9, 25). Moreover, ChREBP was recently reported to be a novel LXR target gene (14), which promotes hepatic conversion of excess carbohydrate to lipid (26, 27). It is essential to identify selective LXR ligands that exert beneficial cholesterol-lowering properties with minimal influence on lipogenesis. Because DMHCA has been shown to increase cholesterol efflux in THP-1 macrophages and targets involved in reverse cholesterol transport without inducing SREBP1c (18), we investigated the consequences of DMHCA on gene expression in wild-type and apoE-deficient mice and its impact on atherogenesis.
Earlier studies revealed that CYP7A1, ABCA1, and ABCG1 mRNA were significantly increased in liver and intestine only in animals dosed by intraperitoneal injection, which led Quinet et al. (18) to suggest that DMHCA may not be well absorbed by mice. The authors concluded that DMHCA had poor bioavailability or might be rapidly metabolized within the enterocyte or shortly thereafter. Although all effects on mRNA activation in livers of wild-type mice were less pronounced when compared with T0901317-treated animals, in our hands DMHCA administration significantly increased CYP7A1 mRNA levels in all our experimental conditions. Concomitantly, fecal bile acid concentrations were increased compared with controls; again this increase was less pronounced than in T0901317-treated mice (data not shown). Moreover, we measured the concentration of DMHCA in the plasma as described in the supplementary information. The high dose (80 mg/kg body weight/day) resulted in DMHCA plasma levels of 2–27 ng/ml, while the lower dose was undetectable (detection limit 0.1 ng/ml). Further experiments evaluating DMHCA concentrations in various tissues and feces should provide better insight into DMHCA metabolism.
Although we observed an approximately 2-fold increase in hepatic SREBP1c mRNA, the protein level of the mature peptide was not increased. In addition, mRNA expression of FAS and ChREBP were only slightly increased upon DMHCA treatment for 15 days. These data are in line with unchanged liver weights and only slightly increased hepatic TG concentrations in mice fed high concentrations of DMHCA. After 4 or 15 days of treatment, Oil Red O staining of liver sections showed minor lipid accumulation, not comparable to T0901317 treatment, the latter resulting in severe steatosis. This pathological effect of T0901317 has been shown to be related to increased de novo lipogenesis in combination with an increased free fatty acid uptake by the liver (25). This discrepancy in the action on lipogenesis of DMHCA and T0901317 might be due to the fact that T0901317 also induces pregnane X receptor target genes such as CYP3A4 and the free fatty acid transporter CD36 (28). The authors proposed that some of the effects observed with T0901317 may reflect pregnane X receptor activation, which then results in the observed induction of lipogenic genes. Further studies will be necessary to elucidate whether DMHCA activates or suppresses other nuclear receptors, which in turn leads to an unaltered SREBP1c expression. Experiments in macrophages isolated from LXRα-, LXRβ-, and LXRα/β-knockout mice indicate that DMHCA does not selectively regulate LXRβ (Noam Zelcer and Peter Tontonoz, personal communication), which has been proposed to be a potential target for the treatment of atherosclerosis (29). Because DMHCA shows unique gene regulation not observed with other LXR agonists, the mechanism of DMHCA action is likely to be different to the ones shown for T0901317 or GW3965. Additionally, DMHCA might not act as an oxysterol in suppressing SREBP2 proteolytic processing, which has been shown for 7-ketocholesterol, a LXR ligand that induces ABCA1 mRNA expression but suppresses SREBP2 activity (30). Although one should expect increased expression of LDLR and HMGCR as a compensatory reaction, both mRNA concentrations were unaltered in livers of DMHCA-treated wild-type (data not shown) and apoE-deficient mice.
