

Abstract
Using predictions from heme – quinoline antimalarial complex structures, previous modifications of chloroquine (CQ), and hypotheses for chloroquine resistance (CQR), we synthesize and assay CQ analogues that test structure – function principles. We vary side chain length for both monoethyl and diethyl 4N CQ derivatives. We alter the pKa of the quinolyl N by introducing alkylthio or alkoxy substituents into the 4 position, and vary side chain length for these analogues. We introduce an additional titratable amino group to the side chain of 4O analogues with promising CQR strain selectivity and increase activity while retaining selectivity. We solve atomic resolution structures for complexes formed between representative 4N, 4S and 4O derivatives vs. μ-oxo dimeric heme, measure binding constants for monomeric vs. dimeric heme, and quantify hemozoin (Hz) formation inhibition in vitro. The data provide additional insight for the design of CQ analogues with improved activity vs. CQR malaria.
INTRODUCTION
Promising progress on live sporozoite based vaccines notwithstanding 1,2, new antimalarials active against drug resistant malaria are urgently needed. Moreover, due to economic issues, such compounds need to be relatively inexpensive to produce and distribute. Ideally, the compounds should be stable without refrigeration, act on multiple stages of the malarial parasite, have prophylactic as well as therapeutic utility, and be equally active vs. multiple species of Plasmodia (e.g. P. falciparum and P. vivax).
Mono and combination therapies that include natural, synthetic and semi-synthetic drugs based on the iron-activated 1,2,4-trioxane artemisinin show significant promise in this regard 3-7. A spiroadamantane trioxolane (compound OZ277) optimized by Vennerstrom and colleagues 3 is now in advanced stages of development, and other promising derivatives based on artemisinin 4-7 are also emerging options for improved drug therapy. However, compared to other antimalarial pharmacophores, most of these syntheses are relatively expensive and resistance to compounds exhibiting the endoperoxide pharmacophore of artemisinin has already been noted 8. Unfortunately, resistance to artemisinin derivatives will likely continue to evolve and spread, so until effective vaccines are developed and implemented, a long term strategy to rapidly provide new, inexpensive drugs and drug combinations active against current and emerging drug resistant malaria is critical. In this regard, simultaneous further development of multiple successful pharmacophores provides the best strategy for “staying ahead of the resistance curve” over the next decade or more.
Three of the most successful antimalarial drugs ever used (quinine [QN], chloroquine [CQ] and mefloquine [MQ]) are quinoline derivatives, and numerous additional lead compounds with improved activity vs. CQ resistant (CQR) malaria have been discovered via a variety of recent synthetic modifications of these structures 9-13. It is important to note that even in the face of widespread resistance to CQ, similar compounds (MQ, QN, amodiaquine [AQ] and isoquine [IQ]) remain active against laboratory strains of CQR parasites. This could be in part due to the fact that the principle target of CQ and related quinolines is believed to be one or more forms of uncrystallized heme (ferriprotoporphyrin IX; FPIX) found within the digestive vacuole of the intraerythrocytic malarial parasite 14. FPIX is released from proteolyzed red blood cell hemoglobin (Hb) within the parasite digestive vacuole (DV) as the parasite very rapidly grows within the human red blood cell. As such, the principle target (heme made by the host) cannot be mutated or alternatively expressed by the parasite in order to confer resistance. The CQR mechanism is therefore unique, complex, and has taken decades to appear on a large scale even in the face of massive CQ use 15.
Quinolines, as well as acridines, xanthones, and other classes of antimalarial drugs are believed to exert some component of their toxic action via preventing the crystallization of FPIX to hemozoin (Hz) 14,16,17. A detailed molecular mechanism for this process remains elusive, but likely includes binding of the drugs to either (or both) pre-crystalline soluble heme or (and) growing faces of the crystal 16,18,19. In both cases, multiple modes of interaction are possible. For example, pre-crystalline FPIX exists in multiple chemical forms within the DV (various monomeric and dimeric forms) that are in pH-dependent equilibrium at currently unknown ratios. Yet, it is known that these multiple forms are each capable of binding CQ and other quinoline based drugs. The relative affinities for FPIX monomer vs. dimer are not the same for various quinoline antimalarials, and these affinities also differ strongly depending on pH and other variables. The point being that even subtle modifications to CQ can confer preference for one chemical form of FPIX vs. another (i.e. Fe[III] monomer vs. μ-oxo dimer or vice versa) making these compounds more or less active than CQ in the presence of a given FPIX composition. Changes in FPIX composition are a key prediction of the altered DV physiology known to exist for CQR parasites 20,21.
Until very recently, design and synthesis of additional CQ analogues guided by experimentally determined, atomic level resolution drug – drug target structures has not been possible. Many previous modifications have been addressed by combinatorial synthetic strategies, but how those modifications might influence formation of a drug-target complex has been difficult to rationalize or predict. Also, comparison between activity for many of the CQ derivatives that have been made is difficult because different strains of malaria have been used, and activity has been assayed in multiple ways (e.g. via 3H hypoxanthine incorporation; lactate dehydrogenase activity; DNA staining; etc.). These assays measure growth inhibition and / or death of parasite cultures in different ways and thus do not report identical IC50 data. Furthermore, most published modifications to CQ have not included systematic, subtle alterations (e.g. iterative addition or substitution of single atoms) that can provide clues to important structure – function principles.
In this study, we have analyzed a series of specific modifications to CQ that are predicted to be relevant for CQ activity vs. CQR parasites based on four considerations: 1) the geometry of quinoline vs. FPIX μ-oxo dimer structures we have recently solved 16,18 ; 2) the existence of coordinate CQ – FPIX monomer complexes under conditions that mimic those of the DV 19 ; 3) differences in DV pH that have been deduced for CQS and CQR parasites 20,21 ; 4) previous observations that the CQ side chain length alters selectivity for CQR vs. CQS parasites 9,11. We have measured the activity of these rationally designed compounds vs. two CQ sensitive (CQS) and two CQ resistant (CQR) strains using a standardized, easily validated and inexpensive new assay based on SYBR Green I intercalation that has recently been adopted and validated by a number of laboratories 22 - 24. Taken together, our results suggest important new structure – function principles for quinoline antimalarial drug design based on chloroquine, including 1) replacement of the terminal tertiary amino function by a secondary moiety reduces the potency vs. CQR strains which suggests the side chain amino group is recognized by the CQ resistance mechanism; 2) substitution of S or O for N at position 4 significantly alters the quinolyl N basicity and lowers the antimalarial potency while improving the selectivity index (defined as the ratio of CQR strain IC50 / CQS strain IC50); 3) introduction of an additional basic amino group to the side chain of 4O CQ derivatives can improve the potency while retaining an improved selectivity index; 4) surprisingly, no straightforward relationships between the ability to bind FPIX μ-oxo dimer vs. inhibition of Hz formation and antimalarial potency exists for this series of CQ derivatives.
RESULTS
Recently this consortium was the first to experimentally define atomic level resolution structures for CQ, QN, quinidine (QD) and AQ vs. μ-oxo dimer FPIX 16,18, as well as the existence of a coordinate CQ – monomeric FPIX complex under acidic aqueous conditions 19. Other data suggest that DV pH may differ for CQS vs. CQR parasites 20. As previously suggested 16,18,19,20 these data led to several structure - function predictions for CQ analogues that can now be systematically tested via strategic modifications of the CQ structure. These include that simultaneously fine tuning both basicity of the quinolyl N and the length of the CQ side chain may be important for optimizing interactions with FPIX, and that basicity of the tertiary aliphatic N for CQ is important for accumulation within the parasite DV, but not for previously predicted ionic stabilization of CQ – FPIX structures. Along with these principles, previous studies 25,26 have demonstrated that desethyl CQ has similar activity relative to CQ for CQS strains, but lower activity vs. some CQR strains of P. falciparum. In addition, shortening or lengthening the aliphatic side chain of CQ has in general been shown to have little effect on the activity vs. CQS strains, but to increase activity vs. CQR strains9,11,26. However, these two modifications have not previously been systematically varied in tandem, which might result in additive or opposing effects. Below we report data that test the above structure – function predictions for CQ analogues.
Compounds 1 – 10
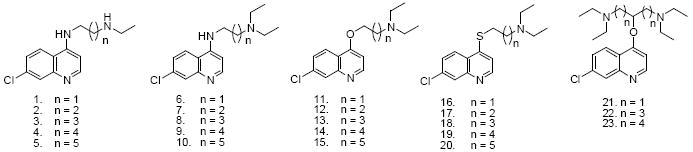
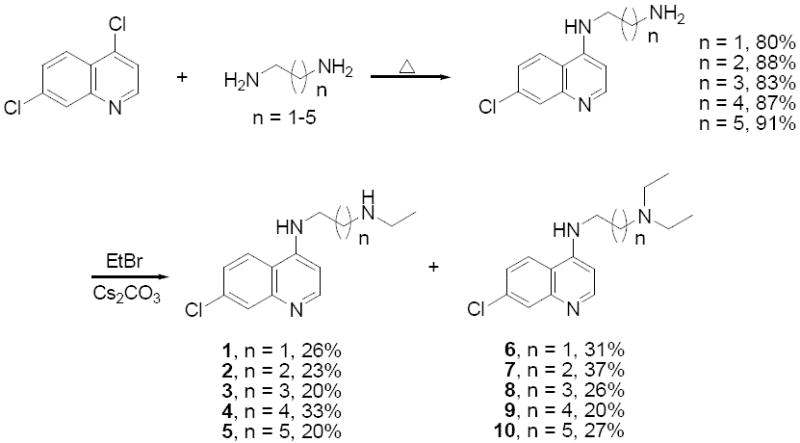
Compounds 1 – 10 were designed to systematically explore the relationship between mono- vs. diethyl substitution at the terminal aliphatic N and the length of the aliphatic side chain vs. activity against CQS and CQR parasites (Table 1). These compounds were prepared in two steps from 4,7-dichloroquinoline and a series of α,ω-diamines. The amination reaction proceeded at elevated temperatures with high yields and the subsequent alkylation with ethyl bromide gave a mixture of approximately 50 % of the desired secondary and tertiary amine leaving about 50 % of remaining starting materials were recovered in all cases (see Scheme 1). Aminoquinolines 1-10 were then analyzed for activity vs. two CQS and two CQR laboratory strains of P. falciparum using a new semi high-throughput SYBR Green I based assay. This assay was developed independently in two laboratories 22,23, is easily standardized, and was recently validated vs. a large collection of antimalarial compounds by the Walter Reed Army Institute 24. As described below and in the Discussion section, standardization of the activity of candidate antimalarials against different strains and species of Plasmodium is essential for future progress, and the SYBR Green I assay offers one inexpensive route that should be accessible to most laboratories engaged in malaria research.
