

Abstract
We describe a method for identifying genes encoding proteins with stereospecific intracellular localizations in the fission yeast Schizosaccharomyces pombe. Yeast are transformed with a gene library in which S. pombe genomic sequences are fused to the gene encoding the Aequorea victoria green fluorescent protein (GFP), and intracellular localizations are subsequently identified by rapid fluorescence screening in vivo. In a model application of these methods to the fission yeast nucleus, we have identified several novel genes whose products are found in specific nuclear regions, including chromatin, the nucleolus, and the mitotic spindle, and sequence similarities between some of these genes and previously identified genes encoding nuclear proteins have validated the approach. These methods will be useful in identifying additional components of the S. pombe nucleus, and further extensions of this approach should also be applicable to a more comprehensive identification of the elements of intracellular architecture in fission yeast.
The fission yeast Schizosaccharomyces pombe, with its well-developed genetics (1, 2), has been an excellent model organism for genetical analyses of fundamental problems of cell biology, most notably regulation the cell cycle (3). Recently we have initiated a study of cell morphogenesis in fission yeast, isolating mutants altered in cell shape (4–6). These mutants have identified genes that play important roles in defining how the cell organizes itself in space and have been useful for initially characterizing the regulatory pathways involved in these processes. For cell-biological investigations of this nature, however, another useful point of entry is the identification and characterization of specific subcellular structures. To complement our studies of morphogenesis we would therefore like to identify gene products that are organized in specific regions of the cell and thus might represent structural elements under the control of these regulatory pathways. With this objective in mind we have developed a screening procedure to identify proteins that are localized to specific subcellular regions in fission yeast. A starting point for this approach was provided by the work of Burns et al. (7), who identified open reading frames in Saccharomyces cerevisiae on the basis of the spatial and temporal expression of randomly inserted LacZ gene fusions into the genome, using large-scale monitoring of β-galactosidase expression and immunofluorescent screening. Our method uses random fusions between S. pombe genomic fragments encoding open reading frames and the green fluorescent protein (GFP) gene, followed by rapid screening in vivo. In principle, this method allows the identification of new components of known subcellular structures, as well as the identification of altogether new structures. It should also increase the number of useful molecular markers available for fission yeast cytology, which will assist studies of cell morphogenesis. In the present paper we describe this procedure and its application to fission yeast nuclear structure; this work has identified new markers of previously recognized nuclear domains as well as markers of previously unrecognized domains.
MATERIALS AND METHODS
Standard molecular biology techniques were used throughout (8). The parent plasmid for library construction, pSGA, was constructed by two-step PCR amplification of the Aequorea victoria GFP coding sequence (kindly provided on plasmid TU65 by M. Chalfie, Columbia University, New York), which was then cloned into the thiamine-regulatable S. pombe expression vector REP3X (9, 10) (Fig. 1). The 5′ primer sequence in both steps of PCR was GGGGCTCGAGCCACCATGAGTAAAGGAGAAGAAC and the 3′ primer sequence was CTCACTCACTCAGTCGACGCGGCCGCGGTACCTTTGTATAGTTCATCCATGCCATG in the first step and GGGGATCCATGCATGTTTAAACTCACTCACTCAGTCGACGCG in the second step. The final PCR product leaves GFP without a termination codon, followed by restriction sites for KpnI, SacII, NotI, and SalI, termination codons (UGA) in all three reading frames, and then additional sites for PmeI, NsiI, and BamHI. This was digested with XhoI and BamHI and cloned into REP3X digested with the same enzymes.
Figure 1.
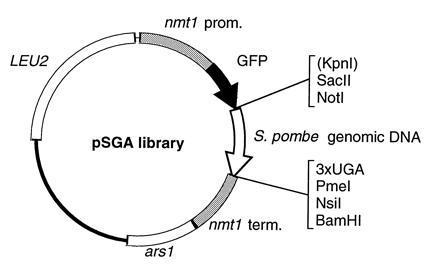
Diagram of the pSGA plasmid library used for screening (see Materials and Methods). Shown flanking the genomic DNA insert are unique restriction sites (KpnI is non-unique) and three stop codons, each in a different reading frame.
To construct a genomic library in pSGA, S. pombe genomic DNA was isolated (2) and further purified by CsCl centrifugation. The purified DNA was partially digested with Sau3AI, size-fractionated in the range 0.7–1.5 kb, and partially filled using DNA polymerase I Klenow fragment and dATP and dGTP. pSGA was digested with SalI and partially filled with dCTP and dTTP. By partial filling-in, neither vector nor insert ends should self-anneal during ligation (11). The vector and insert DNA were ligated and transformed into XL2-Blue cells (Stratagene), and approximately 100,000 independent transformant colonies were pooled to prepare library DNA. Analysis of random clones suggests that the library contains approximately 75% recombinants.
