

Abstract
The ionic mechanisms that contribute to general anesthetic actions have not been elucidated, although increasing evidence has pointed to roles for subthreshold ion channels, such as the HCN channels underlying the neuronal hyperpolarization-activated cationic current (Ih). Here, we used conventional HCN1 knockout mice to test directly the contributions of specific HCN subunits to effects of isoflurane, an inhalational anesthetic, on membrane and integrative properties of motor and cortical pyramidal neurons in vitro. Compared with wild-type mice, residual Ih from knockout animals was smaller in amplitude and presented with HCN2-like properties. Inhibition of Ih by isoflurane previously attributed to HCN1 subunit-containing channels (i.e., a hyperpolarizing shift in half-activation voltage [V1/2]) was absent in neurons from HCN1 knockout animals; the remaining inhibition of current amplitude could be attributed to effects on residual HCN2 channels. We also found that isoflurane increased temporal summation of excitatory postsynaptic potentials (EPSPs) in cortical neurons from wild-type mice; this effect was predicted by simulation of anesthetic-induced dendritic Ih inhibition, which also revealed more prominent summation accompanying shifts in V1/2 (an HCN1-like effect) than decreased current amplitude (an HCN2-like effect). Accordingly, anesthetic-induced EPSP summation was not observed in cortical cells from HCN1 knockout mice. In wild-type mice, the enhanced synaptic summation observed with low concentrations of isoflurane contributed to a net increase in cortical neuron excitability. In summary, HCN channel subunits account for distinct anesthetic effects on neuronal membrane properties and synaptic integration; inhibition of HCN1 in cortical neurons may contribute to the synaptically mediated slow-wave cortical synchronization that accompanies anesthetic-induced hypnosis.
INTRODUCTION
Neuronal and ionic mechanisms that underlie behavioral actions of general anesthetics remain to be determined. The actions that define the anesthetic state are immobility, unconsciousness, and amnesia (Eger et al. 1997); it is likely that different cellular and molecular substrates contribute to these distinct anesthetic endpoints and the relevant targets may also be different for individual anesthetic drugs (Franks 2006; Rudolph and Antkowiak 2004; Sonner et al. 2003). There is now good evidence implicating a number of ion channels in anesthetic action, based on their sensitivity to modulation by clinically relevant concentrations of anesthetics and their expression in behaviorally relevant neural systems. This includes a number of ionotropic receptors (e.g., γ-aminobutyric acid type A [GABAA], glycine, and glutamate receptors) and background K+ channels (e.g., K2P2.1, K2P3.1, K2P9.1) (Franks 2006; Linden et al. 2007; Rudolph and Antkowiak 2004; Sonner et al. 2003).
Our recent work indicates that general anesthetics also produce a subunit-selective inhibition of hyperpolarization-activated cyclic-nucleotide gated (HCN) channels containing HCN1 and HCN2 subunits (Chen et al. 2005a,b); these channels are expressed in motoneurons and in projection neurons of the thalamus and cortex where they underlie a hyperpolarization-activated cationic current (Ih) that contributes to intrinsic and integrative neuronal properties and to generation of synchronized oscillatory rhythms generated in the thalamus and cortex (Destexhe and Sejnowski 2003; Pape 1996; Pape and McCormick 1989; Steriade et al. 1993b). Depending on HCN subunit composition in heterologous expression systems, we found that inhalational anesthetics primarily inhibit voltage-dependent activation properties (HCN1 homomers) or maximal current amplitude (HCN2 homomers), with combined effects observed in heteromeric HCN1–HCN2 channels (Chen et al. 2005b). However, since there are no HCN subunit-selective antagonists, support for the conclusion that these particular subunits account for anesthetic effects on neuronal Ih has been indirect and correlative, relying on shared properties of modulation between native currents and cloned channels (Chen et al. 2005b). Moreover, previous studies have not evaluated the relative contributions of the distinct modulatory mechanisms of anesthetics on neuronal properties that are strongly influenced by Ih, such as synaptic integration.
Here, we used HCN1 knockout mice and computer simulations to examine HCN subunit contributions to anesthetic effects on intrinsic and integrative properties in motor neurons and cortical pyramidal neurons in vitro. Our results verify HCN subunit-specific effects of isoflurane and demonstrate that inhibition of Ih by isoflurane causes membrane hyperpolarization and enhanced synaptic summation; the net effect of these changes in cortical neuronal properties is an increase in synaptic excitability. Thus isoflurane-mediated inhibition of HCN1-containing channels may contribute to synaptically driven slow-wave oscillations in thalamocortical circuits (McCormick and Bal 1997; Rudolph and Antkowiak 2004; Steriade et al. 1993a), such as those associated with hypnosis at the onset of anesthesia (Antkowiak 2002).
METHODS
HCN1 knockout mice
Conventional HCN1 channel knockout mouse lines were used for in vitro electrophysiological experiments investigating the role of HCN1-containing channels in neuronal effects of isoflurane. The HCN1 knockout mice were originally prepared and characterized by the Kandel laboratory (Nolan et al. 2003, 2004); they have been deposited for distribution by Jackson Labs (stock #005034). The knockout was prepared by “floxing” the exon containing the pore domain and sixth transmembrane segment, followed by Cre-mediated excision of that exon in embryonic stem cells. Homozygous HCN1 knockout animals were generated by crossing 129SVEV HCN1−/+ animals to C57BL/6 wild-type mice and then intercrossing heterozygous progeny to yield the HCN1−/− knockout animals that are maintained as homozygotes by Jackson Labs. As reported in the original publications (Nolan et al. 2003, 2004), mice homozygous for the targeted mutation are viable, fertile, normal in size and longevity, and they do not display any gross physical or behavioral abnormalities; functional channel deletion was confirmed by electrophysiological analysis of Ih in cerebellar Purkinje neurons (decreased by ∼90%) and hippocampal CA1 pyramidal cells (decreased by ∼60–70%) (Nolan et al. 2003). As controls for HCN1 knockout animals, we used age, sex, and weight-matched B6129SF2/J mice (stock #101045). These animals are hybrids of the parental strains C57BL/6J and 129S1/SvImJ; they are recommended by Jackson Labs as an “approximate” control for strains designated B6;129S, including HCN1 knockout mice, and are appropriate for the cellular studies reported here (although littermate controls derived from the breeding of heterozygotes are advised for behavioral studies).
Electrophysiological recordings
Transverse brain slices from mice of either sex (14–22 days old for cortical cells; 11–15 days old for motoneurons) were prepared, as described previously (Chen et al. 2005a; Talley et al. 2000). Mice were anesthetized (ketamine/xylazine: 200/14 mg/kg, administered intramuscularly) and decapitated; transverse slices (300 μm) were cut with a microslicer in ice-cold Ringer solution containing (in mM): sucrose, 260; KCl, 3; MgCl2, 5; CaCl2, 1; NaH2PO4, 1.25; NaHCO3, 26; glucose, 10; and kynurenic acid, 1. Slices were maintained in Ringer solution containing (in mM): NaCl, 130; KCl, 3; MgCl2, 2; CaCl2, 2; NaH2PO4, 1.25; NaHCO3, 26; and glucose, 10, bubbled with 95% O2-5% CO2. Brain slices were submerged in a recording chamber on a Zeiss Axioskop FS microscope and visualized with Nomarski optics; hypoglossal motor neurons and cortical pyramidal neurons were targeted for recording based on location in the slice and characteristic size and shape (Chen et al. 2005a; Talley et al. 2000).
Electrical recordings were obtained at room temperature in HEPES-based bath solutions containing (in mM): NaCl, 140; KCl, 3; HEPES, 10; CaCl2, 2; MgCl2, 2; and glucose, 10. For voltage recording, pipettes (2–4 MΩ) were filled with (in mM): KCH3O3S, 120; NaCl, 4; MgCl2, 1; CaCl2, 0.5; HEPES, 10; EGTA, 10; MgATP, 3; and GTP-Tris, 0.3 (pH 7.2); for current clamp, pipette solutions contained (in mM): KCl, 17.5; potassium gluconate, 122.5; HEPES, 10; EGTA, 0.2; NaCl, 9; MgCl2, 1; MgATP, 3; and GTP-Tris, 0.3 (pH 7.2). Bath solutions were bubbled vigorously and inhalational anesthetics were added to the perfusate (∼2 ml/min) via calibrated vaporizers (Ohmeda); anesthetic concentrations were determined by gas chromatography of perfusate samples. All data were corrected for a measured liquid junction potential of 8 mV.