Table 1.
IC50 values for 4-Amino-, 4-alkoxy- and 4-alkylthioquinoline derivatives.
 | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Compound | Experimental IC50 (μM) | Previously reported IC50 (μM) | ||||||||||
| HB3 | Dd2 | SI* | GCO3 | FCB | SI* | NF5426 | K126 | SI* | Haití 13529 | Indochina I29 | SI* | |
| CQ | 0.011 | 0.116 | 10.50 | 0.010 | 0.157 | 15.70 | 0.016 | 0.315 | 19.60 | 0.007 | 0.095 | 13.60 |
| 1 | 0.012 | 0.081 | 6.750 | 0.012 | 0.089 | 7.420 | 0.021 | 0.133 | 1.250 | - | - | - |
| 2 | 0.007 | 0.115 | 16.40 | 0.020 | 0.085 | 4.250 | 0.015 | 0.365 | 24.30 | - | - | - |
| 3 | 0.018 | 0.649 | 36.10 | 0.026 | 0.854 | 32.80 | - | - | - | - | - | - |
| 4 | 0.021 | 0.786 | 37.40 | 0.062 | 1.270 | 20.50 | - | - | - | - | - | - |
| 5 | 0.019 | 0.982 | 31.30 | 0.052 | 0.987 | 10.00 | - | - | - | - | - | - |
| 6 | 0.013 | 0.026 | 2.000 | 0.013 | 0.030 | 2.300 | 0.021 | 0.049 | 2.330 | 0.007 | 0.005 | 0.714 |
| 7 | 0.006 | 0.026 | 4.330 | 0.007 | 0.029 | 4.140 | 0.018 | 0.060 | 3.330 | 0.005 | 0.006 | 1.200 |
| 8 | 0.012 | 0.199 | 16.60 | 0.049 | 0.181 | 3.690 | - | - | - | 0.005 | 0.051 | 10.20 |
| 9 | 0.029 | 0.357 | 12.30 | 0.066 | 0.509 | 7.170 | - | - | - | 0.006 | 0.058 | 9.670 |
| 10 | 0.097 | 0.085 | 0.876 | 0.016 | 0.102 | 6.380 | - | - | - | 0.005 | 0.056 | 11.20 |
| 11 | 5.800 | 4.570 | 0.788 | 5.290 | 5.080 | 0.960 | - | - | - | - | - | - |
| 12 | 5.100 | 2.800 | 0.549 | 2.340 | 2.330 | 0.996 | - | - | - | - | - | - |
| 13 | 2.920 | 1.180 | 0.404 | 1.660 | 1.100 | 0.663 | - | - | - | - | - | - |
| 14 | 1.870 | 1.010 | 0.540 | 1.490 | 0.930 | 0.624 | - | - | - | - | - | - |
| 15 | 3.060 | 4.160 | 1.360 | 2.270 | 1.190 | 0.524 | - | - | - | - | - | - |
| 16 | 9.530 | 5.500 | 0.577 | 5.740 | 6.020 | 1.040 | - | - | - | - | - | - |
| 17 | 5.600 | 2.660 | 0.475 | 3.460 | 1.610 | 0.465 | - | - | - | - | - | - |
| 18 | 7.390 | 4.440 | 0.601 | 5.630 | 3.500 | 0.622 | - | - | - | - | - | - |
| 19 | 2.330 | 1.080 | 0.464 | 1.610 | 0.814 | 0.506 | - | - | - | - | - | - |
| 20 | 6.020 | 4.110 | 0.683 | 4.450 | 3.300 | 0.742 | - | - | - | - | - | - |
| 21 | 1.490 | 1.290 | 0.866 | 2.250 | 3.830 | 1.700 | - | - | - | - | - | - |
| 22 | 0.094 | 0.409 | 4.350 | 0.074 | 0.352 | 4.750 | - | - | - | - | - | - |
| 23 | 0.073 | 0.316 | 4.310 | 0.045 | 0.604 | 13.40 | - | - | - | - | - | - |
Selectivity Index; ratio of the IC50 for a given drug shown by a CQ - resistant strain vs. IC50 for the companion CQ – sensitive strain. Column 4 shows SI computed as Dd2 IC50 / HB3 IC50, column 7 shows FCB IC50 / GCO3 IC50, column 10 is K1 / NF54, and column 13 is IndoI/ Haiti 135.
Scheme 1.
Synthesis of chloroquine derivatives 1 – 10
Aminoquinolines 3 – 5 are novel and have not previously been analyzed vs. malarial parasites, whereas 1, 2 and 6 – 10 have been synthesized previously 27-30 using similar but not identical methods (Scheme 1) and tested vs. less commonly used laboratory strains of P. falciparum. Assessment of the activities of all of these related CQ analogues has not previously been standardized using the same strains, culture conditions, and malarial growth inhibition assays. HB3 (CQS, Honduras) and Dd2 (CQR, Indochina) are parents of a genetic cross that produced a collection of progeny (GC03 [CQS] being one) for which a very large amount of data has been collected regarding the biochemistry and genetics of chloroquine drug resistance 15,31,32. Strain FCB (CQR, SE Asia) expresses similar CQR – causing PfCRT mutations relative to Dd215 yet in most laboratories shows 50 – 100 % higher levels of CQR relative to Dd2. As such, these strains are valuable reference points for future quinoline based antimalarial drug design guided by ongoing elucidation of the CQR mechanism(s).
We measured similar, but not identical, IC50 for 1 2 vs. CQS (HB3, GC03) and CQR (Dd2, FCB) parasites, consistent with earlier work that assayed CQS strains NF54 and Haiti 135 or CQR strains K1 and Indochina I 26. Differences in precise IC50 are likely due to strain variation, our use of synchronized culture vs. asynchronous culture by others, the use of 3H hypoxanthine incorporation assays vs. the present SYBR Green I approach, or some combination. Regardless, we expanded this analysis to include compounds bearing 4 to 6 methylene groups between the two amino moieties (compounds 3 - 5) to explore the role of deethylation (as occurs as a consequence of human metabolism) vs. side chain length could be analyzed in more depth (compare the structure of compounds 1 – 5 vs. 6 - 10).
Previously, Krogstad and colleagues 9 as well as Ridley et al. 26 observed that the desethyl CQ derivatives 1 or 2 still exhibit high IC50 vs. CQR strains, whereas the diethyl analogues 6 or 7 show substantially lower IC50. One hypothesis for the trend in the diethyl side chain series that has been offered previously is that both longer and shorter side chain analogues are less well recognized by the resistance mechanism (for example, drug binding to the mutated PfCRT protein responsible for CQR33 could be weaker for long and short chain CQ analogues). If this is indeed the case, then these data suggest that removal of one alkyl group reverses this effect quite dramatically for longer chain analogues. For example, the selectivity index (SI, defined in Table 1 caption) obtained for 3 – 5 is 3 fold higher than for CQ, while IC50 of 3 – 5 remains near that seen for CQ in CQS strains. That is, the basic tertiary side chain amino group likely contributes to recognition by the CQ resistance mechanism (see Discussion).