Fission yeast media and procedures were as described (2). For screening, the pSGA genomic library was introduced into strain PN43 (leu1-32 h-) by electroporation and plated onto minimal medium containing 5 μg/ml thiamine to repress expression from the nmt1 promoter. Because many inserts would be expected to be either out-of-frame with or in the opposite orientation to the GFP coding sequence, we enriched for in-frame inserts by replica-plating transformant colonies to minimal medium/phloxin B indicator plates lacking thiamine. This allowed us to identify colonies containing higher-than-normal proportions of sick or dying cells upon expression of GFP fusion proteins from the nmt1 promoter. The rationale behind this procedure was that these dying cells would predominantly be a consequence of hyperexpression of an in-frame fusion protein in a small percentage of cells in a given colony due to cell-to-cell variation in copy number. This turned out to be an effective way to enrich for in-frame insertions, and 90% of colonies chosen in this manner were still able to form colonies on media lacking thiamine (data not shown). Corresponding colonies on master plates containing thiamine were picked with toothpicks and placed into 384-well plates (Nunc), and then replica-spotted in the same 16 × 24 grid pattern to 150-mm round plates of minimal medium, plus and minus thiamine, using a custom-designed 384-pin spotter.
Approximately 24 hr after replica-spotting to minimal medium lacking thiamine (about 3–4 generations of growth after switching on the nmt1 promoter), colonies were screened by fluorescence microscopy as follows. A 1 × 2 inch rectangle of Whatman no. 1 filter paper was placed over a 4 × 8 array of colonies for about 30 sec. The filter was then removed and placed on a microscope slide and the colonies were transferred to the microscope slide by pressure from a second, overlying slide. After addition of 5 μl of minimal medium, colonies were covered with a 24 × 40 mm coverslip and taken to the microscope for observation. Although colonies were not fixed to the slide, cells remained mostly stationary for up to 30 min after making a replica, allowing about 1 min observation time per colony. For screening with longer observation times, a 4 × 4 array was used to allow more time between samples.
Screening was done on a Zeiss Axioplan microscope equipped with differential interference–contrast optics and a 100×/1.4 Pol Plan-Neofluar objective, and HQ fluorescein filter sets (Chroma Technology, Brattleboro, VT). Plasmids from colonies showing interesting localizations were isolated from yeast and shuttled once through Escherichia coli (either strains XL2-Blue or JA226) before retransformation and rescreening of yeast to confirm that the correct plasmid had been isolated.
For fixed-cell microscopy, log-phase cells grown under selective conditions in minimal medium containing thiamine were washed extensively in minimal medium lacking thiamine and then grown for 12–20 hr, with careful monitoring to obtain earliest visible expression levels. Cultures were fixed by addition of 4% formaldehyde/0.1% glutaraldehyde to the growth medium, and fixed for 15–30 min. Triton X-100 was added to 0.1% to permeabilize cells, which were then washed extensively with TBS. Cells (5 × 106) in 1 ml were then treated with 200 μg/ml RNase A for 2–4 hr at 37°C, and then stained with 4 μg/ml propidium iodide (PI). Cells were then centrifuged through a TBS/20% glycerol cushion onto polylysine-coated coverslips (12) and mounted in 80% glycerol/20 mM Tris, pH 8, containing 4 μg/ml PI. Series of optical sections were taken with a Bio-Rad model MRC-600 confocal microscope and a Nikon DM60×/1.4 objective and projected to single images using Bio-Rad software. Photomontages were created using Adobe Systems (Mountain View, CA) photoshop software.
Automated sequencing of plasmids was done using an ALF system (Pharmacia) and fluorescently labeled primers GCATGGATGAACTATAC from the C terminus of GFP and GGCTTCCATAGTTTGAAAGA from the 3′ terminator region of nmt1. Some sequencing was done using an ABI377 automated sequencer (Perkin–Elmer), using the same primers in unlabeled form. Database searches were done against GenBank data bases using Genetics Computer Group (Madison, WI) tfasta searches and also using tfasta and blast searches against Pombase at the Sanger Centre (Hinxton, U.K.; http://www.sanger.ac.uk/yeast/S.pombe_blast_server.html).