To block Ih in some experiments, CsCl (3 mM) was added to the bath, or 4-(N-ethyl-N-phenylamino-1,2-dimethyl-6-methylamino)pyrimidinium chloride (ZD-7288) was included either in the pipette solution or the bath (50 μM; Tocris Cookson). In other experiments, cAMP was added to the pipette solution to activate HCN channels (at 50 μM; Sigma). In voltage-clamp recordings of Ih from motoneurons, we inhibited the prominent anesthetic-activated TASK-like currents by lowering bath pH to 6.5 (Sirois et al. 2000, 2002); we also used a pipette solution containing 25 mM HEPES (substituted for KCH3O3S) to minimize the effects of bath acidification on the pH-sensitive motoneuronal Ih (Sirois et al. 2002). We included BaCl2 (200 μM) in voltage-clamp experiments to inhibit inwardly rectifying K+ currents. Where noted, tetrodotoxin (TTX at 0.5 μM; Alomone Labs) was added to the perfusate to block action potentials and a bicuculline/strychnine cocktail (both at 30 μM; Sigma) was added to block GABAA and glycine receptor channels.
Electrophysiological recordings were acquired with an Axopatch 200B amplifier via a Digidata 1322A digitizer using pCLAMP software (all Axon Instruments). For voltage-clamp recording, time-dependent hyperpolarization-activated currents (Ih) were evoked with incrementing (Δ −10 mV) hyperpolarizing pulses (3–4 s) from a holding potential of −48 mV, followed immediately by a step to fixed potential (−98 mV) to obtain tail currents. Current amplitude at each potential was taken as the time-dependent current at the end of hyperpolarizing voltage steps; maximal available current was determined at −128 mV. Tail currents were normalized, plotted as a function of the preceding hyperpolarization step voltage, and fitted with Boltzmann curves for derivation of half-activation voltage (V1/2) by using a least-squares analysis and the “Solver” add-in of Excel (Microsoft). Time constants (τ) were determined by fitting currents evoked during hyperpolarizing steps to a biexponential function (Clampfit). For current-clamp analysis, we measured resting membrane potential, input resistance, and depolarizing “sag” during rectangular current steps. Because voltage “sag” is influenced by membrane potential and input resistance as well as the magnitude of Ih, we compared voltage “sag” between cells from the same membrane potential (i.e., during current steps in which initial voltage deflections approached approximately −88, −98, and −108 mV). We monitored changes in Ih or membrane potential continuously during isoflurane application and measurements were made after steady state was achieved (usually within 5 min).
Synaptic integration and computer simulation
A stimulating electrode, connected to a Grass stimulator (S48) via a stimulus isolation unit (Grass SIU5), was filled with standard external solution containing Lucifer yellow and placed in the superficial layers of the cortex. For recordings of evoked EPSPs, the bath solution contained a bicuculline/strychnine cocktail (each at 30 μM). Synaptic responses were evoked by applying 40-Hz, 5- to 10-V pulses (corresponding to 2 to 10 μA) and CNQX-sensitive EPSPs were recorded under whole cell current clamp in cortical pyramidal neurons. To relate measured effects on EPSP summation to anesthetic-induced changes in Ih properties, a computer simulation was performed using the NEURON software package (Hines and Carnevale 1997) and a previously published pyramidal neuron model (Day et al. 2005) kindly provided by Drs. J. Held and J. Surmeier (Northwestern University). With this computer model, we compared effects on EPSP summation of imposing shifts in V1/2 of Ih activation, decreases in current amplitude, or both.
Neuronal morphology
For morphological identification of recorded cells, 0.2% biocytin (Sigma) was included in the patch electrode solution, as described previously (Chen et al. 2005a). In brief, slices were fixed in 4% buffered paraformaldehyde solution and then rinsed and incubated with 1% H2O2 and 0.5% Triton X-100. After incubation with avidin-biotin complex and diaminobenzidine substrate kit (both from Vector Labs, Burlingame, CA), slices were mounted and dehydrated through graded ethanol and xylene and embedded in DPX mounting medium (BDH Laboratory Supplies, Bolton, UK); biocytin-stained neurons were visualized and photographed using an Axioskop microscope (Zeiss), equipped with a digital camera (Retiga 1300C, QImaging) and IPLab software (Scanalytics). A digital camera lucida system was used for reconstruction and quantitative analysis of biocytin-filled cortical neurons (NeuroLucida, MicroBrightfield). In some experiments (e.g., during field stimulation), Lucifer yellow (0.2%) was included in the pipette solution to identify the soma and primary dendrite of recorded cortical neurons to aid in placement of the stimulating pipette.
Quantitative real-time PCR
Cortical HCN subunit expression was determined in wild-type and knockout mice by using quantitative real-time polymerase chain reaction (qRT-PCR). The cortex was microdissected for RNA isolation from juvenile and adult mice (n ≥ 3 animals per genotype at each age) and qRT-PCR was performed from each sample in quadruplicate, using an ICycler (Bio-Rad, Hercules, CA); each animal contributed a single data point for a given HCN channel subunit. We used the following primer sets: HCN1: GenBank accession number AJ225123, upper: AGG TTA ATC AGA TAC ATA CACC, lower: GAG TGC GTA GGA ATA TTG TTTT, 231-bp amplicon; HCN2: GenBank accession number AJ225122, upper: CGG CTC ATC CGA TAT ATC CA, lower: AGC GCG AAC GAG TAG AGC TC, 230-bp amplicon; HCN3: GenBank accession number NM008227, upper: GAT ACT GCA GCG GAA ACG CTC, lower: AGA TAC CTG GGA ACG CCC TGT; 482-bp amplicon; HCN4: GenBank accession number XM287905, upper: CCC GCC TCA TTC GAT ACA TTC AT, lower: CCC GCC TCA TTC GAT ACA TTC AT, 232-bp amplicon; and PCR conditions: 3 min 95°C; 40 cycles: 10 s 95°C, 40 s 60°C, 40 s 72°C that were optimized in preliminary experiments to yield ≥97% efficiency. The identity of PCR products was verified in initial experiments by agarose gel electrophoresis (which yielded amplicons of appropriate size) and in all experiments by melt curve analysis (which yielded a single peak at appropriate Tm). Cyclophilin served as an internal standard (GenBank accession number NM_005038; upper: GGC TCT TGA AAT GGA CCC TTC, lower: CAG CCA ATG CTT GAT CAT ATT CTT, 91-bp amplicon (Lauritzen et al. 2003), and control samples with no added template were included with every experiment. We analyzed qRT-PCR data by using a modification of the so-called ΔΔCt normalization procedure to obtain HCN subunit mRNA levels for each genotype, relative to cyclophilin (Pfaffl 2001).
Data acquisition and analysis
Results are presented as means ± SE. Data were analyzed statistically using one-way and two-way ANOVAs or Student's t-test; post hoc pairwise comparisons used Bonferroni's correction of the t-test (Excel and/or SigmaStat). Differences in mean values were considered significant if P < 0.05.
RESULTS
HCN1 expression and contribution to membrane properties of cortical pyramidal neurons are diminished in HCN1 knockout mice
To verify HCN1 subunit deletion in HCN1 knockout mice, we examined HCN subunit expression by qRT-PCR. As shown in Fig. 1, A and B, qRT-PCR using primers for HCN1, HCN2, HCN3, and HCN4 revealed a complete deletion of HCN1 channels in cortex from adult HCN1 knockout mice, with little change in expression of other HCN subunits. A substantial expression of HCN2 was evident in both control and HCN1 knockout animals, whereas levels of HCN3 and HCN4 mRNA were about 50- to 100-fold lower in cortex of either wild-type or HCN1 knockout mice. We obtained essentially identical results using cortical tissue from juvenile mice (postnatal day 20 [P20]), with the exception that, for both genotypes, HCN2 mRNA levels were approximately twofold higher in cortex of younger mice than in adults (data not shown; n = 3 and 4 for control and HCN1 knockout, respectively). These results are consistent with previous reports that HCN1 and HCN2 are the predominant HCN subunits expressed in cortical neurons (Monteggia et al. 2000; Santoro et al. 2000) and they indicate that there is little change in expression of other HCN subunits to compensate for deletion of HCN1 (Nolan et al. 2003, 2004).
FIG. 1.