Compounds 11 - 20
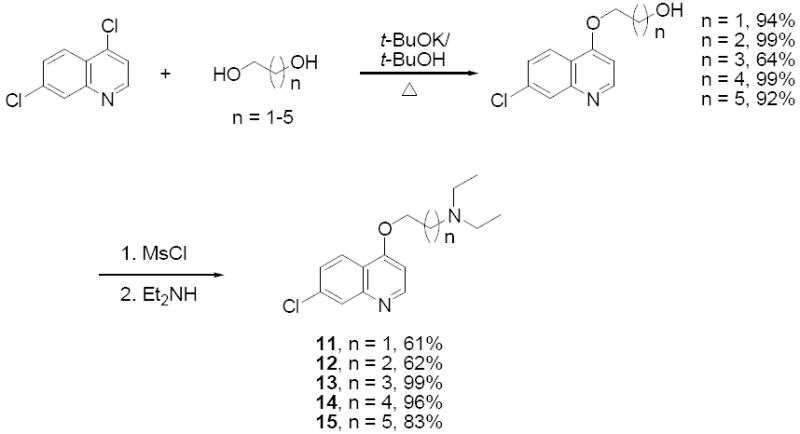
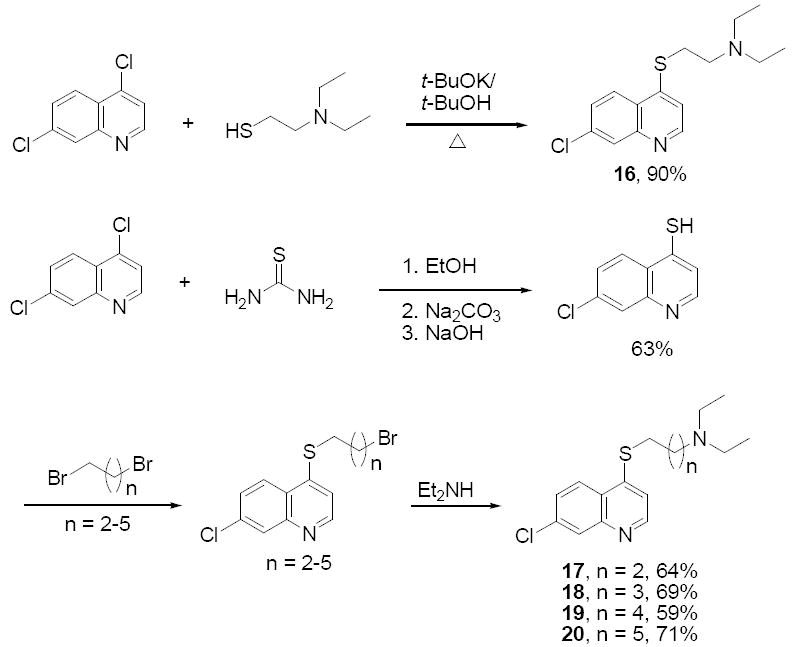
Compounds 11-15 were synthesized from 4,7-dichloroquinoline and α,ω–alkanediols via consecutive nucleophilic displacements (Scheme 2). Compound 16 was synthesized in a single step from 4,7-dichloroquinoline and 2-diethylaminoethanethiol (Scheme 3), and compounds 17 - 20 were prepared from 7-chloroquinolyl-4-thiol and α,ω–dibromoalkanes via two consecutive SN2 displacements (Scheme 3). These compounds were synthesized in order to inspect the combined effects of heteroatom substitution at the 4 position and the side chain modifications described above. This strategy was pursued (in part) because recent solid state NMR studies have shown that CQ may form a covalent coordinate complex with monomeric FPIX (via a heme Fe – quinoline N bond 19) under acidic conditions that mimic those of the parasite digestive vacuole (DV). Thus, assuming other structural features remain constant, altering the nucleophilicity of the quinolyl N (as predicted for 4O and 4S substitutions) would influence reactivity of the drug vs. monomeric heme without necessarily altering the structure required for non-covalent association to μ-oxo dimer heme 16, and hence preference for drug – monomer vs. drug heme dimer complexes16,18. Yet, none of these compounds showed heightened activity relative to CQ, and in fact exhibited only modest antimalarial activity, with IC50 values in the μM range (Table 1). However, we note that the selectivity index (SI; c.f. Table 1) is substantially improved for several of these compounds. Thus, the 4S and 4O CQ analogue structures are valid starting points for quinoline based antimalarial drug design wherein the goal is improved activity vs. CQR strains (e.g. lower SI, see results for compounds 21 - 23, below).
Scheme 2.
Synthesis of CQ – derived ethers
Scheme 3.
Synthesis of CQ – derived sulfides
To further probe the molecular basis of these trends in relative activity and selectivity index, we analyzed other features of quinoline based drugs that are believed to be critical with regard to their antimalarial potency. We examined in detail a structurally related set that best mimics the overall structure of CQ (namely, the members of this set include those compounds that contain side chains of similar length relative to CQ; 8, 13, 18). The pKa of titratable N were calculated and measured (Table 2), binding constants for μ-oxo dimeric and monomeric heme were measured in aqueous and 40 % DMSO solutions, respectively (Table 3), and the ability to inhibit Hz formation in vitro at DV pH measured for CQS (5.6) and CQR (5.2) parasites are tabulated (Table 4). In addition, we performed inversion recovery experiments at varied drug : dimer heme ratios and solved the atomic level structures of complexes formed between these drugs and μ-oxo dimer heme (Figure 1A & 1B).
Table 2.
Calculated and Measured pKa for representative compounds
| SPARC Approx.a | Experimentalc | ||||
|---|---|---|---|---|---|
| Compound | pKa1b | pKa2b | pKa3b | pKa1b | pKa2b |
| CQ | 6.3 | 9.3 | - | 8.6 | 9.8 |
| 8 | 6.6 | 9.3 | - | 8.5 | 9.8 |
| 13 | 3.7 | 9.4 | - | 4.5 | 9.8 |
| 18 | 5.0 | 9.3 | - | 4.1 | 9.5 |
| 22 | 5.0 | 9.9 | 9.9 | nd | nd |
. SPARC is an online pKa approximation program developed at the University of Georgia (S.W. Karickhoff, L.A.Carreira and S.H. Hilal)
. pKa1 or pKa3 represent the pKa of side chain tertiary N and pKa2 represents the pKa of quinolyl N
. The pKa measurements represent an average of three determinations performed by acid/base titrations at room temperature. See also ref 61 for previous determination of CQ pKa.
nd = not determined.
Table 3.
Measured binding constants for monomeric (pH 3.9) and μ-oxo dimeric (pH 7.5) heme
| Experimental Binding Constants |
||||
|---|---|---|---|---|
| pH 3.9 | pH 7.5 | |||
| Compound | Ka (M-1) | Log Ka | Ka (M-1) | Log Ka |
| CQ | ND | - | 1.75 × 105 | 5.2 |
| 8 | ND | - | 1.65 × 105 | 5.2 |
| 13 | ND | - | 8.82 × 104 | 5.0 |
| 18 | ND | - | 8.22 × 104 | 4.9 |
| 22 | ND | - | 3.47 × 104 | 4.5 |
ND = not detected
Table 4.
Measured Hemozoin (Hz) Inhibition IC50
| IC50 (μM) |
||
|---|---|---|
| Compound | pH 5.2 | pH 5.6 |
| CQ | 35 ± 3 | 16 ± 4 |
| 8 | 24 ± 1 | 12 ± 2 |
| 13 | >1000 | >1000 |
| 18 | 216 ± 19 | 155 ± 8 |
| 22 | 199 ± 50 | 687 ± 213 |
Figure 1.
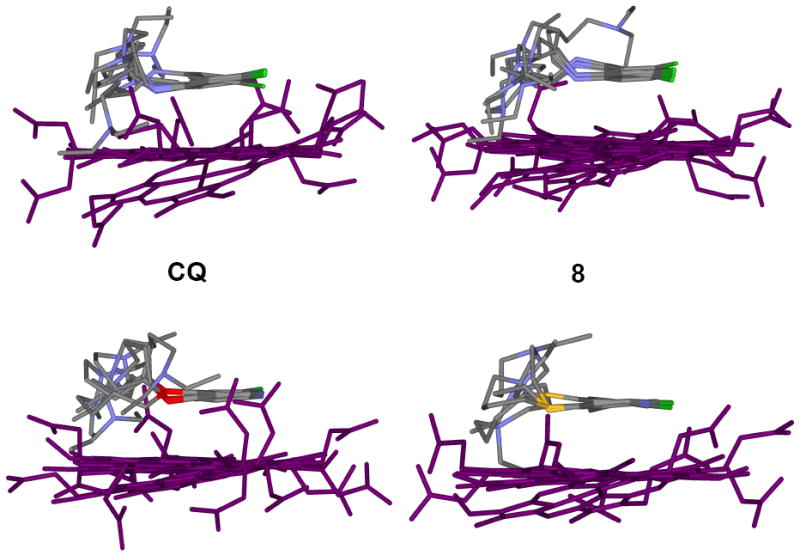
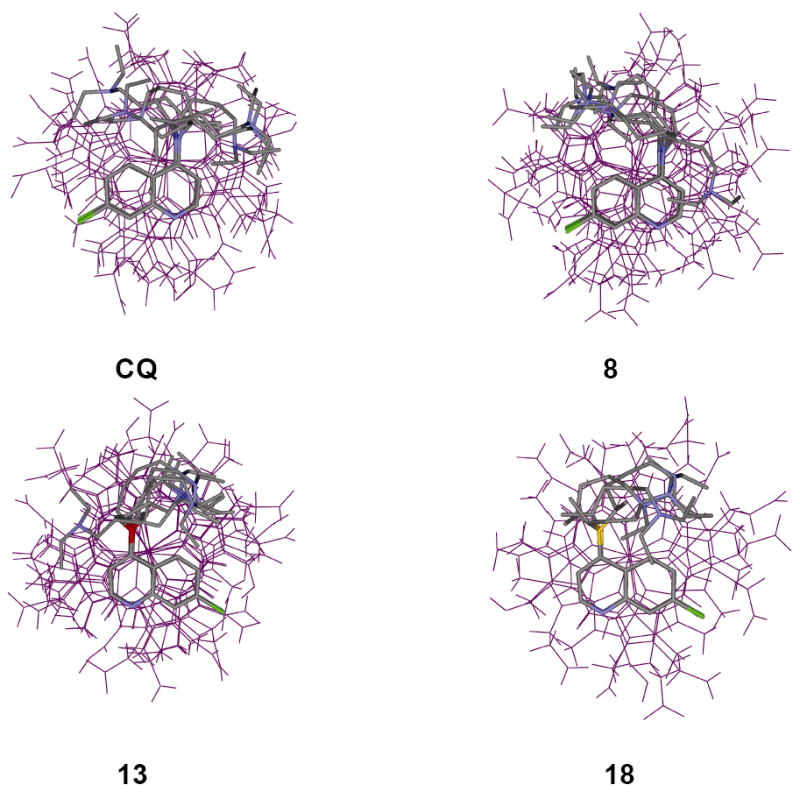
A. Structures of drug – μ-oxo dimer complexes derived from distance geometry calculations using Fe(III)-drug (1H) distance restraints from relaxation measurements. The drug molecules, on average, are approximately 3-4 Å above the plane of the porphyrin ring. Since the distance restraints are drawn from a single point (Fe(III)), the porphyrin plane’s rotational orientation is not unequivocally defined (see Fig 1B). Within the limitations imposed by assumptions made in these calculations and the accuracy of the data, no significant differences in how these drug molecules interact with the μ-oxo dimer are found.