RESULTS
We constructed an S. pombe plasmid expression library in which random genomic sequences were fused to the GFP coding sequence under the control of the thiamine-regulatable nmt1 promoter. Cells expressing GFP from the parent plasmid pSGA produce an even cytoplasmic fluorescence when grown in media lacking thiamine and are essentially nonfluorescent in media containing thiamine (not shown; see also ref. 13). The pSGA genomic library was used to transform leu1-32 cells to leucine prototrophy, and we then enriched for clones likely to contain in-frame fusions (see Materials and Methods). In a trial of ≈384 colonies screened, 75 showed no fluorescence, 164 showed general cytoplasmic fluorescence, 40 showed clumped staining that was not easily interpreted, and 92 showed staining of potential interest. This included localization to the cell surface, vacuoles, and/or other intracellular membrane-bound compartments, cytoplasmic, and spindle microtubules, and to various regions of the cell nucleus. To test the validity of this approach, a selection of plasmids producing nuclear fluorescence were chosen for detailed analysis by confocal microscopy and were rescued from yeast and partially sequenced. These results are described below, grouped according to the localization pattern.
(i) General Nuclear Staining.
Several plasmids produced homogeneous nuclear fluorescence (Fig. 2). This was less bright in the nucleolus, which is easily recognized in fission yeast as the non-DNA-containing region of the nucleus, typically occupying one-third to one-half of total nuclear volume (see Fig. 2A′, arrowhead) (14). Fig. 2 A and A′ shows the localization of one such plasmid, M31; the uniform distribution throughout the nucleus, compared with chromosome staining, is particularly apparent during mitosis. Sequencing of M31 revealed that it resides on S. pombe cosmid c31F12, and a region of the relevant cosmid ORF was found to have a strong homology with a 45-amino acid region in the C-terminal portion of the budding yeast genes ZDS2 and ZDS1/OSS1 (15–17) (Table 1). These two genes have been isolated in a wide variety of distinct genetic screens, and their precise molecular functions, while probably overlapping, remain unclear (15, 16). A second clone, M26, also showed a similar pattern of nuclear fluorescence, but did not have any close relatives in database searches (not shown). A third plasmid, M30, showed a similar localization to M31 and M26 when expressed at low levels (Fig. 2 B and B′). Interestingly, when expressed at higher levels, M30 was found enriched at the nuclear periphery (Fig. 2 C and C′), and cells often became elongated or enlarged (not shown). Sequencing M30 showed it to be on cosmid c2F3, and data base searches using both M30 and its cognate cosmid ORF revealed that this coding sequence is closely related to human transportin (19) (see Table 1 and Discussion).
Figure 2.
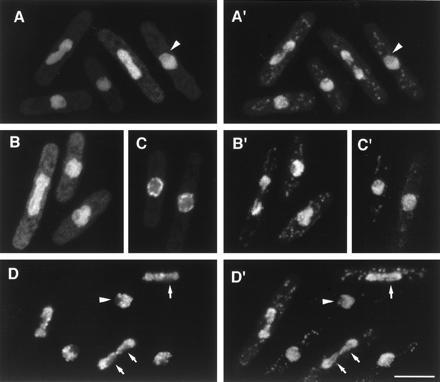
GFP fusion proteins localized to the nucleus. (A–D) GFP fluorescence; (A′–D′) corresponding images of DNA staining by PI. (A and A′) Localization of M31. (B and B′) Localization of M30 at low expression levels. (C and C′) Nuclear “rim” localization of M30 at higher expression levels (see text). (D and D′) Localization of M25 to nuclear chromatin. Note lack of staining in the interphase nucleolus, which stains weakly with PI (arrowhead), and lack of association of M25 with specific strands of segregating chromatin (arrows), which may be embedded in nucleolar material. (Bar = 5 μm.)
Table 1.
Plasmid localizations and sequence homologs
| Plasmid (cosmid)* | Localization | Homologs | Percent identity/similarity (over n amino acids) |
|---|---|---|---|
| M31 (c31F12) | Nucleus | S.c. Zds2p (U32938) | 67%/90% (45) |
| S.c. Oss1p (U63849) | 67%/93% (45) | ||
| M30 (c2F3) | Nucleus, | H.s. TRN (U70322) | 39%/76% (656) |
| nuclear rim | S.c. ORF YBR017c (Z35886) | 35%/76% (806) | |
| M26 | Nucleus | — | — |
| M25 | Chromatin | — | — |
| S32 (c29A4) | Nucleolus | — | — |
| S2 | Nucleolus (subregion?) | — | — |
| S19 (c29A4) | Nucleolar spots | S.c. Cbf5p (L12351) | 71%/95% (466) |
| R.n. NAP57 (Z34922) | 62%/91% (474) | ||
| S26 | Nucleolus, mitotic spindle | chromodomain proteins | (see Fig. 5) |
| S5† | Nuclear spot(s), | S.p. pcr1+(D63667) | 65%/88% (58) |
| mitotic spindle | H.s. ATF-2 (X15875) | 47%/79% (66) |
S.c., S. cerevisiae; H.s., Homo sapiens; R.n., Rattus norvegicus; S.p., S. pombe.