HCN1 subunit deletion results in a smaller and slower hyperpolarization-activated cationic current (Ih) in cortical pyramidal neurons from HCN1 knockout mice. A: average traces from quadruplicate determinations by quantitative RT-PCR of HCN1 and HCN2 subunit expression in cortex of control and HCN1 knockout mice. Note that HCN1 subunit mRNA is undetectable and HCN2 expression is not up-regulated in HCN1 knockout mice. B: expression of the indicated HCN subunits in cortex from control and HCN1 knockout animals (n = 5 each); for each subunit, data are expressed as averaged fold-difference from a cyclophilin control (2−ΔCt) (*, significantly different from control, P < 0.001 by unpaired t-test). C: photomicrograph of biocytin-stained neurons recorded from neocortex in wild-type (top) and HCN1 knockout (bottom) mice. Scale bar represents 50 μm. D: voltage-clamp recordings of Ih in wild-type and HCN1 knockout mice (from cells presented in C). The voltage-clamp protocol depicted is typical of that used throughout these studies (conditioning voltage steps were always 3–4 s in duration). E: time-dependent currents were measured at the end of each voltage step in wild-type (WT) or HCN1-KO (KO) mice and plotted as a function of membrane potential (left). The amplitude of Ih (at −130 mV) was determined from individual cells and plotted as a frequency distribution (middle) and averaged (right, ±SE). Ih was about 70% smaller in cortical cells from HCN1 subunit knockout mice. F: sample traces of Ih activation recorded at −120 mV in cells from WT and KO mice were normalized and overlaid (left); the residual current from the KO animal activated more slowly than Ih from WT mice. Activation kinetics were analyzed by using biexponential fits to currents evoked at −120 mV; time constants (τf) describing the fastest (and largest) current component were obtained from individual cells and plotted as a frequency distribution (middle) and averaged (right, ±SE). Ih activation was markedly slower in cortical neurons from KO mice (*, significantly different from WT, P < 0.001 by unpaired t-test).
We used whole cell patch-clamp recording in brain slices to characterize hyperpolarization-activated currents mediated by HCN subunits in cortical pyramidal neurons from wild-type and HCN1 knockout mice. We examined properties of HCN currents in cortical pyramidal neurons from juvenile control and HCN1 knockout mice (P14–P22); the age distribution was not different between mouse lines and we found no age-dependent changes in Ih density within the age span considered in either group of cells (data not shown).
Biocytin-filled neurons from wild-type and knockout mice, presented in Fig. 1C, display similar morphological features that are typical of cortical pyramidal neurons (i.e., the cells have large apical dendrites that extend upward toward the pial surface, where they ramify extensively). We reconstructed a subset of biocytin-filled cortical neurons from wild-type (n = 4) and HCN1 knockout mice (n = 3); there were no significant differences in length of the apical dendrites in these neurons (596.2 ± 26.7 vs. 648.6 ± 19.9 μm, P = 0.20) nor in the number (36.5 ± 7.1 vs. 35.7 ± 3.3, P = 0.93) or average length (99.2 ± 9.7 vs. 92.4 ± 2.4 μm, P = 0.58) of secondary and tertiary dendritic branches, the values for which were generally consistent with previous descriptions of these properties in cortical pyramidal cells (e.g., see Larsen and Callaway 2006).
There were striking differences in voltage- and time-dependent currents evoked by hyperpolarizing voltage steps (i.e., Ih) between HCN1 knockouts and control animals, as depicted in Fig. 1D. Specifically, Ih was consistently smaller in cells from knockout animals; in addition, the residual current activated more slowly and at more hyperpolarized potentials. In wild-type mice, the maximal time-dependent Ih in cortical neurons was about −320 pA at the end of a hyperpolarizing step to −128 mV (−320.3 ± 26.4 pA, n = 24), whereas in cells from HCN1 knockout mice, the residual Ih at the same potential was only −106.5 ± 10.7 pA (n = 46, Fig. 1E). In cortical cells from control animals, biexponential fits to Ih revealed a variable slow component and a prominent fast component (accounting for >60% of current amplitude) that activated with a time constant (fast tau, τf) of about 80 ms (83.5 ± 6.3 ms at −118 mV, n = 18; Fig. 3D). The slower Ih activation in pyramidal neurons from HCN1 knockout mice is apparent in the scaled exemplar traces of Fig. 1F (left) and in the substantially longer τf values (519.2 ± 66.8 ms, n = 19; Fig. 1F, middle and right). Finally, the half-activation potential (V1/2) for Ih was shifted to more hyperpolarized potentials in pyramidal neurons from HCN1 knockout mice (−91.2 ± 1.6 vs. −106.0 ± 2.0 mV in control and knockout, respectively; n = 10 and 12, P < 0.05; see Fig. 3D). The differences in amplitude, kinetics, and voltage dependence of Ih obtained in cortical pyramidal neurons from control and HCN1 knockout mice are entirely consistent with removal of fast-activating HCN1 subunits, leaving a small residual current mediated primarily by slowly activating HCN2 subunits that operate over a more hyperpolarized voltage range (Franz et al. 2000; Santoro et al. 2000).
The differences in Ih characteristics observed in cortical neurons from HCN1 knockout mice were reflected in altered membrane properties of those cells, as determined by voltage recordings obtained under current clamp (Fig. 2). Compared with wild-type mice, cortical neurons from HCN1 knockout animals presented with a more hyperpolarized resting membrane potential (RMP), a higher peak input resistance (RN), and they showed less voltage “sag” in response to hyperpolarizing current steps, particularly at more depolarized membrane potentials. Moreover, whereas blocking Ih with CsCl (3 mM) caused a marked hyperpolarization and increased peak RN of control cells, it had little effect on RMP and peak RN in pyramidal neurons from HCN1 knockout mice; furthermore, genotype-dependent differences in RMP, RN, and “sag” largely disappeared after blocking Ih with 3 mM CsCl.
FIG. 2.

Cortical neurons from HCN1 channel knockout mice have a more hyperpolarized membrane potential, higher input resistance, and display less “sag.” A: voltage recordings in cortical pyramidal neurons from WT and KO mice during current injection (from 60 to −100 pA; see inset). Under control conditions (left), the cortical neuron from the KO animal was more hyperpolarized (−74 vs. −66 mV) and had higher input resistance than that of the cell from a WT animal; also note that the depolarizing “sag” associated with Ih activation was less evident in KO mice, especially at less hyperpolarized potentials (e.g., see indicated traces that reach −88 mV). In the presence of CsCl, the depolarizing sag was essentially eliminated. Also note that the membrane hyperpolarization associated with Cs+-mediated inhibition of Ih in the WT cell was not present in the HCN1-KO cell. B: frequency distributions (top) and averaged data (bottom) for resting membrane potential (RMP, left), input resistance (RN, middle), and voltage “sag” (right, from −88 mV) obtained in cortical neurons from WT and KO mice. Knocking out the HCN1 subunit or blocking all Ih in cortical neurons by using Cs+ induced a hyperpolarization in membrane potential and an increase in input resistance (*, significantly different from WT control by one-way ANOVA, P < 0.05). The Cs+-sensitive voltage “sag” is plotted against peak membrane potential obtained during the current injection; the sag in HCN1-KO mice was nearly absent at −88 mV (see arrow) and was diminished relative to WT animals at all potentials. All recordings were performed in the continued presence of bicuculline (30 μM), strychnine (30 μM), tetrodotoxin (TTX, 0.5 μM), and barium (0.2 mM).
Overall, this analysis reveals differences in Ih characteristics and intrinsic membrane properties of cortical pyramidal neurons that are expected from deletion of the HCN1 subunit. In addition, our results are remarkably similar to those reported previously for hippocampal pyramidal neurons of HCN1 knockout and wild-type mice (Nolan et al. 2004).
Effects of isoflurane on Ih attributed to HCN1 subunits were absent in cortical cells from HCN1 knockout animals
In previous work, we correlated anesthetic actions on cloned HCN channels and native neuronal Ih to assign distinct features of anesthetic modulation to particular HCN subunits. Specifically, we found that modulation of homomeric HCN1 channels by inhalational anesthetics was associated primarily with hyperpolarizing shifts in V1/2, whereas modulation of homomeric HCN2 channels was associated with decreased current amplitude; with heteromeric HCN1–HCN2 channels, inhibition by inhalational anesthetics included effects on both current amplitude and voltage dependence (Chen et al. 2005b). Our next set of experiments took advantage of HCN1 knockout mice, and the demonstrated absence of HCN1-like currents in cortical neurons, to test directly the subunit-specific effects proposed for these anesthetics.
The effects of the inhalational anesthetic isoflurane on cortical pyramidal neuron Ih are presented in Fig. 3. For these experiments, we used supraclinical concentrations of isoflurane (0.9 mM) to ensure full modulation of neuronal currents in cells from both wild-type and knockout mice. Isoflurane caused inhibition of maximal current amplitude (−455.5 ± 89.4 vs. −238.9 ± 51.0 pA, n = 9, P < 0.001, paired t-test) and a negative shift in voltage dependence of Ih activation (V1/2: −94.3 ± 2.3 vs. −106.0 ± 2.5 mV, P < 0.001) in neurons from wild-type animals (Fig. 3, A and B, top). In cells from HCN1 knockout mice, isoflurane decreased the amplitude of the small residual Ih (see inset in Fig. 3B, bottom left) but had no effect on voltage dependence of activation (Fig. 3B, bottom right); current amplitude at −128 mV was reduced by isoflurane from −120.2 ± 18.1 to −58.8 ± 19.1 pA (n = 6, P < 0.05, paired t-test), but V1/2 remained unchanged in the presence of isoflurane (−106.4 ± 1.7 vs. −106.9 ± 1.9 mV, P = 0.69). Summary data indicate that, whereas isoflurane diminished Ih by about 50% in cells from both wild-type and knockout animals (Fig. 3C; 48.2 ± 3.5 vs. 50.3 ± 13.5%, n = 9 and 6), the shift in V1/2 induced by isoflurane (−11.7 ± 2.1 mV, n = 9) in cortical neurons from wild-type mice was not observed in cells from HCN1 knockout mice (Fig. 3D).