B. As in 1A, but top-down view. The relaxation rates of the alipathic protons are likewise enhanced by the addition of heme and as shown in these structures, the side chains do not extend away from Fe(III), but trace the perimeter of the porphyrin ring (see also references 16 and 18).
The pKa data show that incorporation of alkoxy and alkylthio substituents into position 4 affords CQ analogues that are effectively monoprotic weak bases at physiologic pH. CQ and the 4N CQ derivatives have pKa’s of approximately 10 and 8.5 (Table 2) and are effectively diprotic weak bases and thus concentrate within the acidic parasite DV proportional to the square of the net pH gradient (DV interior to outside). However, concentration of the effectively monoprotic 4S and 4O analogues will be linearly related to the net pH gradient 14 as shown in Table 5, which summarizes our calculations for DV accumulation for each compound. Thus one possible explanation for the reduced activity of these compounds is a lowered ability to concentrate within the DV (site of hemoglobin digestion and release of free heme).
Table 5.
Computed Vacuolar Accumulation Ratios (VAR)
| Compound/Vacuolar Accumulation Ratio (VAR) | |||||
|---|---|---|---|---|---|
| DV pH | CQ | 8 | 13 | 18 | 22 |
| CQR (pH 5.2) | 2.3 × 104 | 2.3 × 104 | 1.9 × 102 | 1.7 × 102 | 7.9 × 104 |
| CQS (pH 5.6) | 3.7 × 103 | 3.7 × 103 | 6.7 × 101 | 6.4 × 101 | 5.0 × 103 |
VAR is calculated using the Henderson – Hasselbach equation and knowing cytosolic pH = 7.4, DV pH for CQR parasites = 5.2, DV pH for CQS = 5.620,2 and assuming: 1) that charged (protonated) drugs are essentially membrane impermeable; 2) net accumulation is not affected by binding to drug target. Although these are both simplifications, the calculated differences for (effectively) mono vs. diprotic drugs are orders of magnitude apart, whereas binding effects are expected to be (at most) several fold.
To test whether binding to Hz precursors is also altered for the 4S and 4O derivatives, binding constants were measured as previously described 34-36 for both μ-oxo dimeric and monomeric heme (Table 3). Due to the instability of monomeric heme in aqueous solution, the affinity to monomer measured by conventional absorbance experiments can only be estimated using 40% DMSO in water as solvent 36. At appropriately acidic solution pH (≤ 5.0) FPIX heme is primarily monomeric, whereas at pH 7, appreciable dimer is formed. As shown (Table 3), CQ and compounds 8, 13, 18 all have poor affinity for monomeric heme in acidic 40% DMSO. To further test if CQ interacts with monomeric heme, T1 measurements of the CQ protons were made with samples containing 10 mM CQ and 2 mM hemin chloride in 40% DMSO at pH 5.0. Although the lines are broadened in this solution due to paramagnetic susceptibility, the measured T1’s indicate only weak paramagnetic relaxation. For example, the measured T1 for CQ proton 1 in this sample is 0.70 s, whereas at pH 7.0 the T1 for the same proton is 0.039 s. The longer T1’s in the lower pH sample indicate that CQ does not interact appreciably with monomeric heme.
Table 3 also tabulates similar measured affinities for compounds 8, 13, 18 vs. μ-oxo dimer in aqueous solution. Inspection of the side and top – down views of the noncovalent solution structures formed between these drugs and μ-oxo dimeric heme solved via T1 measurements (Fig. 1A & 1B) shows that the overall geometries (and hence calculated binding energies) are quite similar. Thus, to a first approximation, interactions between either CQ, 8, 13, or 18 and monomeric or dimeric heme are all similar.
Surprisingly then, the ability of 13, 18 to inhibit Hz formation was found to be significantly lower vs. that measured for CQ and 8 (Table 4). These results, viewed alongside data in Fig. 1 and Table 3, suggest that noncovalent complexation with μ-oxo dimer heme is unlikely to be the primary mode of inhibition of Hz formation. Interactions between these compounds and other heme aggregates or the growing faces of Hz must play an important role, since the relative μ-oxo dimer binding constants and complex geometries (energies) do not correlate with the relative ability of these compounds to perturb Hz growth in vitro (Table 4 vs. Table 3, Fig. 1).
Compounds 21 – 23
Since the SI was improved for several of the 4O CQ derivatives, but since accumulation of these effectively monobasic drugs into the DV of the parasite is predicted to be lower than CQ and the 4N derivatives (Table 5), we designed and synthesized “symmetrically branched”, dibasic 4O CQ analogues 21 – 23 (Scheme 4). Starting from either 5-oxoazelaic acid or 4-ketopimelic acid, two α,ω-bis(diethylamido)alkanones were synthesized by coupling with diethylamine in the presence of PyBop (benzotriazol – 1 – yl – oxytripyrrolidinophosphonium hexafluorophosphate). As expected, reduction with lithium aluminum hydride gave the corresponding α,ω-bis(diethylamino)alkanols. Deprotonation with potassium tert-butoxide and subsequent nucleophilic aromatic substitution at position 4 of 4,7-dichloroquinoline then yielded 7-chloro-4-(1’,7’-bis(diethylamino)-4’-heptoxy) quinoline, 22, and 7-chloro-4-(1’,9’-bis(diethylamino)-5’-nonoxy) quinoline, 23, respectively. Since 1,3-bis(diethylamino)-2-propanol is readily available, we were able to prepare 21 in a single step. Unfortunately, attempts to extend this synthetic strategy to symmetrically branched 4S CQ derivatives via thiation of α,ω-bis(diethylamido)alkanones with Lawesson’s reagent and subsequent reduction towards secondary thiols were not successful. As a result, our optimization efforts were restricted to 4O CQ analogues carrying two basic terminal amino groups. One candidate was designed based on the monobasic 4O compound (13) that showed good activity (μM IC50) vs. both CQR strains. Compound 22 harbors one extended aliphatic chain of similar length relative to 13 (4 methylenes between the 4N-quinolyl unit and the tertiary aliphatic amino group) such that it is predicted to wrap around the periphery of the protoporphyrin ring when forming a non-covalent complex with dimeric FPIX as previously observed for 13 (Fig. 1B), and a second aliphatic chain of appropriate length for possible ion pairing with a free FPIX propionate (Fig. 2). Interestingly, this compound and its homologue 23 showed improved activity in vivo vs. both CQR and CQS strains relative to 13 (Table 1), whereas the shorter chain analogue (21) that cannot ion pair with free propionate, remained significantly less active. Also, importantly, although we initially expected that improved activity would be due merely to increased accumulation (addition of the diamino branched side chain converts the monoprotic derivative 13 to a diprotic weak base at physiologic pH; (c.f. Table 2, Table 5), Table 4 shows that 22 is also a more potent inhibitor of Hz formation relative to monobasic 4O derivative 13. Interestingly, this is in spite of similar affinity for heme (Table 3), further emphasizing the lack of a simple relationship between heme monomer or heme μ-oxo dimer binding affinity and the ability of a drug to inhibit the formation of Hz. We note however that we measure Hz formation using an in vitro assay that may not completely mimic Hz formation in vivo.
Scheme 4.
Introduction of α,ω–diaminoalkoxy branched sidechains to 7-chloroquinoline
Figure 2.
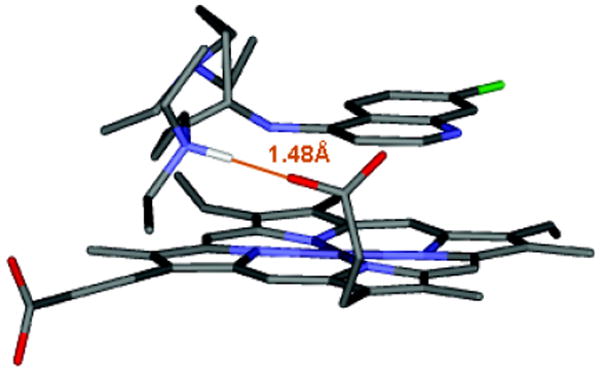
A suggested structure for a drug – μ-oxo dimer complex, in which the drug has a branched side chain. One of the branches is placed along the perimeter of the porphyrin ring, as seen in Figure 1B and for previously solved CQ, QN, QD, and AQ structures16,18, while the other branch extends away from the ring. In this arrangement, it is possible that this terminal amino group then forms an a hydrogen bonding pair with the propionate side chain of heme. A minimal distance (> 4 methylenes between terminal amino and the branch point) for both maximal π – π interaction and hydrogen bonding is defined in this structure.
DISCUSSION
The collection of compounds analyzed in this study introduce systematic modifications of the CQ side chain structure and cover a range of antiplasmodial activities. These modifications were suggested by detailed analysis of CQ – heme target structures that have recently been solved, as described elsewhere 16, 19 and, taken in their entirety, are more subtle and systematic than most previous quinoline antimalarial drug design studies. Such structure – function based analysis of candidate antimalarials is relatively rare but required since inexpensive antimalarial drugs active against CQR malaria are desperately needed. From these experiments we draw several new and important conclusions relevant to inexpensive quinoline antimalarial drug design:
Substitution of the amino function by a secondary derivative at the terminus of the side chain of 4-amino-7 chloroquinolines generally reduces the potency against CQR strains, but shows little effect on the antimalarial activity vs. CQS strains.
Replacement of the 4 – position nitrogen atom of the 7-chloroquinoline by either sulfur or oxygen substantially decreases the basicity of the quinolyl nitrogen which correlates with a general decrease in antimalarial activity. Thus, without further modification (see point 3 below) the basicity of the quinolyl N is crucial to antiplasmodial activity.