To examine the effects of DMHCA administration on the development of atherosclerosis, we evaluated lesion formation in apoE-deficient mice fed WTD supplemented with DMHCA. Body weight, plasma cholesterol, and TG concentrations were followed over the whole period of treatment. After 11 weeks, we observed reduced plasma TG and TC levels, which were more pronounced in overnight fasted animals. DMHCA significantly reduced plaque formation in male and female apoE-deficient mice. Alterations in the lipoprotein profiles might contribute to this beneficial effect; however, HDL cholesterol levels were only slightly increased. Comparable to the results shown in T0901317 treated apoE-deficient mice (16), our data suggest that DMHCA in macrophages within the artery wall directly contributes to its anti-atherogenic effect because DMHCA is able to highly stimulate ABCA1 expression in foam cells in vitro. DMHCA did not significantly increase ABCA1 expression in liver, macrophages, or ileum of apoE-deficient mice after 11 weeks of feeding. Yet, hepatic ABCG1 and CYP7A1 mRNA levels were increased, which might have led to enhanced cholesterol efflux and bile acid production, similar to what we observed in C57Bl/6 mice. In accordance, short-term administration of DMHCA resulted in enhanced ABCA1 mRNA levels in aorta, macrophages, and ileum, while ABCA1 in the liver remained unaltered. In macrophages, upregulation of ABCA1 and ABCG1 promotes reverse cholesterol transport of unesterified cholesterol and phospholipids to produce HDL (31–33). HDL cholesterol is then delivered to the liver where an LXR-dependent increase in CYP7A1 expression results in enhanced bile acid synthesis and cholesterol elimination. We observed an increase in ABCA1, ABCG1, and ABCG5 in ileum of DMHCA-treated apoE-deficient mice. It has been shown that increased biliary excretion and decreased intestinal uptake through the LXR-mediated regulation of ABC transporters allow for the elimination of excess body cholesterol (34–36). Similar to previous work that has shown that LXR ligands build heterodimers with retinoic X receptor and reduce cholesterol absorption (3), we observed reduced cholesterol absorption in DMHCA-treated wild-type mice (unpublished results). Very recently, Tang et al. (37) showed that intestinal cholesterol absorption was required for T0901317 to raise plasma HDL-C in mice. Furthermore, ezetimibe, a potent cholesterol absorption inhibitor, was shown to reduce plasma cholesterol levels as reviewed by Davis and Veltri (38). The consequences of inhibiting cholesterol absorption by ezetimibe included reduced cholesterol delivery to the liver, which resulted in reduced hepatic cholesterol stores. These data are in line with our observations that DMHCA reduces cholesterol absorption with the consequences that i) HDL-C is only slightly but not significantly increased by DMHCA, and ii) hepatic cholesterol levels are decreased. Most interestingly, our results differ from previous studies feeding other LXR ligands in that SREBP1c, FAS, or acetyl-CoA carboxylase were unaltered or even decreased in liver, macrophages, and ileum. All effects on mRNA regulation were more pronounced when a high dose of DMHCA was provided in chow diet for 4 days compared with longer time of treatment with a smaller dose applied in WTD.
Taken together, our results obtained with different diets, dosage, and length of DMHCA treatment in wild-type and apoE-deficient mice indicate that independent of the dietary status, DMHCA regulates mRNA expression of genes involved in cholesterol catabolism and efflux in the liver, reverse cholesterol transport in macrophages, and cholesterol absorption in the intestine in a beneficial way. Higher concentration of DMHCA in the diet results in significantly higher differences in mRNA expression levels. In conclusion, we have demonstrated that even a small dose of DMHCA fed for 11 weeks was able to reduce atherosclerosis in apoE-deficient mice without inducing hepatic and plasma TG levels, which are independent risk factors for insulin resistance and cardiovascular disease. Therefore, DMHCA may be a promising agent for the treatment of atherosclerosis.
Supplementary Material
Acknowledgments
The authors thank S. Povoden, E. Stanzer, A. Ibovnik, H. Reicher, A. Blaschitz, and S. Schauer for excellent technical assistance, and A. Hermann for her invaluable help with mice care. Furthermore, the authors appreciate the additional supply of DMHCA by E.M. Quinet from Wyeth Pharmaceuticals, Collegeville, PA.
Published, JLR Papers in Press, September 23, 2008.
Footnotes
This work was supported by the Austrian Federal Ministry of Science and Research (Genomforschung Austria Project G.O.L.D.II, Genomics of Lipid-associated Disorders II), the Austrian Science Fund FWF (P19186, SFB-LIPOTOX F3004 and F3001 and SFB F3210-HEART), and the Austrian Nationalbank (10797).
The online version of this article (available at http://www.jlr.org) contains supplementary information and two supplementary figures.