FIG. 3.

Subunit-selective effects of isoflurane on native Ih are revealed in cortical pyramidal neurons from HCN1-KO mice. A: sample voltage-clamp recordings illustrating Ih in cortical pyramidal neurons from WT (upper) and KO (lower) mice under control conditions (left) and in the presence of 0.9 mM isoflurane. B: effects of isoflurane on Ih properties in cortical neurons from WT (top) and HCN1-KO mice (bottom). Time-dependent currents were measured at the end of hyperpolarizing voltage steps under control conditions (squares) and in the presence of isoflurane (triangles) and plotted as a function of membrane potential (left). Likewise, normalized tail currents were obtained following incrementing hyperpolarizing steps, averaged, and fitted with Boltzmann curves in control and in the presence of isoflurane (right). In cells from WT animals, isoflurane induced a shift in half-activation voltage (V1/2) together with inhibition of maximal current amplitude. In cortical neurons from HCN1-KO mice, isoflurane retained the ability to inhibit current amplitude (see inset for data on expanded scale in B, bottom) but was without effect on V1/2 (*, significantly different from control by paired t-test, P < 0.05). C: averaged isoflurane-evoked inhibition of maximal current amplitude (at −130 mV) in cells from WT and HCN1-KO mice, recorded with or without 50 μM cAMP in the recording pipette. The inhibition of Ih amplitude by isoflurane was preserved in cells from HCN1-KO mice, but amplitude inhibition was reduced by cAMP. D: voltage dependence of Ih activation was determined in WT and KO cells (with or without cAMP) by using normalized tail currents fitted with Boltzmann curves; averaged V1/2 (left) and effects of isoflurane on activation gating (ΔV1/2, right) indicate that initial V1/2 was shifted to more hyperpolarized potentials in HCN1-KO cortical neurons and the hyperpolarizing shift in V1/2 typically associated with isoflurane was absent in those KO neurons. In the presence of cAMP, however, isoflurane caused a shift in V1/2 from a more depolarized initial value in cells from HCN1-KO mice (*, significantly different by one-way ANOVA or 2-way repeated-measures ANOVA, P < 0.05).
Inhalational anesthetics modulate HCN2 channels in a cAMP-dependent manner; when intrinsic allosteric inhibition of HCN2 channels is relieved by cAMP, modulation by anesthetics is associated with less amplitude inhibition and a more pronounced shift in V1/2 (Chen et al. 2005b). As shown in Fig. 3D, the V1/2 of Ih activation was about 10 mV more depolarized in cortical cells from HCN1 knockout mice recorded with 50 μM cAMP in the patch pipette, indicative of relief from intrinsic allosteric inhibition. Anesthetic modulation of the residual, cAMP-stimulated current in cortical cells from HCN1 knockout mice was as expected for HCN2 channels; the anesthetic-induced decrease in current amplitude was reduced to 18.5 ± 6% (vs. ∼50% without cAMP; P < 0.05) and a clear shift in V1/2 was now obtained (−6.6 ± 0.7 mV; from −96.9 ± 1.3 to −102.8 ± 1.6 mV; n = 5, P < 0.001).
We also compared effects of isoflurane on membrane potential, input resistance, and “sag” in cortical pyramidal neurons from wild-type and HCN1 knockout mice under current-clamp conditions (Fig. 4). All recordings were performed in the continued presence of bicuculline and strychnine (both at 30 μM), TTX (0.5 μM), and barium (0.2 mM). Representative traces from a wild-type cell are shown in Fig. 4A and averaged data from wild-type and HCN1 knockout mice are provided in Fig. 4, B and C. Isoflurane caused membrane hyperpolarization of neurons from wild-type animals, with subsequent application of 3 mM CsCl, further hyperpolarizing the cells (Fig. 4, A and B). Anesthetic actions were associated with an increase in RN (∼12% increase, from 197.8 ± 28.7 to 223.7 ± 24.4 MΩ; n = 5, both P < 0.05) and a decrease in voltage “sag” that was evident when compared using current steps that hyperpolarized the cell to the same peak potential (i.e., to −88 mV; Fig. 4C). These effects of isoflurane on membrane properties were mediated by Ih since they were eliminated in neurons from HCN1 knockout animals (Fig. 4, B and C).
FIG. 4.
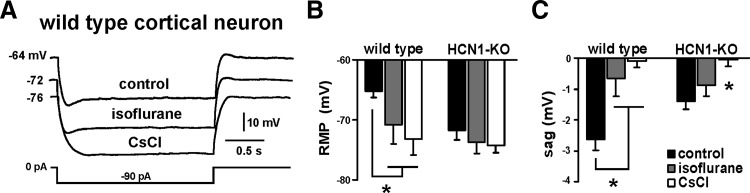
Isoflurane effects on membrane properties are diminished in cortical neurons from HCN1-KO mice. A: sample voltage responses to injected current in WT cortical neurons under control conditions and in the presence of isoflurane (0.9 mM), followed by 3 mM CsCl. B: isoflurane induced a membrane hyperpolarization in WT cells, whereas in neurons from HCN1-KO mice, initial membrane potential was more hyperpolarized and isoflurane did not cause further hyperpolarization. C: voltage-dependent “sag” was determined during current steps that reached the same peak voltage (∼ −88 mV). In WT neurons, isoflurane significantly decreased voltage “sag.” In HCN1-KO animals the initial “sag” was reduced and unaffected by isoflurane. All recordings were performed in the continued presence of bicuculline (30 μM), strychnine (30 μM), TTX (0.5 μM), and barium (0.2 mM) (*, significantly different from control, P < 0.05 by 2-way ANOVA).
Deletion of HCN1 also reduces Ih in motoneurons, leaving a residual HCN2-like current modulation by anesthetics
We also verified HCN subunit-selective modulation of Ih by isoflurane in motoneurons; these cells are implicated in immobilizing anesthetic actions (Franks 2006; Rudolph and Antkowiak 2004; Sonner et al. 2003) and they express HCN1 and HCN2 subunits (Monteggia et al. 2000; Santoro et al. 2000). Hypoglossal motoneurons were recorded in brain stem slices using an acidified bath (pH 6.5) to inhibit motoneuronal anesthetic-sensitive TASK-like currents, and with elevated HEPES in the pipette (25 mM) to minimize changes in intracellular pH (Sirois et al. 2002); Ih properties were not affected by these manipulations (data not shown; see Sirois et al. 2002).
Recordings were performed in motoneurons from neonatal control and HCN1 knockout mice (P11–P15); the age distribution was not different between mouse lines and we found no age-dependent changes in Ih density within the age span considered, in either group of cells (data not shown). However, the magnitude and properties of motoneuronal Ih were different between cells from the two mouse lines. Sample voltage-clamp records in Fig. 5A depict Ih in representative motoneurons from control (top) and HCN1 knockout mice (bottom). Compared with cells from wild-type animals, Ih was reduced in amplitude (by ∼32% at −128 mV; Fig. 5B, left) and activated about fivefold more slowly in motoneurons from HCN1 knockout mice (Fig. 5B, right). In addition, the initial V1/2 of Ih activation was more hyperpolarized in motoneurons from HCN1 knockout mice (−108.9 ± 1.2 vs. −113.5 ± 1.4 mV in control and knockout, respectively; n = 15 and 14, P < 0.05). These data are as expected for deletion of fast activating HCN1 subunits from motoneurons and retention of a slower HCN2-like current that activates at more hyperpolarized potentials.
FIG. 5.