However, introduction of an additional basic amino group to the side chain of 4O CQ derivatives improves potency vs. both CQS and CQR strains while preserving an improved selectivity index, and also substantially increases the ability of 4O CQ analogues to inhibit formation of Hz while not altering the affinity to either monomeric or μ-oxo dimeric FPIX.
Surprisingly, and in contrast to many assumptions in the literature, we find no straightforward relationship between the ability to bind FPIX μ-oxo dimer and inhibition of Hz formation, nor any simple relationship between either of these drug characteristics and antimalarial potency.
With regard to conclusion 1, the well known observation that either shortening or lengthening the aliphatic side chain of CQ specifically improves activity vs. CQR parasites 9,11 only holds for diethyl derivatives. Surprisingly, longer chain monoethyl analogues with otherwise identical side chains (e.g. 5 vs. 10) show relatively high IC50 vs. CQR parasites, whereas either mono or diethyl short chain analogues are improved (e.g. 1, 6). The observed trends for 1 – 10 are important for two reasons; first, the data suggest that substituents at the terminal aliphatic N may interact with the CQ resistance mechanism15,29. Current models for the CQ resistance mechanism propose direct interaction of CQ with mutant PfCRT protein 15, 33. If correct, then results with 1 – 5 vs. 6 – 10 suggest a secondary amino group at the terminus of the side chain allows for better binding to PfCRT. Along with guiding additional modifications of quinolines, this concept should be useful for determining the nature of PfCRT CQ binding sites 33 via the design of azido – drug analogues or other probes. Second, these results suggest that metabolism to desethyl derivatives will impair the activity of longer chain CQ analogues vs. CQR parasites much more so than is the case for short chain analogues.
With regard to conclusion 2, we note another recent report on one compound in which carbon is substituted for nitrogen at position 437. This CQ isotere has the same length side chain as does CQ (as is the case for 13, 18) and showed a similarly reduced quinolyl N pKa (measured to be 4.8 vs. 4.5 and 4.1 for 13 and 18, respectively). However, the 4C CQ isotere showed even more significantly reduced potency, with no affect on parasite growth measured up to 3 μM drug. Thus, substitution with sulfur or oxygen at position 4 is not analogous to substitution with carbon even though all three affect quinolyl N pKa to a similar extent. Specifically, the improved SI of the analogues with oxygen at position 4, the ~ 1 μM IC50 vs. CQR parasites for some compounds (e.g. 13,14) and the ability to further titrate potency without fully reversing improved SI via addition of additional basic N to the aliphatic side chain (e.g. compounds 22, 23) suggests the 4O CQ pharmacophore is an attractive scaffold for drug design schemes to circumvent CQR.
Conclusions 3 and 4 have several important implications for antimalarial drug design, and force a rethinking of recent proposals for the action of CQ and related quinoline antimalarials 16,18,19. Importantly, we find that binding to heme (either the μ-oxo or monomeric forms) is not necessarily correlated with the ability of a CQ analogue to inhibit Hz formation. Association constants, Ka, (μ-oxo dimer) are quite similar for a representative set of compounds with side chain length similar to CQ (8, 13, 18), whereas IC50 for Hz inhibition among the same group of compounds varies by 100 fold. This is particularly impressive since 8,13,18 differ only at position 4 but are otherwise identical. Association to monomer (in 40 % DMSO) is similarly very weak for all compounds, and lowest energy geometries for 8, 13 or 18 μ-oxo dimer complex structures deduced by inversion recovery experiments are very similar. We note that although the biologically relevant dimer for Hz crystallization is the tethered head–to–tail dimer and not the μ-oxo, noncovalent association with this dimer is likely quite similar and governed by similar π–π and van der Waals interactions as described 16,18. Thus these data suggest that quinoline compounds inhibit Hz formation via some other mechanism. Possibilities include binding to one or more growing crystal faces, or by association with monomeric heme that cannot be measured in 40% DMSO solution. We also suggest that the lipophilicity of the noncovalent complex, which depends on the protonation state of the quinolyl N, needs to be accounted for, since recent work suggests Hz formation at a rate commensurate with what is observed in vivo is catalyzed by a lipid environment17,35,36.
Observed trends in this rationally designed series of compounds point out that even subtle variation in the quinoline structure can very significantly influence the ability to inhibit Hz formation, and that complex relationships between heme affinity and Hz inhibition exist for even very closely related quinoline antimalarials. We note that the improved activity of 22 relative to 13 is due to both an unanticipated improved ability to inhibit Hz as well as increased accumulation within the DV due to an improved VAR (vacuolar accumulation ratio). In addition, we note that the relative ability of these compounds to inhibit Hz formation at either pH 5.6 (approximate DV pH measured for CQS parasites20,21) or pH 5.2 (approximate DV pH measured for CQR parasites20,21) is not well correlated with their antimalarial activity vs. CQS or CQR strains. This conclusion is in contrast to previous work with other quinoline-based antimalarials38. For example, 22 has a 10 fold lower IC50 vs. strains Dd2 and FCB relative to 18, but a nearly identical IC50 for Hz inhibition at pH 5.2. More dramatically, 13 shows a 4 fold lower IC50 vs. strain Dd2 relative to 18, but roughly 5 - fold higher IC50 for Hz inhibition. In the previously reported trend38, only one CQS strain (NF54) was tested and the drugs examined were not as structurally similar as those in this study. Importantly then, either the chemistry of drug inhibition of Hz formation differs in some interesting way for CQR vs. CQS parasites, or DV accumulation for many of these compounds is also influenced by substitution at the 4 position and differs significantly for CQS vs. CQR parasites. Perhaps both concepts are relevant, since we also now find differences in Hz inhibition IC50 at pH 5.2 vs. 5.6 for CQ and other members of this series. Although the concept remains controversial, several reports have noted that mutant PfCRT found in the DV membrane of CQR parasites confers lower endosomal pH20,21,39,40, and that the pH for CQR DV is about 5.2 whereas for CQS it is closer to 5.620,21. Also, the volume of the DV, and apparent Cl- - dependent volume regulatory processes differ for CQR vs. CQS parasites21, with DV volume for CQR parasites recently measured to be significantly larger21. Assuming a similar rate of hemoglobin metabolism (and hence liberation of free heme) within the DV as suggested21, then these simple changes in the chemical environment for heme within the DV (i.e., bulk pH and heme concentration) likely affect the ability of a given quinoline compound to exert toxic effects via the production of heme – drug complexes16,18,19.
In summary, we have prepared a set of CQ structural modifications based upon simple predictions from recent atomic – level elucidation of drug – heme complexes 16,18,19. Overall, the results suggest additional modifications to CQ that can promote improved selectivity vs. CQR parasites and illustrate that relationships between heme binding, Hz inhibition, and antimalarial activity are more complex than previously thought. The data also show that additional modification of compounds with an improved SI, that work to promote improved bioavailability, can provide valuable new leads for further development of inexpensive quinoline antimalarials with good activity vs. CQR parasites (e.g. compounds 22, 23).
EXPERIMENTAL
I. General Methods and Synthesis
All reagents and solvents were commercially available and used without further purification. Flash chromatography was performed on Kieselgel 60, particle size 0.032-0.063 mm. NMR spectra were obtained on a 300 MHz (1H-NMR) and 75 MHz (13C-NMR) Varian FT-NMR spectrometer using CDCl3 as solvent unless indicated otherwise. The purity of all products was verified by two orthogonal HPLC methods with ODS-AQ and Nucleosil NH2 columns. Electrospray mass spectra (ESI-MS) were collected on a Thermo Finnigan LCQ mass spectrometer.
1. Representative procedure for the synthesis of N-(7-chloro-4-quinolyl)-1,n-diaminoalkanes
A mixture of 4,7-dichloroquinoline (1.0 g, 5.1 mmol) and ethylenediamine (1.7 mL, 25.3 mmol, 5 equiv.) was heated to 110 °C for 6 h under inert atmosphere and then cooled to room temperature. Aqueous NaOH (1N, 10 mL) was then added and the mixture was extracted with CH2Cl2. The organic layers were washed with water, brine, dried over anhydrous Na2SO4 and evaporated under reduced pressure. N-(7-Chloro-4-quinolyl)-1,2-diaminoethane (1.04 g, 4.4 mmol, 87% yield) was obtained as pale yellow crystals and used without further purification.
N-(7-Chloro-4-quinolyl)-1,2-diaminoethane.41-48
1H-NMR (300 MHz, CDCl3) δ = 1.26 (bs, 2H), 3.07-3.16 (m, 2H), 3.25-3.36 (m, 2H), 5.60-5.80 (m, 1H), 6.42 (d, J = 5.7 Hz, 1H), 7.37 (dd, J = 2.4 Hz, 8.7 Hz, 1H), 7.72 (d, J = 8.7 Hz, 1H), 7.96 (d, J = 2.4 Hz, 1H), 8.54 (d, J = 5.7 Hz, 1H); 13C-NMR (75 MHz, CDCl3) δ = 39.2, 44.9, 97.7, 116.6, 122.1, 123.3, 126.8, 133.1, 148.1, 149.5, 150.8.
N-(7-Chloro-4-quinolyl)-1,3-diaminopropane.13,41-45,49,50
Employing 1.0 g (5.1 mmol) of 4,7-dichloroquinoline in the procedure described above gave 1.05 g (4.5 mmol, 88% yield) of pale yellow crystals. 1H-NMR (300 MHz, CDCl3) δ = 1.48 (bs, 2H), 1.84-1.96 (m, 2H), 3.00-3.10 (m, 2H), 3.38-3.48 (m, 2H), 6.33 (d, J = 5.4 Hz, 1H), 7.30 (dd, J = 2.1 Hz, 9.0 Hz, 1H), 7.72 (d, J = 9.0 Hz, 1H), 7.92 (d, J = 2.1 Hz, 1H), 8.50 (d, J = 5.4 Hz, 1H); 13C-NMR (75 MHz, CDCl3) δ = 29.5, 40.8, 42.8, 97.8, 117.1, 122.0, 124.2, 127.6, 133.9, 148.6, 150.0, 151.5.