For plasmids found to reside on sequenced S. pombe cosmids, the cosmid identity is shown; in these cases the full cosmid ORF was used for sequence comparisons.
S5 is identical to atf21+ (Ref. 18); the comparison shown here is based on our sequence data only.
(ii) Association with the Chromatin-Containing Region.
Fig. 2 D and D′ show the localization of M25, which was also found in the nucleus, strongly associated with chromatin in both interphase and mitosis, and specifically absent from nucleolar regions (see Fig. 2 D and D′, arrowheads). During mitosis, M25 was often found to be absent from specific strands in the segregating chromatin mass (Fig. 2 D and D′, arrows); these may represent rDNA (which is nucleolar during interphase). High expression of M25 inhibited cell growth and resulted in very bright punctate nuclear staining, which appeared to superimpose with chromatin, now aberrantly organized (data not shown). M25 was partially sequenced, but no related sequences were found.
(iii) General Nucleolar Staining.
Four GFP fusion proteins showed distinct localizations in the nucleolus (Figs. 3 and 4). The fusion S32 (Fig. 3 A–E) appears to be a general nucleolar marker in both interphase and mitosis. DNA sequencing has shown that S32 is on S. pombe c29A4; we have not found any related sequences in the data bases. Another protein, S2, was also found to be nucleolar during interphase but unlike S32, remained more compact during mitosis, tending toward one side of the dividing nucleus (Fig. 3 F–H). At this level of resolution it is difficult to determine whether S2 is specifically associated with a particular region of the segregating chromatin (for example, rDNA), but it seems likely that S2 reveals segregation of nucleolar material to the two daughter nuclei. Another nucleolar marker, S38, showed a similar distribution in mitosis (not shown), and sequencing S38 revealed it to be identical to S2. Thus far we have not found any homologies with S2 in data base searches.
Figure 3.
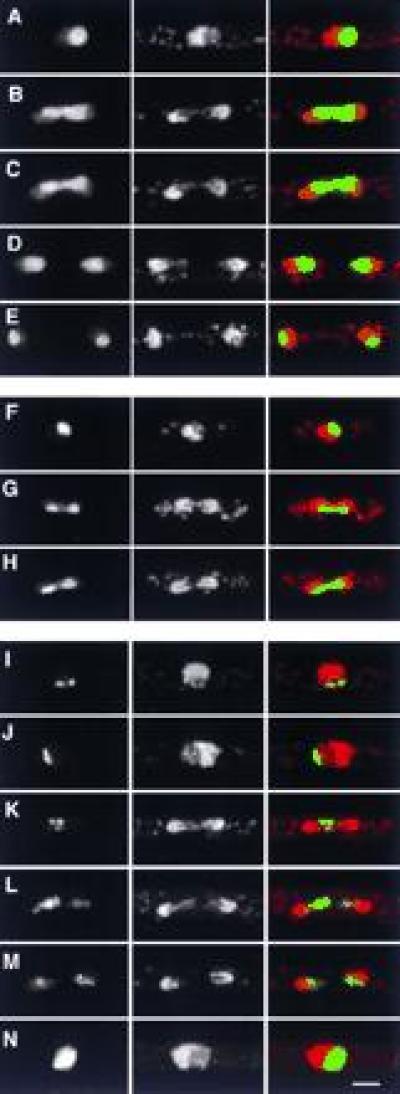
GFP fusion proteins with nucleolar localizations. (Left) GFP; (Center) DNA (PI stain); (Right) merged image of GFP (green) and DNA (red). (A–E) General nucleolar localization of S32, during interphase (A) and different stages of mitosis (B–E). (F–H) Localization of S2 in interphase (F) and mitosis (G and H). Note the more compact organization of S2 in mitosis relative to S32. (I–N) Punctate nucleolar localization of S19 in interphase (I and J) and mitosis (K–M), and more general nucleolar localization when highly overexpressed (N). Note the specific association of S19 and trailing chromatin strands in L. (Bar = 2 μm.)