Ih is reduced in amplitude and isoflurane effects on Ih are HCN2-like in motoneurons from HCN1-KO mice. A: sample voltage-clamp recordings depict Ih in hypoglossal motoneurons from WT (top) and HCN1-KO mice (bottom) under control conditions (left) and in the presence of isoflurane (0.9 mM; right). Note that residual Ih in the KO motoneuron activates more slowly and is reduced in amplitude compared with WT; also note that inhibition of Ih by isoflurane remains substantial in HCN1-KO motoneurons. Cells were recorded in an acidified bath (pH 6.5) with raised intracellular HEPES (25 mM) to minimize effects on voltage-clamp conditions secondary to anesthetic activation of motoneuronal TASK-like currents. B, left: averaged amplitude of Ih (at −130 mV) was determined in individual motoneurons from WT and HCN1-KO mice; Ih was reduced by about 33% in the KO cells. Right: averaged fast activation time constants (τf at −120 mV) for Ih in motoneurons from control and HCN1-KO mice; current activation was about 5-fold slower in KO motoneurons (*, significantly different from WT by unpaired t-test, P < 0.05). C: effects of isoflurane on Ih in motoneurons from WT and HCN1-KO mice. Left: averaged isoflurane-evoked inhibition of maximal current amplitude (at −130 mV) was not different in cells from WT and HCN1-KO mice. Right: the isoflurane-induced hyperpolarizing shift in V1/2 was absent in motoneurons from HCN1-KO mice (*, significantly different from WT by one-way ANOVA, P < 0.05). D: basic membrane properties and effects of isoflurane in motoneurons from WT and HCN1-KO mice. Left: averaged RMP was more hyperpolarized and isoflurane had no effect on RMP in HCN1-KO mice. Right: averaged initial RN was greater in HCN1-KO mice and isoflurane inhibition of residual Ih led to a further increase in RN (*, significantly different from WT by one-way ANOVA, P < 0.05).
As shown in Fig. 5, A and C, we also found that anesthetic effects on Ih in motoneurons from control and HCN1 knockout mice were consistent with HCN subunit-selective anesthetic actions (Chen et al. 2005b). Isoflurane (0.9 mM) decreased current amplitude in both control (% inhibition: 40.1 ± 5.7%, n = 6; P < 0.05, paired t-test) and HCN1 knockout motoneurons (% inhibition: 32.1 ± 5.4%, n = 6; P < 0.05); however, the shift in V1/2 of activation typically evoked by isoflurane in motoneurons from control animals (ΔV1/2: −7.0 ± 1.3 mV; P < 0.05) was absent in cells from HCN1 knockout mice (ΔV1/2: −0.9 ± 1.5 mV; P = 0.56).
The smaller Ih in motoneurons from HCN1 knockout mice was associated with a more hyperpolarized membrane potential and an increased RN, as determined under whole cell current clamp (Fig. 5D). In motoneurons from wild-type mice, isoflurane caused hyperpolarization from the resting membrane potential and increased RN. In cells from HCN1 knockouts, isoflurane had no effect on resting membrane potential, where residual HCN2 currents show little activity; however, an isoflurane-induced increase in RN was apparent with hyperpolarizing current injection, as expected given the decrease in maximal Ih amplitude with isoflurane that we observed at hyperpolarized potentials.
In sum, the properties and anesthetic modulation of motoneuronal Ih were consistent with removal of HCN1 subunit-containing channels from those cells. In addition, results from motoneurons were generally similar to those obtained in cortical pyramidal cells. It is noteworthy, however, that Ih was considerably larger in motoneurons than that in cortical cells from wild-type mice (∼ −470 vs. −320 pA) and HCN1 subunit deletion had a smaller effect on motoneuronal Ih (i.e., in motoneurons of HCN1 knockout mice, residual Ih was about −300 pA, comparable to Ih in cortical neurons from wild-type mice). Thus by comparison to cortical neurons, HCN1 subunits provide a relatively smaller contribution to the larger Ih in motoneurons and the residual isoflurane-sensitive Ih in motoneurons from HCN1 knockout mice remains substantial in amplitude.
Isoflurane enhances temporal summation in cortical pyramidal neurons from wild-type mice, but not from HCN1 knockouts
HCN channel expression and Ih density increase with distance from the soma in pyramidal neurons (Magee 1999, 2000) and it has become clear that dendritic Ih acts to shunt synaptic currents and thereby decrease temporal summation of synaptic inputs (Desjardins et al. 2003; Magee 1999). This suggests the possibility that modulation of dendritic Ih and synaptic integration in cortical pyramidal neurons may also be a consequence of anesthetic action.
We first used a computer simulation in a cortical pyramidal neuron model (Day et al. 2005) to test independently the subunit-specific actions of anesthetics on Ih and assess their effects on EPSP temporal summation, incorporating: 1) 50% inhibition of peak current amplitude (e.g., mimicking effects of isoflurane on HCN2 homomeric channels); 2) −10-mV shift in V1/2 of Ih activation (e.g., mimicking effects of isoflurane on HCN1 homomeric channels); or 3) a combination of 50% amplitude inhibition with a −10-mV shift in V1/2 (e.g., mimicking effects of isoflurane on heteromeric HCN1–HCN2 channels). We performed the computer simulation from two initial membrane potentials. The first approximated resting membrane potential of cortical neurons (∼ −70 mV); the second, from about −90 mV, was chosen to maximize effects of Ih on EPSP summation, minimize effects of other voltage-dependent channels, and match conditions used in our experimental tests (see following text).
As depicted in Fig. 6, A and B, simulating anesthetic-induced inhibition of Ih caused membrane hyperpolarization and enhanced EPSP summation from either starting membrane potential; this was true regardless of whether the inhibition took the form of a decrease in current amplitude, a shift in V1/2, or both. As expected, the initial shunting effect of Ih on EPSP summation was most pronounced in the simulation performed at the more hyperpolarized potential, where Ih is most active (EPSP5:EPSP1 was 2.1 at −70 mV and 1.5 at −90 mV); likewise, inhibition of Ih also had a more prominent effect on membrane potential and EPSP summation in the simulation performed at −90 mV. In that case, simulating decreased Ih amplitude, a −10-mV shift in V1/2, and both produced a hyperpolarization of the neuron model (from −90.8 to −93.4, −95.1, and −98.4 mV, respectively) and enhanced temporal summation of EPSPs (EPSP5:EPSP1 increased from 1.5 to 1.7, 2.2, and 2.4, respectively). Note that the shift in V1/2, by itself, produced a large relative enhancement of EPSP summation (48% increase) that was much greater than that accompanying the decrease in current amplitude (15% increase) and only slightly less than that obtained with the combination of both amplitude inhibition and a shift in V1/2 (59% increase). Note, also, that the enhanced EPSP summation accompanying Ih inhibition was not due to the change in membrane potential since hyperpolarizing the neuron model to those same initial potentials with simulated DC current injection, in the absence of Ih inhibition, actually depressed the EPSP5:EPSP1 ratio (data not shown).
FIG. 6.

Isoflurane enhances excitatory postsynaptic potential (EPSP) temporal summation in cortical pyramidal neurons from WT mice, but not from HCN1-KO mice. A and B: computer simulations in a cortical neuron model from 2 different initial potentials (−70 and −90 mV) illustrate effects on EPSP summation accompanying a 50% decrease in Ih amplitude, −10-mV shift in V1/2 of activation, or both. Aligned traces illustrate the enhanced summation observed in the model with both a decrease in Ih amplitude and a shift in V1/2, and histogram plots show effects on EPSP summation of independently or coordinately changing these 2 parameters in the model. C: sample voltage traces show EPSP recordings obtained from cortical pyramidal neuron in response to 40-Hz, 7-V stimulation under control conditions and after incubation in 0.4 mM isoflurane (right). To enhance basal dendritic Ih, current injection was used to produce an initial somatic membrane potential of −90 mV; EPSPs are aligned to initial membrane potential and normalized to the amplitude of the first EPSP in the train to highlight enhanced temporal summation of EPSPs. D: summation was quantified as the amplitude ratio of the 5th to 1st EPSP and plotted with respect to time; isoflurane enhanced temporal summation. Note that DC injection to hyperpolarize the neuron caused a decrease of EPSP summation, consistent with a contribution of Ih to the sublinear summation under control conditions. E: averaged percentage increase of EPSP summation (ratio of EPSP5/EPSP1, relative to control) induced by 0.4 mM isoflurane or 50 μM ZD-7288 or in WT and HCN1-KO mice; isoflurane and ZD-7288 enhanced EPSP summation in cortical neurons from WT animals (*P < 0.05 vs. control), but had no effect in cells from HCN1-KO mice.