N-(7-Chloro-4-quinolyl)-1,4-diaminobutane.13,41-43,45, 49,51
Employing 2.0 g (10.1 mmol) of 4,7-dichloroquinoline in the procedure described above gave 2.09 g (8.4 mmol, 83% yield) of pale yellow crystals. 1H-NMR (300 MHz, CDCl3) δ = 1.28 (bs, 2H), 1.56-1.68 (m, 2H), 1.76-1.90 (m, 2H), 2.79 (t, J = 6.6 Hz, 2H), 3.22-3.32 (m, 2H), 6.09 (bs, 1H), 6.34 (d, J = 5.4 Hz, 1H), 7.29 (dd, J = 2.7 Hz, 9.0 Hz, 1H), 7.72 (d, J = 9.0 Hz, 1H), 7.91 (d, J = 2.7 Hz, 1H), 8.49 (d, J = 5.4 Hz, 1H); 13C-NMR (75 MHz, CDCl3) δ = 25.9, 30.6, 41.4, 43.0, 98.6, 117.3, 121.6, 124.8, 128.3, 134.5, 149.0, 150.0, 151.9.
N-(7-Chloro-4-quinolyl)-1,5-diaminopentane.29,52
Employing 1.0 g (5.1 mmol) of 4,7-dichloroquinoline in the procedure described above gave 1.16 g (4.4 mmol, 87% yield) of pale yellow crystals. 1H-NMR (300 MHz, CDCl3) δ = 1.15 (bs, 2H), 1.40-1.60 (m, 4H), 1.66-1.86 (m, 2H), 2.71 (t, J = 6.6 Hz, 2H), 3.20-3.38 (m, 2H), 5.49 (t, J = 4.8 Hz, 1H), 6.36 (d, J = 5.4 Hz, 1H), 7.28 (dd, J = 2.1, 9.0 Hz, 1H), 7.72 (d, J = 9.0 Hz, 1H), 7.92 (d, J = 2.1 Hz, 1H), 8.50 (d, J = 5.4 Hz, 1H); 13C-NMR (75 MHz, CDCl3) δ = 24.3, 28.5, 33.1, 41.9, 43.0, 98.9, 117.0, 120.9, 125.0, 128.6, 134.6, 149.0, 149.6, 151.9.
N-(7-Chloro-4-quinolyl)-1,6-diaminohexane.41,43,51
Employing 1.0 g (5.1 mmol) of 4,7-dichloroquinoline in the procedure described above gave 1.28 g (4.6 mmol, 91% yield) of pale yellow crystals. 1H-NMR (300 MHz, DMSO-d6) δ = 1.30-1.42 (m, 6H), 1.60-1.72 (m, 2H), 2.48-2.56 (m, 2H, overlapping with DMSO signal), 3.20-3.34 (m, 2H, overlapping with water signal), 6.46 (d, J = 5.4 Hz, 1H), 7.29 (t, J = 4.8 Hz, 1H), 7.44 (dd, J = 2.1 Hz, 9.0 Hz, 1H), 7.77 (d, J = 2.1 Hz, 1H), 8.27 (d, J = 9.0 Hz, 1H), 8.39 (d, J = 5.4 Hz, 1H); 13C-NMR (75 MHz, CD3OD) δ = 27.7, 28.0, 29.3, 33.5, 42.3, 43.9, 99.5, 118.7, 120.2, 124.3, 125.8, 127.5, 136.2, 149.6, 152.3.
2. Representative procedure for the synthesis of N-(7-chloro-4-quinolyl)-N’,N’-diethyl-1,(n)-diaminoalkanes and N-(7-chloro-4-quinolyl)-N’-ethyl-1,n-diaminoalkanes
To a solution of N-(7-chloro-4-quinolyl)-1,2-diaminoethane (3.8 g, 17.1 mmol) in anhydrous DMF was added Cs2CO3 (16.8 g, 51.4 mmol, 3 equiv.). The solution was stirred at 25 °C for 0.5 h and ethyl bromide (1.28 mL, 17.1 mmol, 1 equiv.) was added and stirred at 25 °C for 24 h. DMF was removed in vacuo. The residue was dissolved in CH2Cl2, extracted with water, dried over anhydrous Na2SO4, and the solvents were removed under reduced pressure. Flash chromatography (0.25%-1% Et3N in EtOH) allowed isolation of 1.73 g (6.2 mmol, 26% yield) of N-(7-chloro-4-quinolyl)-N’,N’-diethyl-1,2-diaminoethane and 1.78 g (7.1 mmol, 31% yield) of N-(7-chloro-4-quinolyl)-N’-ethyl-1,2-diaminoethane as pale yellow crystals.
N-(7-Chloro-4-quinolyl)-N’,N’-diethyl-1,2-diaminoethane 6.29,53
1H-NMR (300 MHz, CDCl3) δ = 1.07 (t, J = 7.2 Hz, 6H), 2.60 (q, J = 7.2 Hz, 4H), 2.81 (t, J = 6.0 Hz, 2H), 3.20-3.30 (m, 2H), 6.09 (bs, 1H), 6.36 (d, J = 5.4 Hz, 1H), 7.36 (dd, J = 2.1 Hz, 9.0 Hz, 1H), 7.65 (d, J = 9.0 Hz, 1H), 7.94 (d, J = 2.1 Hz, 1H), 8.52 (d, J = 5.4 Hz, 1H); 13C-NMR (75 MHz, CD3OD) δ = 11.6, 41.2, 47.9, 51.7, 99.6, 118.7, 124.0, 126.0, 127.6, 136.3, 149.5, 152.4.
N-(7-Chloro-4-quinolyl)-N’-ethyl-1,2-diaminoethane 1.26
1H-NMR (300 MHz, CDCl3) δ = 1.14 (t, J = 7.2 Hz, 3H), 1.24 (bs, 1H), 2.70 (q, J = 7.2 Hz, 2H), 2.98-3.07 (m, 2H), 3.27-3.37 (m, 2H), 5.89 (bs, 1H), 6.38 (d, J = 5.4 Hz, 1H), 7.34 (dd, J = 2.1 Hz, 9.0 Hz, 1H), 7.70 (d, J = 9.0 Hz, 1H), 7.93 (d, J = 2.1 Hz, 1H), 8.51 (d, J = 5.4 Hz, 1H); 13C-NMR (75 MHz, CDCl3) δ = 15.3, 41.9, 43.4, 47.1, 99.0, 117.2, 121.3, 124.9, 128.4, 134.5, 149.0, 149.8, 151.9.
N-(7-Chloro-4-quinolyl)-N’,N’-diethyl-1,3-diaminopropane 7.29,54
Employing 3.0 g (12.7 mmol) of N-(7-chloro-4-quinolyl)-1,3-diaminopropane in the procedure described above and purification by flash chromatography (0.25%-1% Et3N in EtOH) gave 0.85 g (2.9 mmol, 23% yield) of pale yellow crystals. 1H-NMR (300 MHz, CDCl3) δ = 1.09 (t, J = 7.2 Hz, 6H), 1.86-1.97 (m, 2H), 2.58-2.72 (m, 6H), 3.33-3.42 (m, 2H), 6.28 (d, J = 5.4 Hz, 1H), 7.31 (dd, J = 1.8 Hz, 8.7 Hz, 1H), 7.68 (d, J = 8.7 Hz, 1H), 7.91 (d, J = 1.8 Hz, 1H), 8.15 (bs, 1H), 8.49 (d, J = 5.4 Hz, 1H); 13C-NMR (75 MHz, CDCl3) δ = 11.3, 24.0, 44.4, 46.8, 53.3, 98.0, 117.5, 122.0, 124.4, 128.3, 134.2, 149.0, 150.4, 151.9.
N-(7-Chloro-4-quinolyl)-N’-ethyl-1,3-diaminopropane 2.26,28
Employing 3.0 g (12.7 mmol) of N-(7-chloro-4-quinolyl)-1,3-diaminopropane in the procedure described above and purification by flash chromatography (0.25%-1% Et3N in EtOH) gave 1.27 g (4.8 mmol, 37% yield) of pale yellow crystals. 1H-NMR (300 MHz, CDCl3) δ = 1.22 (t, J = 7.2 Hz, 3H), 1.35 (bs, 1H), 1.87-1.98 (m, 2H), 2.74 (q, J = 7.2 Hz, 2H), 2.90-2.98 (m, 2H), 3.34-3.43 (m, 2H), 6.29 (d, J = 5.4 Hz, 1H), 7.31 (dd, J = 2.1 Hz, 9.3 Hz, 1H), 7.74 (d, J = 9.3 Hz, 1H), 7.91 (d, J = 2.1 Hz, 1H), 7.98 (bs, 1 H), 8.49 (d, J = 5.4 Hz, 1H); 13C-NMR (75 MHz, CDCl3) δ = 15.2, 27.0, 43.9, 44.0, 49.3, 98.0, 117.5, 122.2, 124.4, 128.2, 134.3, 149.0, 150.4, 151.9.