References
- 1.Janowski B. A., P. J. Willy, T. R. Devi, J. R. Falck, and D. J. Mangelsdorf. 1996. An oxysterol signalling pathway mediated by the nuclear receptor LXR alpha. Nature. 383 728–731. [DOI] [PubMed] [Google Scholar]
- 2.Costet P., Y. Luo, N. Wang, and A. R. Tall. 2000. Sterol-dependent transactivation of the ABC1 promoter by the liver X receptor/retinoid X receptor. J. Biol. Chem. 275 28240–28245. [DOI] [PubMed] [Google Scholar]
- 3.Repa J. J., S. D. Turley, J. A. Lobaccaro, J. Medina, L. Li, K. Lustig, B. Shan, R. A. Heyman, J. M. Dietschy, and D. J. Mangelsdorf. 2000. Regulation of absorption and ABC1-mediated efflux of cholesterol by RXR heterodimers. Science. 289 1524–1529. [DOI] [PubMed] [Google Scholar]
- 4.Schwartz K., R. M. Lawn, and D. P. Wade. 2000. ABC1 gene expression and apoA-I-mediated cholesterol efflux are regulated by LXR. Biochem. Biophys. Res. Commun. 274 794–802. [DOI] [PubMed] [Google Scholar]
- 5.Venkateswaran A., J. J. Repa, J. M. Lobaccaro, A. Bronson, D. J. Mangelsdorf, and P. A. Edwards. 2000. Human white/murine ABC8 mRNA levels are highly induced in lipid-loaded macrophages. A transcriptional role for specific oxysterols. J. Biol. Chem. 275 14700–14707. [DOI] [PubMed] [Google Scholar]
- 6.Venkateswaran A., B. A. Laffitte, S. B. Joseph, P. A. Mak, D. C. Wilpitz, P. A. Edwards, and P. Tontonoz. 2000. Control of cellular cholesterol efflux by the nuclear oxysterol receptor LXR alpha. Proc. Natl. Acad. Sci. USA. 97 12097–12102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brunham L. R., J. K. Kruit, T. D. Pape, J. S. Parks, F. Kuipers, and M. R. Hayden. 2006. Tissue-specific induction of intestinal ABCA1 expression with a liver X receptor agonist raises plasma HDL cholesterol levels. Circ. Res. 99 672–674. [DOI] [PubMed] [Google Scholar]
- 8.Repa J. J., K. E. Berge, C. Pomajzl, J. A. Richardson, H. Hobbs, and D. J. Mangelsdorf. 2002. Regulation of ATP-binding cassette sterol transporters ABCG5 and ABCG8 by the liver X receptors alpha and beta. J. Biol. Chem. 277 18793–18800. [DOI] [PubMed] [Google Scholar]
- 9.Repa J. J., G. Liang, J. Ou, Y. Bashmakov, J. M. Lobaccaro, I. Shimomura, B. Shan, M. S. Brown, J. L. Goldstein, and D. J. Mangelsdorf. 2000. Regulation of mouse sterol regulatory element-binding protein-1c gene (SREBP-1c) by oxysterol receptors, LXRalpha and LXRbeta. Genes Dev. 14 2819–2830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yoshikawa T., H. Shimano, M. Amemiya-Kudo, N. Yahagi, A. H. Hasty, T. Matsuzaka, H. Okazaki, Y. Tamura, Y. Iizuka, K. Ohashi, et al. 2001. Identification of liver X receptor-retinoid X receptor as an activator of the sterol regulatory element-binding protein 1c gene promoter. Mol. Cell. Biol. 21 2991–3000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang Y., J. J. Repa, K. Gauthier, and D. J. Mangelsdorf. 2001. Regulation of lipoprotein lipase by the oxysterol receptors, LXRalpha and LXRbeta. J. Biol. Chem. 276 43018–43024. [DOI] [PubMed] [Google Scholar]
- 12.Joseph S. B., B. A. Laffitte, P. H. Patel, M. A. Watson, K. E. Matsukuma, R. Walczak, J. L. Collins, T. F. Osborne, and P. Tontonoz. 2002. Direct and indirect mechanisms for regulation of fatty acid synthase gene expression by liver X receptors. J. Biol. Chem. 277 11019–11025. [DOI] [PubMed] [Google Scholar]