We next examined experimentally the effect of isoflurane on temporal summation of EPSPs evoked in cortical pyramidal neurons by using local field stimulation (40 Hz, 5–10 V) in superficial cortical layers. In cells from wild-type mice, we found that isoflurane (0.4 mM) caused membrane hyperpolarization and enhanced EPSP summation by >60% (Fig. 6, C–E), producing results similar to those obtained with the neuron simulation. Again, the enhanced EPSP summation was observed in spite of the hyperpolarization, which by itself diminished summation (e.g., see effect of DC injection in Fig. 6D). It should also be noted that individual EPSP amplitudes were decreased by isoflurane at these concentrations (Fig. 6C, right), as previously reported by others (Berg-Johnsen and Langmoen 1992; Maclver et al. 1996; Wu et al. 2004); to the extent possible, we adjusted for decreased EPSP amplitudes by increasing stimulus intensity in the presence of isoflurane. The enhanced EPSP summation by isoflurane could be attributed to Ih inhibition since it was mimicked by ZD-7288 (50 μM), a potent Ih channel antagonist, and diminished in neurons from HCN1 knockout mice (Fig. 6E). Note that although isoflurane decreased maximal Ih in cortical neurons from HCN1 knockout mice (see Fig. 3), this likely had only relatively modest effects on EPSP summation in the knockout since the initial shunting action of the small residual HCN currents would be minimal.
During slow-wave sleep and in light planes of anesthesia, a cortically derived rhythm underlies synchronous neuronal firing at low frequencies (<1 Hz), with coherent cortical activity driven by synaptic inputs (Steriade et al. 1993b). To determine how effects of isoflurane on Ih and EPSP summation influence overall cortical neuron excitability we exposed cortical pyramidal neurons to a low, hypnotic concentration of isoflurane (0.15 mM) while applying the 40-Hz stimulus train every second, mimicking an approximately 1-Hz rhythm of cortical synaptic inputs. An example is shown in Fig. 7. Under control conditions, the cell occasionally fired an action potential triggered by summating EPSPs (Fig. 7, A, left and C). Exposure to isoflurane caused a small membrane hyperpolarization (∼2–3 mV) and enhanced excitability such that many more stimulus events were associated with action potential discharge (Fig. 7, A, middle and C); after a brief wash, the effect on excitability partially reversed (Fig. 7, A, right and C). Examination of individual events revealed that isoflurane caused an increase in EPSP summation that was sufficient to trigger action potential discharge despite the attendant membrane hyperpolarization (Fig. 7B). We obtained similar results in two additional pyramidal neurons. Thus at concentrations of isoflurane expected to support synaptically driven coherent cortical activity in vivo, we found a net increase in synaptic excitability that could be attributed to effects of isoflurane on Ih (i.e., membrane hyperpolarization and increased EPSP summation).
FIG. 7.

Inhibition of Ih by isoflurane increases synaptic excitability in cortical neurons. A: effects on WT cortical pyramidal neurons of 40-Hz electrical stimulation delivered at a rate of 1 Hz to the superficial cortex under control conditions (left), in the presence of 0.15 mM isoflurane (middle), and following wash (right). (Spikes are truncated in this panel and in B.) B: an expanded view of 2 stimulus events under control conditions (blue) and during exposure to isoflurane (red); note the slight hyperpolarization and enhanced EPSP summation in isoflurane, when both events were associated with spike firing. C: quantification of data from this cell, illustrating the increased likelihood that the synaptic stimulation triggered a spike in the presence of isoflurane (as a percentage of all stimulation events in a 2-min epoch under the indicated conditions). These results are representative of those obtained in the 2 other cells studied with this protocol.
DISCUSSION
Inhalational anesthetics induce subunit-specific inhibition of HCN channels that is evident at clinically relevant concentrations; this appeared to be reproduced in recordings of Ih from neurons that natively express different HCN subunits (Chen et al. 2005b). In the present study, we used whole cell recording in motoneurons and cortical pyramidal neurons from HCN1 knockout mice to verify subunit-specific actions of inhalational anesthetics on native Ih. In these neurons from HCN1 knockout mice, isoflurane did not cause the typical HCN1-like hyperpolarizing shift in V1/2 of activation, but it inhibited maximal amplitude of a substantially smaller and slower Ih, an HCN2-like action; by comparison to cortical neurons, the larger fraction of residual current in motoneurons suggests a relatively greater contribution from HCN2 channel subunits in those cells. Under current clamp, we found that anesthetic-induced inhibition of Ih caused membrane hyperpolarization and enhanced temporal summation of evoked EPSPs in cortical cells; in NEURON simulations, changes in Ih that were reminiscent of isoflurane actions on HCN1-containing channels (i.e., hyperpolarizing shift in V1/2) had the most pronounced effect on synaptic summation and, accordingly, the isoflurane-enhanced EPSP summation was absent in HCN1 knockout mice. In wild-type mice, the enhanced synaptic summation evoked by isoflurane increased the probability of cortical neuron discharge. These data indicate that isoflurane acts primarily by inhibition of HCN1-containing channels to cause membrane hyperpolarization and enhanced synaptic summation, conditions that favor generation of synchronized cortical slow wave oscillations that are associated with sleep and anesthetic-induced hypnosis (Bazhenov et al. 1998; Hill and Tononi 2005).
HCN subunit-specific effects of general anesthetics on Ih
Anesthetic modulation of Ih causes membrane hyperpolarization, increased RN, and a decrease in voltage-dependent “sag.” The anesthetic-induced changes in membrane properties were absent in neurons from HCN1 knockout mice, suggesting that those effects primarily reflect inhibition of HCN1-containing channels. This result is consistent with the idea that HCN1 homomeric and/or HCN1–HCN2 heteromeric channels provide a substantial contribution to Ih near neuronal resting membrane potential by virtue of their relatively depolarized V1/2 of activation (Chen et al. 2001; Franz et al. 2000; see also Nolan et al. 2004, 2007).
In earlier work, we attributed distinct effects of inhalational anesthetics on neuronal Ih to specific HCN subunits (Chen et al. 2005b); those conclusions are now strongly supported by this work. We previously found that different forms of Ih inhibition by inhalational anesthetics—shift in V1/2 and decrease in current amplitude—were associated, respectively, with homomeric HCN1 and HCN2 channels; in heteromeric HCN1–HCN2 channels, inhalational anesthetics caused both a shift in V1/2 and a decrease in current (Chen et al. 2005b). We could attribute the subunit-specific effects of anesthetics to differences in allosteric inhibition imposed by HCN subunit C-terminal domains, as reflected in the initial V1/2 of Ih activation (Chen et al. 2005b). Accordingly, we found here that isoflurane shifted V1/2 and decreased current amplitude in cortical pyramidal neurons from wild-type animals, where Ih included both an HCN1 and HCN2 subunit contribution and presented with a relatively depolarized initial V1/2 (∼91 mV). In cells from HCN1 knockout mice, where residual Ih was slower and activated at more hyperpolarized potentials (V1/2 ≈ −106 mV), as expected for HCN2 homomeric channels, the isoflurane-induced inhibition of Ih included only a decrease in current amplitude, with no concomitant shift in V1/2. In cells from HCN1 knockout animals recorded in the presence of cAMP, the initial V1/2 of activation of the residual Ih was shifted to depolarized levels and isoflurane modulation included a prominent hyperpolarizing shift in V1/2 and a smaller inhibition of current amplitude. This effect of cAMP on anesthetic modulation is also consistent with previous results obtained with cloned HCN2 channels.
Anesthetics enhance EPSP summation in cortical neurons by inhibiting Ih
It is now clear that dendritic Ih in cortical pyramidal neurons, which increases in density with distance from the soma, serves as a current shunt to reduce temporal summation of synaptic inputs; inhibition of Ih decreases the dendritic shunt, resulting in enhanced summation of synaptic inputs (Magee 1999, 2000). Our results indicate that isoflurane enhances EPSP summation; these effects were predicted by simulating anesthetic-induced Ih inhibition in a cortical neuron computer model and they were mimicked by application of the Ih blocker ZD-7288. Moreover, the anesthetic-induced increase in temporal summation observed in wild-type mice was absent in HCN1 knockout mice, further verifying a role for inhibition of Ih. Interestingly, our computer simulation revealed that a shift in V1/2 (an HCN1-like effect) induced a relatively larger enhancement of EPSP summation than a decrease in current amplitude (an HCN2-like effect), suggesting that anesthetic modulation of HCN1-containing channels may potentiate synaptic inputs more effectively than inhibition of HCN2 homomeric channels. The combination of synaptic enhancement (via effects on dendritic Ih) with membrane hyperpolarization (via effects on somatic Ih) provides a background in which isolated synaptic inputs are less effective, whereas effects of synchronous excitatory synaptic potentials are enhanced. In general, this situation would favor coherent oscillatory behavior (Carr et al. 2007) and, indeed, decreases in Ih have been observed in a number of epilepsy models in rodents (Jung et al. 2007; Kole et al. 2007; Shah et al. 2004).
Possible neural mechanism for HCN1-mediated actions of isoflurane
Could the isoflurane-mediated, HCN1 subunit-dependent membrane hyperpolarization and enhanced synaptic summation that we have described in cortical neurons contribute to behavioral actions of isoflurane?