N-(7-Chloro-4-quinolyl)-N’,N’-diethyl-1,4-diaminobutane 8.29,55
Employing 1.6 g (6.4 mmol) of N-(7-chloro-4-quinolyl)-1,4-diaminobutane in the procedure described above and purification by flash chromatography (0.25%-1% Et3N in EtOH) gave 0.4 g (1.3 mmol, 20% yield) of pale yellow crystals. 1H-NMR (300 MHz, CDCl3) δ = 1.03 (t, J = 7.2 Hz, 6H), 1.61-1.74 (m, 2H), 1.78-1.90 (m, 2H), 2.50 (t, J = 6.9 Hz, 2H), 2.57 (q, J = 7.2 Hz, 4H), 3.25-3.34 (m, 2H), 5.96 (bt, 1H), 6.38 (d, J = 5.4 Hz, 1H), 7.34 (dd, J = 2.1 Hz, 9.0 Hz, 1H) 7.70 (d, J = 9.0 Hz, 1H), 7.93 (d, J = 2.1 Hz, 1H), 8.52 (d, J = 5.4 Hz, 1H); 13C-NMR (75 MHz, CDCl3) δ = 11.1, 25.1, 26.7, 43.2, 46.6, 52.0, 98.8, 117.2, 121.6, 124.6, 128.4, 134.5, 149.0, 150.0, 151.9.
N-(7-Chloro-4-quinolyl)-N’-ethyl-1,4-diaminobutane 3
Employing 1.6 g (6.4 mmol) of N-(7-chloro-4-quinolyl)-1,4-diaminobutane in the procedure described above and purification by flash chromatography (0.25%-1% Et3N in EtOH) gave 0.45 g (1.6 mmol, 26% yield) of pale yellow crystals. 1H-NMR (300 MHz, CDCl3) δ = 1.16 (t, J = 7.2 Hz, 3H), 1.60-1.74 (m, 3H), 1.77-1.89 (m, 2H), 2.64-2.77 (m, 4H), 3.30 (t, J = 6.4 Hz, 2H), 6.18 (bs, 1H), 6.36 (d, J = 5.4 Hz, 1H), 7.33 (dd, J = 2.4 Hz, 9.0 Hz, 1H), 7.72 (d, J = 9.0 Hz, 1H), 7.93 (d, J = 2.4 Hz, 1H), 8.51 (d, J = 5.4 Hz, 1H); 13C-NMR (75 MHz, CDCl3) δ = 15.0, 26.0, 27.7, 42.9, 43.9, 48.8, 98.5, 117.2, 121.7, 124.5, 128.1, 134.4, 148.9, 150.0, 151.7; MS (ESI) m/z calcd for C15H20ClN3 277.1. Found (M + H)+: 278.1.
N-(7-Chloro-4-quinolyl)-N’,N’-diethyl-1,5-diaminopentane 9.29
Employing 2.48 g (9.4 mmol) of N-(7-chloro-4-quinolyl)-1,5-diaminopentane in the procedure described above and purification by flash chromatography (0.25%-1% Et3N in EtOH) gave 0.99 g (3.1 mmol, 33% yield) of pale yellow crystals. 1H-NMR (300 MHz, CDCl3) δ = 1.02 (t, J = 7.2 Hz, 6H), 1.41-1.61 (m, 4H), 1.72-1.85 (m, 2H), 2.41-2.48 (m, 2H), 2.53 (q, J = 7.2 Hz, 4H), 3.26-3.38 (m, 2H), 4.99 (bt, 1H), 6.41 (d, J = 5.4 Hz, 1H), 7.36 (dd, J = 2.4 Hz, 9.0 Hz, 1H), 7.65 (d, J = 9.0 Hz, 1H), 7.95 (d, J = 2.4 Hz, 1H), 8.53 (d, J = 5.4 Hz, 1H); 13C-NMR (75 MHz, CD3OD) δ = 11.1, 26.0, 26.6, 28.9, 43.6, 47.3, 53.2, 99.2, 118.3, 123.8, 125.5, 127.4, 135.8, 149.2, 151.9, 152.1.
N-(7-Chloro-4-quinolyl)-N’-ethyl-1,5-diaminopentane 4
Employing 2.48 g (9.4 mmol) of N-(7-chloro-4-quinolyl)-1,5-diaminopentane in the procedure described above and purification by flash chromatography (0.25%-1% Et3N in EtOH) gave 0.55 g (1.9 mmol, 20% yield) of pale yellow crystals. 1H-NMR (300 MHz, CDCl3) δ = 1.10 (t, J = 7.2 Hz, 3H), 1.22 (bs, 1H), 1.44-1.64 (m, 4H), 1.72-1.84 (m, 2H), 2.61-2.70 (m, 4H), 3.24-3.38 (m, 2H), 5.04 (bt, 1H), 6.39 (d, J = 5.4 Hz, 1H), 7.34 (dd, J = 2.1 Hz, 8.7 Hz, 1H), 7.65 (d, J = 8.7 Hz, 1H), 7.94 (d, J = 2.1 Hz, 1H), 8.52 (d, J = 5.4 Hz, 1H); 13C-NMR (75 MHz, CDCl3) δ = 15.0, 24.6, 28.3, 29.6, 42.8, 43.9, 49.3, 98.6, 117.0, 121.3, 124.7, 128.1, 134.4, 148.8, 149.7, 151.6; MS (ESI) m/z calcd for C16H22ClN3 291.2. Found (M + H)+: 292.2.
N-(7-Chloro-4-quinolyl)-N’,N’-diethyl-1,6-diaminohexane 10.29,56
Employing 4.0 g (14.4 mmol) of N-(7-chloro-4-quinolyl)-1,6-diaminohexane in the procedure described above and purification by flash chromatography (0.25%-1% Et3N in EtOH) gave 0.96 g (2.9 mmol, 20% yield) of pale yellow crystals. 1H-NMR (300 MHz, CDCl3) δ = 1.01 (t, J = 7.2 Hz, 6H), 1.31-1.56 (m, 6H), 1.71-1.84 (m, 2H), 2.38-2.45 (m, 2H), 2.52 (q, J = 7.2 Hz, 4H), 3.26-3.36 (m, 2H), 4.92 (bt, 1H), 6.41 (d, J = 5.4 Hz, 1H), 7.36 (dd, J = 2.1 Hz, 9.0 Hz, 1H), 7.65 (d, J = 9.0 Hz, 1H) 7.95 (d, J = 2.1 Hz, 1H), 8.53 (d, J = 5.4 Hz, 1H); 13C-NMR (75 MHz, CDCl3) δ = 11.2, 26.4, 26.6, 26.8, 28.1, 42.6, 46.3, 52.2, 98.3, 116.9, 121.5, 124.3, 127.6, 134.1, 148.6, 149.7, 151.2.
N-(7-Chloro-4-quinolyl)-N’-ethyl-1,6-diaminohexane 5
Employing 4.0 g (14.4 mmol) of N-(7-chloro-4-quinolyl)-1,6-diaminohexane in the procedure described above and purification by flash chromatography (0.25%-1% Et3N in EtOH) gave 1.28 g (4.2 mmol, 27% yield) of pale yellow crystals. 1H-NMR (300 MHz, CDCl3) δ = 1.10 (t, J = 7.2 Hz, 3H), 1.36-1.60 (m, 7H), 1.70-1.84 (m, 2H), 2.58-2.70 (m, 4H), 3.26-3.36 (m, 2H), 4.95 (bt, 1H), 6.40 (d, J = 5.4 Hz, 1H), 7.35 (dd, J = 2.1 Hz, 9.0 Hz, 1H), 7.65 (d, J = 9.0 Hz, 1H), 7.95 (d, J = 2.1 Hz, 1H), 8.53 (d, J = 5.4 Hz, 1H); 13C-NMR (75 MHz, CDCl3) δ = 15.0, 26.8, 26.8, 28.3, 29.8, 42.8, 43.9, 49.4, 98.6, 117.0, 121.3, 124.6, 128.0, 134.3, 148.8, 149.7, 151.6; MS (ESI) m/z calcd for C17H24ClN3 305.2. Found (M + H)+: 306.2.
3. Representative procedure for the synthesis of α,ω-(7-chloro-4-quinolyl)alkanediols
To a solution of 4,7-dichloroquinoline (0.2 g, 1.0 mmol, 1 equiv.) in ethylene glycol (2.0 mL, 35.9 mmol, 35.5 equiv.) under inert atmosphere was added a 1.0 M solution of potassium t-butoxide in t-butyl alcohol (1.5 mL, 1.5 mmol, 1.5 equiv.). The reaction proceeded with good stirring at 80° C for 18 h and was then quenched with saturated NaHCO3. The mixture was extracted with CH2Cl2, dried over anhydrous MgSO4, concentrated in vacuo, and purified by recrystallization from CHCl3 to yield 0.21 g of white crystals (0.95 mmol, 94% yield).
O-(7-Chloro-4-quinolyl)ethylene glycol
1H-NMR (300 MHz, CDCl3) δ = 2.17 (bs, 1H), 4.16 (bt, 2H), 4.33 (t, J = 4.5 Hz, 2H), 6.74 (d, J = 5.1 Hz, 1H), 7.45 (dd, J = 2.2 Hz, 8.9 Hz, 1H), 8.03 (d, J = 2.2 Hz, 1H), 8.15 (d, J = 8.9 Hz, 1H), 8.74 (d, J = 5.1 Hz, 1H); 13C-NMR (75 MHz, CDCl3) δ = 61.4, 70.8, 101.6, 120.2, 124.0, 127.1, 128.1, 136.5, 150.0, 152.9, 162.1.
O-(7-Chloro-4-quinolyl)-1,3-propanediol
Employing 0.2 g (1.0 mmol) of 4,7-dichloroquinoline in the procedure described above and recrystallization from CHCl3 gave 0.25 g (1.0 mmol, 99% yield) of white crystals. 1H-NMR (300 MHz, CDCl3) δ = 2.18 (m, 2H), 3.03 (bs, 1H), 3.98 (t, J = 5.9 Hz, 2H), 5.27 (t, J = 5.9 Hz, 2H), 6.55 (d, J = 5.5 Hz, 1H), 7.36 (dd, J = 2.1 Hz, 8.7 Hz, 1H), 7.96 (d, J = 2.1 Hz, 1H), 7.97 (d, J = 8.7 Hz, 1H), 8.59 (d, J = 5.5 Hz, 1H); 13C-NMR (75 MHz, CDCl3) δ = 31.7, 57.8, 64.8, 100.5, 119.3, 123.1, 126.2, 126.8, 135.7, 148.7, 151.8, 161.3.