Figure 4.
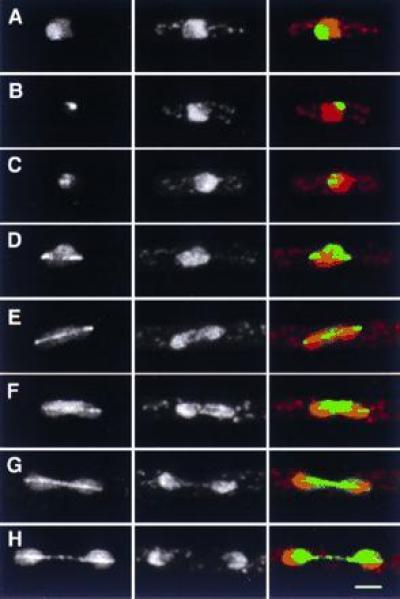
Localization of S26 to the nucleolus during interphase, and to the nucleolus and mitotic spindle during mitosis. (Left) GFP; (Center) DNA (PI stain); (Right) merged image of GFP (green) and DNA (red). (A–C) Interphase cells. Note heterogeneity of fluorescence, which in all cases is confined to the nucleolus. (D–H) Different stages of mitosis. During mitosis, S26 remains associated with nucleolar regions, albeit to varying degrees in different cells (compare E with F, and G with H). (Bar = 2 μm.)
(iv) Specific Localization in the Nucleolus.
A third nucleolar marker, S19, was restricted to small spots in more peripheral regions of the nucleolus (Fig. 3 I and J), although when highly expressed, S19 was found throughout the nucleolus (Fig. 3N), which swelled roughly to the size of the nucleus itself. This also produced long cells due to an inhibition of cell cycle progression (not shown). Because rDNA repeats of chromosome III protrude into the nucleolus, producing two little feet (20, 21), we looked carefully to see if the S19 spots might be superimposed upon the nucleolar rDNA; however, analysis of many interphase cells argued against a consistent association. During mitosis, the punctate localization of S19 was preserved (Fig. 3 K and L), and in late mitosis it appeared to be associated with trailing strands of chromatin (Fig. 3L). This trailing may have been enhanced by expression of the S19 fusion protein, as it was infrequently seen under other circumstances. Sequencing of S19 localized it to cosmid c29A4 (like S32, but clearly distinct and divergently transcribed). Interestingly, S19 was found to be closely related to both S. cerevisiae Cbf5p and rat NAP57. NAP57 was identified as a binding partner to Nopp140, a protein involved in mRNA trafficking in the nucleolus (22); Cbf5p has been suggested to be a component of the budding yeast centromere-binding complex (23), but its function in this regard is unclear. In rat, NAP57 is specifically localized to the dense fibrillar component of the nucleolus as well as to coiled bodies (22), and thus it is possible that the small S19 spots in S. pombe have a similar localization.
(v) Association with the Mitotic Spindle.
During interphase, fluorescence from the plasmid S26 had a nucleolar distribution intermediate between the broad distribution of S2 and S32 and the more punctate staining of S19 (Fig. 4) Surprisingly, during mitosis the GFP–S26 fusion protein was associated with not only nucleolar regions but also the mitotic spindle. Spindle localization appeared to be mitosis-stage specific, in that S26 was enriched at spindle poles during early mitosis, and more evenly distributed later. Interestingly, partial sequence of S26 revealed that it is likely to contain the second half of a chromodomain (Fig. 5), a conserved region found in many proteins involved in organizing heterochromatin (24).
Figure 5.

Comparison of amino acid sequence from the beginning of clone S26 (from the fusion joint with GFP) with chromodomains from Drosophila melanogaster HP1 (GenBank accession no. M57574M57574), a human homolog of HP1 (accession no. L07515L07515), and mouse Modifier 1 protein (accession no. P23197P23197). Residues conserved among all four proteins are shown in bold. The alignment of the latter three sequences is based on that of Aasland and Stewart (24) and further details and comparisons with other chromodomain proteins can be found therein.
A second nuclear GFP fusion, S5, was also localized to the spindle during mitosis (Fig. 6), which was confirmed by anti-tubulin immunofluorescence (not shown). During interphase, localization was confined to a spot or small number of spots in the nucleus, typically at the border between chromatin and the nucleolus (Fig. 6 A–C). Because of the strong spindle association, we suspected that this spot might be associated with the interphase spindle pole body, which is also the site of interphase centromeres (25). However, immunofluorescence using antibodies to the spindle pole body antigen Sad1p (25, 26) showed that localizations of S5 and the spindle pole body are clearly distinct (Fig. 6K). Sequence analysis of S5 revealed it to be a member of the bZIP family of transcription factors (see Table 1), and identical to the recently identified atf21 gene in S. pombe, which was cloned as a multicopy suppressor of an spc1-(sty1-) protein kinase mutant (18).