At low concentrations of isoflurane, the anesthetic induces a hypnosis that is accompanied by EEG oscillations that resemble slow waves of deep sleep (Antkowiak 2002). In thalamic and cortical neurons, Ih contributes to intrinsic mechanisms that contribute to slow rhythms (McCormick and Bal 1997; Rudolph and Antkowiak 2004; Steriade et al. 1993a) and in thalamocortical network models, two major changes in cortical neuron properties are necessary for transitions into synchronized, oscillating cortical activity: membrane hyperpolarization and enhanced excitatory synaptic transmission (Bazhenov et al. 1998; Hill and Tononi 2005). As mentioned earlier, both of these effects accompany isoflurane-induced inhibition of Ih in cortical neurons. Moreover, we showed directly that, at hypnotic concentrations (0.1–0.2 mM), isoflurane acted to increase postsynaptic excitability in cortical neurons by enhancing EPSP summation (see Fig. 7). So, we suggest that inhibition of Ih by anesthetics contributes to the membrane hyperpolarization that supports oscillatory activity in cortical neurons while also reducing dendritic shunt to promote synaptically mediated synchronization of rhythmic cortical activity. Insofar as oscillations and cortical synchronization are critical components of slow-wave EEG activity, we suggest that these effects of isoflurane on HCN1-containing channels may contribute to anesthetic-induced hypnosis in wild-type animals.
It is now widely accepted that decreased motoneuronal excitability accounts for immobilizing effects of anesthetics (Franks 2006; Rudolph and Antkowiak 2004; Sonner et al. 2003) and we previously noted that isoflurane-mediated inhibition of Ih plays a part in decreasing motoneuronal excitability via its contribution to membrane hyperpolarization (Sirois et al. 1998). Our results from HCN1 knockout mice indicate that the hyperpolarizing shift in V1/2 of Ih caused by isoflurane in motoneurons, as in cortical cells, is due largely to effects on HCN1 subunits; likewise the hyperpolarizing actions of isoflurane mediated by Ih inhibition can also be attributed to HCN1-containing channels. Unlike in cortical cells, however, there is no polarized distribution of HCN1 channels in motoneurons, which instead are expressed strongly on both somatic and dendritic membranes (Milligan et al. 2006). Thus it is uncertain whether any synaptic enhancement that might result from dendritic HCN1 channel inhibition can overcome the concurrent somatic membrane hyperpolarization to increase excitability in motoneurons, as we observed in cortical pyramidal cells. Moreover, activation of TASK-like background K+ currents by isoflurane largely accounts for the depressed somatic excitability of motoneurons by causing membrane hyperpolarization and increased input conductance (Sirois et al. 1998, 2000), effects that would further limit any increased excitability due to synaptic enhancement; such a countervailing effect on excitability was not observed in cortical pyramidal cells, where we found little evidence for K+ channel modulation by isoflurane (Chen and Bayliss, unpublished observations). It is also worth noting that we found a more substantial isoflurane-sensitive HCN2-like residual Ih in motoneurons from HCN1 knockout mice (∼30% inhibition of a current that remained ∼70% of wild-type) that could contribute to net effects of isoflurane on motoneuronal excitability (e.g., by increasing RN and enhancing synaptic integration). So, if inhibition of Ih plays a role in immobilizing effects of isoflurane at the level of motoneurons, this likely involves actions on channels including both HCN1 and HCN2 subunits.
The cellular data we obtained from HCN1 knockout mice are consistent with the idea that HCN1 subunits are a viable target for isoflurane action and may contribute to its anesthetic actions in wild-type mice. However, this does not imply that HCN1 knockout mice are themselves a good model for assessing the behavioral consequences of HCN1 deletion; indeed, a number of caveats must be considered. First, isoflurane inhibits both HCN1 and HCN2 subunits, and only HCN1 is deleted in these knockouts. As we discuss with respect to effects on motoneurons and immobilization, a substantial residual HCN2-mediated Ih may continue to provide an effective target. Also, in thalamus, a brain region linked to sleep and hypnosis (McCormick and Bal 1997; Rudolph and Antkowiak 2004; Steriade et al. 1993a), HCN2 is the predominant subunit in both thalamocortical and thalamic reticular neurons (Ludwig et al. 2003; Santoro et al. 2000; Ying et al. 2007); effects of isoflurane on Ih would be preserved in those cells from HCN1 knockout mice. For cortical neurons, where HCN1 subunits are largely responsible for Ih, a second complicating issue is that of compensation; in those cells, we find a markedly enhanced (approximately threefold) isoflurane-activated tonic GABAA current (Chen and Bayliss, unpublished observations). The presence of this confounding isoflurane-sensitive current in the knockout mice precludes simple interpretation of behavioral data in terms of HCN1 deletion from those cells.
In conclusion, by using HCN1 knockout mice, we verified HCN1 subunit-specific contributions to effects of isoflurane on Ih in motoneurons and cortical pyramidal neurons. We also showed that inhibition of HCN1-containing channels largely accounts for isoflurane-induced hyperpolarization and increased temporal summation of EPSPs in cortical pyramidal neurons, two cellular effects associated with slow-wave thalamocortical oscillations similar to those observed in light planes of anesthesia. A definitive test of a behavioral role for inhibition of Ih by isoflurane may require experiments with knockout of both HCN1 and HCN2 and/or development of HCN knockout mice without confounding compensatory currents (e.g., by using GABAA and HCN1 double knockout mice).
GRANTS
This work was supported by American Heart Association Beginning Grant-in-Aid 0665349U to X. Chen and GM66181 to D. A. Bayliss.
Acknowledgments
We thank Drs. Patrice Guyenet and Ruth Stornetta for access to and instruction on Neurolucida software.
The costs of publication of this article were defrayed in part by the payment of page charges. The article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
REFERENCES
- Antkowiak 2002.Antkowiak B In vitro networks: cortical mechanisms of anaesthetic action. Br J Anaesth 89: 102–111, 2002. [DOI] [PubMed] [Google Scholar]
- Bazhenov 1998.Bazhenov M, Timofeev I, Steriade M, Sejnowski TJ. Computational models of thalamocortical augmenting responses. J Neurosci 18: 6444–6465, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg-Johnsen 1992.Berg-Johnsen J, Langmoen IA. The effect of isoflurane on excitatory synaptic transmission in the rat hippocampus. Acta Anaesthesiol Scand 36: 350–355, 1992. [DOI] [PubMed] [Google Scholar]
- Carr 2007.Carr DB, Andrews GD, Glen WB, Lavin A. α2-Noradrenergic receptors activation enhances excitability and synaptic integration in rat prefrontal cortex pyramidal neurons via inhibition of HCN currents. J Physiol 584: 437–450, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen 2001.Chen C, Wang C, Siegelbaum SA. Properties of hyperpolarization-activated pacemaker current defined by coassembly of HCN1 and HCN2 subunits and basal modulation by cyclic nucleotide. J Gen Physiol 117: 491–503, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen 2005a.Chen X, Shu S, Bayliss DA. Suppression of Ih contributes to propofol-induced inhibition of mouse cortical pyramidal neurons. J Neurophysiol 94: 3872–3883, 2005a. [DOI] [PubMed] [Google Scholar]
- Chen 2005b.Chen X, Sirois JE, Lei QB, Talley EM, Lynch C, Bayliss DA. HCN subunit-specific and cAMP-modulated effects of anesthetics on neuronal pacemaker currents. J Neurosci 25: 5803–5814, 2005b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day 2005.Day M, Carr DB, Ulrich S, Ilijic E, Tkatch T, Surmeier DJ. Dendritic excitability of mouse frontal cortex pyramidal neurons is shaped by the interaction among HCN, Kir2, and K(leak) channels. J Neurosci 25: 8776–8787, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desjardins 2003.Desjardins AE, Li YX, Reinker S, Miura RM, Neuman RS. The influences of Ih on temporal summation in hippocampal CA1 pyramidal neurons: a modeling study. J Comput Neurosci 15: 131–142, 2003. [DOI] [PubMed] [Google Scholar]