- 13.Chu K., M. Miyazaki, W. C. Man, and J. M. Ntambi. 2006. Stearoyl-coenzyme A desaturase 1 deficiency protects against hypertriglyceridemia and increases plasma high-density lipoprotein cholesterol induced by liver X receptor activation. Mol. Cell. Biol. 26 6786–6798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cha J. Y., and J. J. Repa. 2007. The liver X receptor (LXR) and hepatic lipogenesis. The carbohydrate-response element-binding protein is a target gene of LXR. J. Biol. Chem. 282 743–751. [DOI] [PubMed] [Google Scholar]
- 15.Lehmann J. M., S. A. Kliewer, L. B. Moore, T. A. Smith-Oliver, B. B. Oliver, J. L. Su, S. S. Sundseth, D. A. Winegar, D. E. Blanchard, T. A. Spencer, et al. 1997. Activation of the nuclear receptor LXR by oxysterols defines a new hormone response pathway. J. Biol. Chem. 272 3137–3140. [DOI] [PubMed] [Google Scholar]
- 16.Joseph S. B., E. McKilligin, L. Pei, M. A. Watson, A. R. Collins, B. A. Laffitte, M. Chen, G. Noh, J. Goodman, G. N. Hagger, et al. 2002. Synthetic LXR ligand inhibits the development of atherosclerosis in mice. Proc. Natl. Acad. Sci. USA. 99 7604–7609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Terasaka N., A. Hiroshima, T. Koieyama, N. Ubukata, Y. Morikawa, D. Nakai, and T. Inaba. 2003. T-0901317, a synthetic liver X receptor ligand, inhibits development of atherosclerosis in LDL receptor-deficient mice. FEBS Lett. 536 6–11. [DOI] [PubMed] [Google Scholar]
- 18.Quinet E. M., D. A. Savio, A. R. Halpern, L. Chen, C. P. Miller, and P. Nambi. 2004. Gene-selective modulation by a synthetic oxysterol ligand of the liver X receptor. J. Lipid Res. 45 1929–1942. [DOI] [PubMed] [Google Scholar]
- 19.Louw D. F., G. Strating, and H. J. Backer. 1954. Delta-5-Steroids and provitamins D with branched side chains. III. Preparation and reduction of some delta-5-steroid omega-amines. Recueil des Travaux Chimiques des Pays-Bas et de la Belgique. 73 667–676. [Google Scholar]
- 20.Wang X., R. Sato, M. S. Brown, X. Hua, and J. L. Goldstein. 1994. SREBP-1, a membrane-bound transcription factor released by sterol-regulated proteolysis. Cell. 77 53–62. [DOI] [PubMed] [Google Scholar]
- 21.Lowry O. H., N. J. Rosebrough, A. L. Farr, and R. J. Randall. 1951. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 193 265–275. [PubMed] [Google Scholar]
- 22.Miyazaki M., H. J. Kim, W. C. Man, and J. M. Ntambi. 2001. Oleoyl-CoA is the major de novo product of stearoyl-CoA desaturase 1 gene isoform and substrate for the biosynthesis of the Harderian gland 1-alkyl-2,3-diacylglycerol. J. Biol. Chem. 276 39455–39461. [DOI] [PubMed] [Google Scholar]
- 23.Pfaffl M. W., G. W. Horgan, and L. Dempfle. 2002. Relative expression software tool (REST) for group-wise comparison and statistical analysis of relative expression results in real-time PCR. Nucleic Acids Res. 30 e36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Repa J. J., and D. J. Mangelsdorf. 2000. The role of orphan nuclear receptors in the regulation of cholesterol homeostasis. Annu. Rev. Cell Dev. Biol. 16 459–481. [DOI] [PubMed] [Google Scholar]
- 25.Schultz J. R., H. Tu, A. Luk, J. J. Repa, J. C. Medina, L. Li, S. Schwendner, S. Wang, M. Thoolen, D. J. Mangelsdorf, et al. 2000. Role of LXRs in control of lipogenesis. Genes Dev. 14 2831–2838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Uyeda K., and J. J. Repa. 2006. Carbohydrate response element binding protein, ChREBP, a transcription factor coupling hepatic glucose utilization and lipid synthesis. Cell Metab. 4 107–110. [DOI] [PubMed] [Google Scholar]