Figure 6.
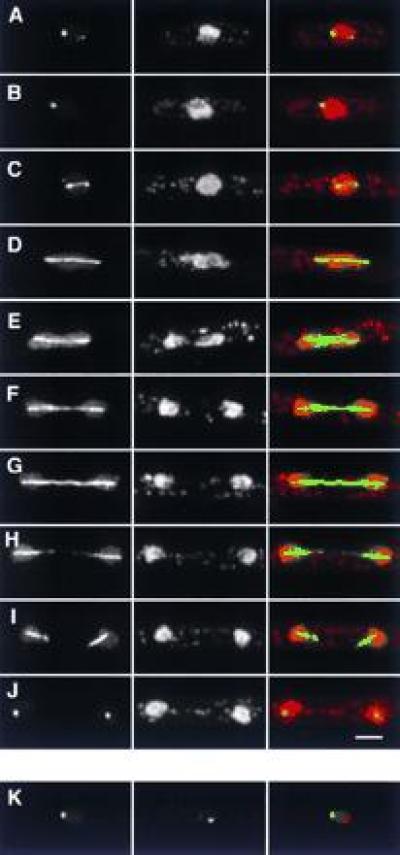
Localization of S5. (A–J) Localization in interphase and in different stages of mitosis. (Left) GFP; (Center) DNA (PI stain); (Right) merged image of GFP (green) and DNA (red). Note the fainter, secondary spots in both interphase (A) and early mitosis (C), the collapse of spindle localization into a single spot at the end of mitosis (J), and the lack of increased S5 fluorescence at spindle poles. (K) Comparison of S5 and spindle pole body localization. (Left) GFP; (Center) anti-sad1p; (Right) merged image of GFP (green) and anti-sad1p staining (red). The two signals are distinct. (Bar = 2 μm.)
DISCUSSION
We have described a relatively simple method for identifying S. pombe genes whose protein products have specific intracellular localizations, using gene fusions with GFP and in vivo fluorescence screening. The advantages of using GFP lie in the speed and ease with which localization can be determined. Live cells can be screened without the potential artifacts of loss or redistribution of signal produced by fixation and/or cell wall digestion, and with these methods a single individual should be able consistently to screen up to 750 colonies per day. The model application of these procedures to the fission yeast nucleus described here illustrates “proof of concept”: several of the proteins we identified are homologous to known nuclear proteins in other organisms, and at the same time the diverse subnuclear localizations of these proteins suggests that they will be useful markers in studying the dynamics of nuclear organization under a variety of conditions, and especially in living cells by time-lapse fluorescence videomicroscopy. A wider application of these methods is thus likely to significantly improve our knowledge of subcellular architecture in fission yeast and will be of considerable importance in studies of cell morphogenesis. Using these methods we have already identified markers defining previously unrecognized domains of the cell cortex, and further technical refinements to the procedure will allow even more efficient large-scale, genome-wide screening (K.E.S., unpublished observations).
Three of the clones described here (M26, M30, and M31) identify markers of the entire nucleus. Both M26 and M31 are distributed throughout the nucleus and thus could be used as nuclear markers in a number of ways, including visual screens for mutants with altered nuclear position or morphology. At higher levels of expression, M30 was found to be concentrated at the nuclear periphery. This localization is of interest given the similarity of the M30 ORF with the recently discovered transportin protein found in mammalian cells. Transportin is thought to be involved in a novel form of nuclear transport which is probably conserved among eukaryotic cells (19), a function consistent with the localization of overexpressed M30.
Four of the clones (S2, S19, S26, and S32) identify markers for the nucleolus. Both S32 and S2 appear to be located in the whole of the nucleolus during interphase, with somewhat different distributions during mitosis. S19 is in a limited number of subnucleolar spots, often close to but not colocalized with the tandem arrays of ribosomal genes. The localization of S26 is more complex, being found in the nucleolus in interphase cells and then also in the spindle during mitosis. Our observations of these clones suggest that there may be some reorganization of the nucleolus during mitosis in fission yeast. In the open mitosis of most Metazoa the nucleolus breaks down, and some nucleolar proteins become cytoplasmic while others remain associated with nucleolar organizer regions and the chromosomal rDNA (27). A related phenomenon may thus also occur in the closed mitosis of S. pombe; that is, some aspects of nucleolar organization may be maintained during mitosis (marked by, for example, S2 and S19), while other components may be more loosely associated and “rebind” at the end of mitosis to rebuild the complete nucleolus (for example, S32 and possibly S26). It is likely that some amount of nucleolar “breakdown” is essential for the proper partitioning of nuclear and nucleolar components during mitosis. This may be emphasized by the effects of the GFP–S19 fusion protein in mitosis, which seemed to “drag” or slow down chromatin segregation.