- Destexhe 2003.Destexhe A, Sejnowski TJ. Interactions between membrane conductances underlying thalamocortical slow-wave oscillations. Physiol Rev 83: 1401–1453, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eger 1997.Eger EI, Koblin DD, Harris RA, Kendig JJ, Pohorille A, Halsey MJ, Trudell JR. Hypothesis: inhaled anesthetics produce immobility and amnesia by different mechanisms at different sites. Anesth Analg 84: 915–918, 1997. [DOI] [PubMed] [Google Scholar]
- Franks 2006.Franks NP Molecular targets underlying general anaesthesia. Br J Pharmacol 147, Suppl. 1: S72–S81, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franz 2000.Franz O, Liss B, Neu A, Roeper J. Single-cell mRNA expression of HCN1 correlates with a fast gating phenotype of hyperpolarization-activated cyclic nucleotide-gated ion channels (Ih) in central neurons. Eur J Neurosci 12: 2685–2693, 2000. [DOI] [PubMed] [Google Scholar]
- Hill 2005.Hill S, Tononi G. Modeling sleep and wakefulness in the thalamocortical system. J Neurophysiol 93: 1671–1698, 2005. [DOI] [PubMed] [Google Scholar]
- Hines 1997.Hines ML, Carnevale NT. The NEURON simulation environment. Neural Comput 9: 1179–1209, 1997. [DOI] [PubMed] [Google Scholar]
- Jung 2007.Jung S, Jones TD, Lugo JN Jr, Sheerin AH, Miller JW, D'Ambrosio R, Anderson AE, Poolos NP. Progressive dendritic HCN channelopathy during epileptogenesis in the rat pilocarpine model of epilepsy. J Neurosci 27: 13012–13021, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kole 2007.Kole MH, Brauer AU, Stuart GJ. Inherited cortical HCN1 channel loss amplifies dendritic calcium electrogenesis and burst firing in a rat absence epilepsy model. J Physiol 578: 507–525, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen 2006.Larsen DD, Callaway EM. Development of layer-specific axonal arborizations in mouse primary somatosensory cortex. J Comp Neurol 494: 398–414, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauritzen 2003.Lauritzen I, Zanzouri M, Honore E, Duprat F, Ehrengruber MU, Lazdunski M, Patel AJ. K+-dependent cerebellar granule neuron apoptosis. Role of task leak K+ channels. J Biol Chem 278: 32068–32076, 2003. [DOI] [PubMed] [Google Scholar]
- Linden 2007.Linden AM, Sandu C, Aller MI, Vekovischeva OY, Rosenberg PH, Wisden W, Korpi ER. TASK-3 knockout mice exhibit exaggerated nocturnal activity, impairments in cognitive functions, and reduced sensitivity to inhalation anesthetics. J Pharmacol Exp Ther 323: 924–934, 2007. [DOI] [PubMed] [Google Scholar]
- Ludwig 2003.Ludwig A, Budde T, Stieber J, Moosmang S, Wahl C, Holthoff K, Langebartels A, Wotjak C, Munsch T, Zong X, Feil S, Feil R, Lancel M, Chien KR, Konnerth A, Pape HC, Biel M, Hofmann F. Absence epilepsy and sinus dysrhythmia in mice lacking the pacemaker channel HCN2. EMBO J 22: 216–224, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maclver 1996.Maclver MB, Mikulec AA, Amagasu SM, Monroe FA. Volatile anesthetics depress glutamate transmission via presynaptic actions. Anesthesiology 85: 823–834, 1996. [DOI] [PubMed] [Google Scholar]
- Magee 1999.Magee JC Dendritic Ih normalizes temporal summation in hippocampal CA1 neurons. Nat Neurosci 2: 508–514, 1999. [DOI] [PubMed] [Google Scholar]
- Magee 2000.Magee JC Dendritic integration of excitatory synaptic input. Nat Rev Neurosci 1: 181–190, 2000. [DOI] [PubMed] [Google Scholar]
- McCormick 1997.McCormick DA, Bal T. Sleep and arousal: thalamocortical mechanisms. Annu Rev Neurosci 20: 185–215, 1997. [DOI] [PubMed] [Google Scholar]
- Milligan 2006.Milligan CJ, Edwards IJ, Deuchars J. HCN1 ion channel immunoreactivity in spinal cord and medulla oblongata. Brain Res 1081: 79–91, 2006. [DOI] [PubMed] [Google Scholar]
- Monteggia 2000.Monteggia LM, Eisch AJ, Tang MD, Kaczmarek LK, Nestler EJ. Cloning and localization of the hyperpolarization-activated cyclic nucleotide-gated channel family in rat brain. Mol Brain Res 81: 129–139, 2000. [DOI] [PubMed] [Google Scholar]
- Nolan 2007.Nolan MF, Dudman JT, Dodson PD, Santoro B. HCN1 channels control resting and active integrative properties of stellate cells from layer II of the entorhinal cortex. J Neurosci 27: 12440–12451, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolan 2004.Nolan MF, Malleret G, Dudman JT, Buhl DL, Santoro B, Gibbs E, Vronskaya S, Buzsáki G, Siegelbaum SA, Kandel ER, Morozov A. A behavioral role for dendritic integration: HCN1 channels constrain spatial memory and plasticity at inputs to distal dendrites of CA1 pyramidal neurons. Cell 119: 719–732, 2004. [DOI] [PubMed] [Google Scholar]
- Nolan 2003.Nolan MF, Malleret G, Lee KH, Gibbs E, Dudman JT, Santoro B, Yin D, Thompson RF, Siegelbaum SA, Kandel ER, Morozov A. The hyperpolarization-activated HCN1 channel is important for motor learning and neuronal integration by cerebellar Purkinje cells. Cell 115: 551–564, 2003. [DOI] [PubMed] [Google Scholar]
- Pape 1996.Pape HC Queer current and pacemaker: the hyperpolarization-activated cation current in neurons. Annu Rev Physiol 58: 299–327, 1996. [DOI] [PubMed] [Google Scholar]
- Pape 1989.Pape HC, McCormick DA. Noradrenaline and serotonin selectively modulate thalamic burst firing by enhancing a hyperpolarization-activated cation current. Nature 340: 715–718, 1989. [DOI] [PubMed] [Google Scholar]
- Pfaffl 2001.Pfaffl MW A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 29: e45, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudolph 2004.Rudolph U, Antkowiak B. Molecular and neuronal substrates for general anaesthetics. Nat Rev Neurosci 5: 709–720, 2004. [DOI] [PubMed] [Google Scholar]
- Santoro 2000.Santoro B, Chen S, Luthi A, Pavlidis P, Shumyatsky GP, Tibbs GR, Siegelbaum SA. Molecular and functional heterogeneity of hyperpolarization-activated pacemaker channels in the mouse CNS. J Neurosci 20: 5264–5275, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah 2004.Shah MM, Anderson AE, Leung V, Lin X, Johnston D. Seizure-induced plasticity of h channels in entorhinal cortical layer III pyramidal neurons. Neuron 44: 495–508, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirois 2000.Sirois JE, Lei Q, Talley EM, Lynch C 3rd, Bayliss DA. The TASK-1 two-pore domain K+ channel is a molecular substrate for neuronal effects of inhalation anesthetics. J Neurosci 20: 6347–6354, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirois 2002.Sirois JE, Lynch C, Bayliss DA. Convergent and reciprocal modulation of a leak K+ current and Ih by an inhalational anaesthetic and neurotransmitters in rat brainstem motoneurones. J Physiol 541: 717–729, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirois 1998.Sirois JE, Pancrazio JJ, Lynch C, Bayliss DA. Multiple ionic mechanisms mediate inhibition of rat motoneurones by inhalation anaesthetics. J Physiol 512: 851–862, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonner 2003.Sonner JM, Antognini JF, Dutton RC, Flood P, Gray AT, Harris RA, Homanics GE, Kendig J, Orser B, Raines DE, Rampil IJ, Trudell J, Vissel B, Eger EI. Inhaled anesthetics and immobility: mechanisms, mysteries, and minimum alveolar anesthetic concentration. Anesth Analg 97: 718–740, 2003. [DOI] [PubMed] [Google Scholar]
- Steriade 1993a.Steriade M, McCormick DA, Sejnowski TJ. Thalamocortical oscillations in the sleeping and aroused brain. Science 262: 679–685, 1993a. [DOI] [PubMed] [Google Scholar]
- Steriade 1993b.Steriade M, Nuñez A, Amzica F. A novel slow (<1 Hz) oscillation of neocortical neurons in vivo: depolarizing and hyperpolarizing components. J Neurosci 13: 3252–3265, 1993b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talley 2000.Talley EM, Lei QB, Sirois JE, Bayliss DA. TASK-1, a two-pore domain K+ channel, is modulated by multiple neurotransmitters in motoneurons. Neuron 25: 399–410, 2000. [DOI] [PubMed] [Google Scholar]
- Wu 2004.Wu XS, Sun JY, Evers AS, Crowder M, Wu LG. Isoflurane inhibits transmitter release and the presynaptic action potential. Anesthesiology 100: 663–670, 2004. [DOI] [PubMed] [Google Scholar]
- Ying 2007.Ying SW, Jia F, Abbas SY, Hofmann F, Ludwig A, Goldstein PA. Dendritic HCN2 channels constrain glutamate-driven excitability in reticular thalamic neurons. J Neurosci 27: 8719–8732, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]