- 27.Yamashita H., M. Takenoshita, M. Sakurai, R. K. Bruick, W. J. Henzel, W. Shillinglaw, D. Arnot, and K. Uyeda. 2001. A glucose-responsive transcription factor that regulates carbohydrate metabolism in the liver. Proc. Natl. Acad. Sci. USA. 98 9116–9121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mitro N., L. Vargas, R. Romeo, A. Koder, and E. Saez. 2007. T0901317 is a potent PXR ligand: implications for the biology ascribed to LXR. FEBS Lett. 581 1721–1726. [DOI] [PubMed] [Google Scholar]
- 29.Bradley M. N., C. Hong, M. Chen, S. B. Joseph, D. C. Wilpitz, X. Wang, A. J. Lusis, A. Collins, W. A. Hseuh, J. L. Collins, et al. 2007. Ligand activation of LXR beta reverses atherosclerosis and cellular cholesterol overload in mice lacking LXR alpha and apoE. J. Clin. Invest. 117 2337–2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schmidt R. J., J. V. Ficorilli, Y. Zhang, K. S. Bramlett, T. P. Beyer, K. Borchert, M. S. Dowless, K. A. Houck, T. P. Burris, P. I. Eacho, et al. 2006. A 15-ketosterol is a liver X receptor ligand that suppresses sterol-responsive element binding protein-2 activity. J. Lipid Res. 47 1037–1044. [DOI] [PubMed] [Google Scholar]
- 31.Oram J. F., R. M. Lawn, M. R. Garvin, and D. P. Wade. 2000. ABCA1 is the cAMP-inducible apolipoprotein receptor that mediates cholesterol secretion from macrophages. J. Biol. Chem. 275 34508–34511. [DOI] [PubMed] [Google Scholar]
- 32.Oram J. F., and A. M. Vaughan. 2000. ABCA1-mediated transport of cellular cholesterol and phospholipids to HDL apolipoproteins. Curr. Opin. Lipidol. 11 253–260. [DOI] [PubMed] [Google Scholar]
- 33.Klucken J., C. Buchler, E. Orso, W. E. Kaminski, M. Porsch-Ozcurumez, G. Liebisch, M. Kapinsky, W. Diederich, W. Drobnik, M. Dean, et al. 2000. ABCG1 (ABC8), the human homolog of the Drosophila white gene, is a regulator of macrophage cholesterol and phospholipid transport. Proc. Natl. Acad. Sci. USA. 97 817–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee M. H., K. Lu, S. Hazard, H. Yu, S. Shulenin, H. Hidaka, H. Kojima, R. Allikmets, N. Sakuma, R. Pegoraro, et al. 2001. Identification of a gene, ABCG5, important in the regulation of dietary cholesterol absorption. Nat. Genet. 27 79–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Berge K. E., H. Tian, G. A. Graf, L. Yu, N. V. Grishin, J. Schultz, P. Kwiterovich, B. Shan, R. Barnes, and H. H. Hobbs. 2000. Accumulation of dietary cholesterol in sitosterolemia caused by mutations in adjacent ABC transporters. Science. 290 1771–1775. [DOI] [PubMed] [Google Scholar]
- 36.Murthy S., E. Born, S. N. Mathur, and F. J. Field. 2002. LXR/RXR activation enhances basolateral efflux of cholesterol in CaCo-2 cells. J. Lipid Res. 43 1054–1064. [DOI] [PubMed] [Google Scholar]
- 37.Tang W., Y. Ma, L. Jia, Y. A. Ioannou, J. P. Davies, and L. Yu. 2008. Niemann-Pick C1-like 1 is required for an LXR agonist to raise plasma HDL cholesterol in mice. Arterioscler. Thromb. Vasc. Biol. 28 448–454. [DOI] [PubMed] [Google Scholar]
- 38.Davis H. R., and E. P. Veltri. 2007. Zetia: inhibition of Niemann-Pick C1 Like 1 (NPC1L1) to reduce intestinal cholesterol absorption and treat hyperlipidemia. J. Atheroscler. Thromb. 14 99–108. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.