It was unexpected to find that S26 may be a chromodomain protein. Such proteins are thought to organize heterochromatin (24, 28), which is expected to be absent from the nucleolus. This may suggest that chromodomains may not be restricted to heterochromatin-associated proteins. A second possibility is that S26 is associated with a small amount of heterochromatin that may be specifically in the nucleolus (but not readily visualized by PI staining), and involved in its organization. In mammalian cells, the human homolog of heterochromatin protein HP-1 is found in subnuclear, nonnucleolar bodies that stain only weakly with DNA binding dyes (29), and it is possible that in S. pombe analogous structures may also be present in the space which we define as “nucleolar”, although they may not be involved with classical nucleolar functions. This has previously been suggested by Tani et al. (30), with reference to mRNA transport. Finally, because S26 is found in the spindle, it is possible that it is involved in heterochromatin organization during mitosis but is sequestered in the nucleolus during interphase.
Two further clones were found to associate with more specific regions of the nucleus. M25 was found to colocalize with chromatin, while S5 was located in both the mitotic spindle and in specific spots within the interphase nucleus, near the border between the nucleolus and chromatin. We have shown that this spot is not associated with the spindle pole body, and by thus by extension also not with interphase centromeres. This site may represent a localized site(s) of active transcription, given that in situ hybridization of the actively transcribed adh gene shows a typical localization for this gene near the nucleolus/chromatin border (30). This possibility is of particular interest given that S5 encodes a bZIP transcription factor. The localization of S5 to the spindle during mitosis raises the possibility that spindle localization provides a means of segregating the protein during mitosis. Chromosome condensation during mitosis might remove transcription factors from the DNA and, although as yet there is no precedent for this kind of mechanism, segregation via the mitotic spindle might be part of a mechanism ensuring that these factors are correctly redistributed during mitosis, and that an appropriate pattern of gene expression is reestablished in the next cell cycle.
Acknowledgments
We thank M. Chalfie for the TU65 plasmid, B. Vallins for advice on automated sequencing, G. Clark and A. Davies for additional sequencing help, I. Goldsmith for oligonucleotide synthesis, I. Hagan for anti-sad1p antibody, P. Jordan for help with confocal microscopy, and especially J. Kitau for construction of the replica-spotting device and coverslip-spin tubes. We also thank J. Mata, B. Stern, and other members of the Cell Cycle Laboratory for helpful discussions. This work was supported by the Imperial Cancer Research Fund and the Helen Hay Whitney Foundation; K.E.S. is a Helen Hay Whitney Postdoctoral Research Fellow.
Footnotes
Abbreviations: GFP, green fluorescent protein; PI, propidium iodide.
Data deposition: The sequences reported in this paper have been deposited in the GenBank data base [accession nos. Y09282Y09282 (M25), Y09283Y09283 (M26), Y09284Y09284 (M30), Y09285Y09285 (M31), Y09286Y09286 (S19), Y09287Y09287 (S2), Y09288Y09288 (S26), Y09289Y09289 (S32), and Y09290Y09290 (S5)].
References
- 1.Hayles J, Nurse P. Annu Rev Genet. 1992;26:373–402. doi: 10.1146/annurev.ge.26.120192.002105. [DOI] [PubMed] [Google Scholar]
- 2.Moreno S, Klar A, Nurse P. Methods Enzymol. 1991;194:795–823. doi: 10.1016/0076-6879(91)94059-l. [DOI] [PubMed] [Google Scholar]
- 3.Nurse P. Nature (London) 1990;344:503–508. doi: 10.1038/344503a0. [DOI] [PubMed] [Google Scholar]
- 4.Snell V, Nurse P. EMBO J. 1994;13:2066–2074. doi: 10.1002/j.1460-2075.1994.tb06481.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nurse P. Mol Biol Cell. 1994;5:613–616. doi: 10.1091/mbc.5.6.613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Verde F, Mata J, Nurse P. J Cell Biol. 1995;131:1529–1538. doi: 10.1083/jcb.131.6.1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Burns N, Grimwade B, Ross M P, Choi E Y, Finberg K, Roeder G S, Snyder M. Genes Dev. 1994;8:1087–1105. doi: 10.1101/gad.8.9.1087. [DOI] [PubMed] [Google Scholar]
- 8.Ausubel F, Brent R, Kingston R, Moore D, Seidman J, Smith J, Struhl K, editors. Current Protocols in Molecular Biology. New York: Wiley; 1996. [Google Scholar]
- 9.Maundrell K. Gene. 1993;123:127–130. doi: 10.1016/0378-1119(93)90551-d. [DOI] [PubMed] [Google Scholar]
- 10.Forsburg S L. Nucleic Acids Res. 1993;21:2955–2956. doi: 10.1093/nar/21.12.2955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zabarovsky E R, Allikmets R L. Gene. 1986;42:119–123. doi: 10.1016/0378-1119(86)90158-7. [DOI] [PubMed] [Google Scholar]
- 12.Evans L, Mitchison T, Kirschner M. J Cell Biol. 1985;100:1185–1191. doi: 10.1083/jcb.100.4.1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Atkins D, Izant J G. Curr Genet. 1995;28:585–588. doi: 10.1007/BF00518173. [DOI] [PubMed] [Google Scholar]
- 14.Robinow C F, Hyams J. In: Molecular Biology of the Fission Yeast. Nasim A, Young P, Johnson B F, editors. San Diego: Academic; 1989. pp. 273–331. [Google Scholar]
- 15.Yu Y, Jiang Y W, Wellinger R J, Carlson K, Roberts J M, Stillman D J. Mol Cell Biol. 1996;16:5254–5263. doi: 10.1128/mcb.16.10.5254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bi E, Pringle J. Mol Cell Biol. 1996;16:5264–5275. doi: 10.1128/mcb.16.10.5264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ma X-J, Lu Q, Grunstein M. Genes Dev. 1996;10:1327–1340. doi: 10.1101/gad.10.11.1327. [DOI] [PubMed] [Google Scholar]
- 18.Shiozaki K, Russell P. Genes Dev. 1996;10:2276–2288. doi: 10.1101/gad.10.18.2276. [DOI] [PubMed] [Google Scholar]
- 19.Pollard V W, Michael W M, Nakielny S, Siomi M C, Wang F, Dreyfuss G. Cell. 1996;86:985–994. doi: 10.1016/s0092-8674(00)80173-7. [DOI] [PubMed] [Google Scholar]
- 20.Toda T, Yamamoto M, Yanagida M. J Cell Sci. 1981;52:271–287. doi: 10.1242/jcs.52.1.271. [DOI] [PubMed] [Google Scholar]
- 21.Uzawa S, Yanagida M. J Cell Sci. 1992;101:267–275. doi: 10.1242/jcs.101.2.267. [DOI] [PubMed] [Google Scholar]
- 22.Meier, U. T. & Blobel, G. (1994) J. Cell Biol. 1505–1514. [DOI] [PMC free article] [PubMed]
- 23.Jiang W, Middleton K, Yoon H J, Fouquet C, Carbon J. Mol Cell Biol. 1993;13:4884–4893. doi: 10.1128/mcb.13.8.4884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Aasland R, Stewart A F. Nucleic Acids Res. 1995;23:3168–3173. doi: 10.1093/nar/23.16.3168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Funabiki H, Hagan I, Uzawa S, Yanagida M. J Cell Biol. 1993;121:961–976. doi: 10.1083/jcb.121.5.961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hagan I, Yanagida M. J Cell Biol. 1995;129:1033–1047. doi: 10.1083/jcb.129.4.1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shaw P, Jordan E G. Annu Rev Cell Biol. 1995;11:93–121. doi: 10.1146/annurev.cb.11.110195.000521. [DOI] [PubMed] [Google Scholar]
- 28.Orlando V, Paro R. Curr Opin Genet Dev. 1995;5:174–179. doi: 10.1016/0959-437x(95)80005-0. [DOI] [PubMed] [Google Scholar]
- 29.Saunders W S, Cooke C A, Earnshaw W C. J Cell Biol. 1991;115:919–931. doi: 10.1083/jcb.115.4.919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tani T, Derby R J, Hiraoka Y, Spector D L. Mol Biol Cell. 1995;6:1515–1534. doi: 10.1091/mbc.6.11.1515. [DOI] [PMC free article] [PubMed] [Google Scholar]