O-(7-Chloro-4-quinolyl)-1,4-butanediol
Employing 0.2 g (1.0 mmol) of 4,7-dichloroquinoline in the procedure described above and recrystallization from CHCl3 gave 0.17 g (0.67 mmol, 66% yield) of white crystals.1H-NMR (300 MHz, CDCl3) δ = 1.64 (bs, 1H), 1.80 (m, 2H), 2.06 (m, 2H), 3.79 (t, J = 6.7 Hz, 2H), 4.24 (t, J = 6.6 Hz, 2H), 6.72 (d, J = 5.3 Hz, 1H), 7.44 (dd, J = 2.0, 8.9 Hz, 1H), 8.02 (d, J = 2.0 Hz, 1H,), 8.14 (d, J = 8.9 Hz, 1H), 8.7 (d, J = 5.3 Hz, 1H); 13C-NMR (75 MHz, CDCl3) δ = 25.8, 29.4, 62.5, 68.5, 100.9, 119.8, 123.3, 126.3, 127.8, 135.5, 149.5, 152.5, 161.6.
O-(7-Chloro-4-quinolyl)-1,5-pentanediol
Employing 0.2 g (1.0 mmol) of 4,7-dichloroquinoline in the procedure described above and recrystallization from CHCl3 gave 0.3 g (1.1 mmol, 99% yield) of white crystals. 1H-NMR (300 MHz, CDCl3) δ = 1.50 (bs, 1H), 1.68 (m, 4H), 1.99 (m, 2H), 3.73 (bt, 2H), 4.20 (t, J = 6.8 Hz, 2H), 6.70 (d, J = 5.3 Hz, 1H), 7.44 (dd, J = 2.1, 9.0 Hz, 1H), 8.01 (d, J = 2.1 Hz, 1H), 8.14 (d, J = 9.0 Hz, 1H), 8.72 (d, J = 5.3 Hz, 1H); 13C-NMR (75 MHz, CDCl3) δ = 22.4, 28.5, 32.3, 62.2, 68.4, 100.8, 119.7, 123.4, 126.3, 127.4, 135.6, 149.3, 152.3, 161.6.
O-(7-Chloro-4-quinolyl)-1,6-hexanediol
Employing 0.2 g (1.0 mmol) of 4,7-dichloroquinoline in the procedure described above and recrystallization from CHCl3 gave 0.34 g (1.2 mmol, 92% yield) of white crystals. 1H-NMR (300 MHz, CDCl3) δ = 1.40-1.69 (m, 6H), 1.99 (m, 2H), 3.68 (m, 3H), 4.24 (t, J = 5.8 Hz, 2H), 6.71 (d, J = 5.3 Hz, 1H), 7.44 (dd, J = 2.1, 8.9 Hz, 1H), 8.01 (d, J = 2.1 Hz, 1H), 8.14 (d, J = 8.9 Hz, 1H), 8.72 (d, J = 5.3 Hz, 1H); 13C-NMR (75 MHz, CDCl3) δ = 25.5, 25.8, 28.7, 32.5, 62.5, 68.5, 100.8, 119.8, 123.4, 126.3, 127.6, 135.6, 149.5, 152.4, 161.6.
4. Representative procedure for the synthesis of O-(7-chloro-4-quinolyl)-N,N-diethylaminoalkanols
To a solution of O-(7-chloro-4-quinolyl)-1,4-butanediol (0.78 g, 3.1 mmol, 1 equiv.) and Et3N (0.94 g, 9.3 mmol, 3 equiv.) in 20 mL of anhydrous THF at room temperature was added dropwise methansulfonyl chloride (1.07 g, 9.3 mmol, 3 equiv.). The reaction proceeded with good stirring for 10 minutes and was then quenched with saturated NaHCO3. The mixture was extracted with CH2Cl2, dried over anhydrous MgSO4, and concentrated in vacuo. The residue was dissolved in anhydrous CH3CN (15.0 mL) under inert atomosphere and N,N-diisopropylethylamine (2.0 g, 15.5 mmol, 5 equiv.) and diethylamine (4.53 g, 62.0 mmol, 20 equiv.) were added. The reaction mixture was stirred at 40° C for 48 h and was quenched with saturated NaHCO3. The mixture was extracted with CH2Cl2, dried over anhydrous MgSO4, and concentrated in vacuo. The product was purified by flash column chromatography using CH2Cl2:EtOH:Et3N (5:1:0.005 v/v) as the mobile phase to give a light yellow oil (0.78 g, 2.5 mmol, 61% yield).
O-(7-Chloro-4-quinolyl)-2-(N,N-diethylamino)ethanol 11.57,58
Employing 0.07 g (0.3 mmol) of O-(7-chloro-4-quinolyl)ethylene glycol in the procedure described above and purification by flash chromatography using CH2Cl2:EtOH (5:1 v/v) containing 0.5% Et3N as the mobile phase gave 0.07 g (0.27 mmol, 61% yield) of the desired product as a light yellow oil. 1H-NMR (300 MHz, CDCl3) δ = 1.10 (t, J = 7.1 Hz, 6H), 2.67 (q, J = 7.1 Hz, 4H), 3.03 (t, J = 5.9 Hz, 2H), 4.25 (t, J = 5.9 Hz, 2H), 6.71 (d, J = 5.1 Hz, 1H), 7.41 (dd, J = 2.2 Hz, 8.9 Hz, 1H), 7.99 (d, J = 2.2 Hz. 1H), 8.10 (d, J = 8.9 Hz, 1H), 8.71 (d, J = 5.1 Hz, 1H); 13C-NMR (75 MHz, CDCl3) δ = 12.6, 48.7, 51.9, 68.2, 101.7, 120.5, 124.1, 127.1, 128.5, 136.3, 150.4, 153.2, 162.2.
O-(7-Chloro-4-quinolyl)-3-(N,N-diethylamino)propanol 12
Employing 0.06 g (0.27 mmol) of O-(7-chloro-4-quinolyl)-1,3-propanediol in the procedure described above and purification by flash chromatography using CH2Cl2:EtOH (5:1 v/v) containing 0.5% Et3N as the mobile phase gave 0.05 g (0.17 mmol, 69% yield) of a light yellow oil. 1H-NMR (300 MHz, CDCl3) δ = 1.06 (t, J = 7.2 Hz, 6H), 2.11 (m, 2H), 2.61 (q, J = 7.2 Hz, 4H), 2.72 (t, J = 6.8 Hz, 2H), 4.27 (t, J = 6.2 Hz, 2H), 6.74 (d, J = 5.3 Hz, 1H), 7.45 (dd, J = 2.2 Hz, 8.8 Hz, 1H), 8.02 (d, J = 2.2 Hz. 1H), 8.13 (d, J = 8.8 Hz, 1H), 8.73 (d, J = 5.3 Hz, 1H); 13C-NMR (75 MHz, CDCl3) δ = 11.7, 26.8, 47.0, 49.1, 66.9, 100.9, 119.8, 123.3, 126.3, 127.8, 135.5, 149.5, 152.5, 161.6; MS (ESI) m/z calcd for C16H21ClN2O 292.1. Found (M + H)+: 293.1.
O-(7-Chloro-4-quinolyl)-4-(N,N-diethylamino)butanol 13
1H-NMR (300 MHz, CDCl3) δ = 1.03 (t, J = 7.2 Hz, 6H), 1.85 (m, 2H), 2.06 (m, 2H), 2.55 (m, 6H), 4.17 (t, J = 6.8 Hz, 2H), 6.72 (d, J = 5.4 Hz, 1H), 7.44 (dd, J = 2.1 Hz, 9.0 Hz, 1H), 8.01 (d, J = 2.1 Hz, 1H), 8.15 (d, J = 9.0 Hz, 1H), 8.72 (d, J = 5.4 Hz, 1H); 13C-NMR (75 MHz, CDCl3) δ = 11.5, 23.7, 26.8, 46.7, 52.4, 68.4, 100.8, 119.8, 123.4, 126.3, 127.7, 135.5, 149.6, 152.4, 161.5; MS (ESI) m/z calcd for C17H23ClN2O 306.2. Found (M + H)+: 307.2.
O-(7-Chloro-4-quinolyl)-5-(N,N-diethylamino)pentanol 14
Employing 0.09 g (0.35 mmol) of O-(7-chloro-4-quinolyl)-1,5-pentanediol in the procedure described above and purification by flash chromatography using CH2Cl2:EtOH (5:1 v/v) containing 0.5% Et3N as the mobile phase gave 0.11 g (0.34 mmol, 96% yield) of a light yellow oil. 1H-NMR (300 MHz, CDCl3) δ = 1.03 (t, J = 7.1 Hz, 6H), 1.56 (m, 4H), 1.96 (m, 2H), 2.55 (m, 6H), 4.24 (t, J = 6.4 Hz, 2H), 6.70 (d, J = 5.3 Hz, 1H), 7.44 (dd, J = 2.1 Hz, 9.0 Hz, 1H), 8.01 (d, J = 2.1 Hz, 1H), 8.15 (d, J = 9.0 Hz, 1H), 8.72 (d, J = 5.3 Hz, 1H); 13C-NMR (75 MHz, CDCl3) δ = 11.2, 24.1, 26.4, 28.7, 46.7, 52.6, 68.4, 100.9, 119.9, 123.4, 126.4, 127.8, 135.6, 149.7, 152.5, 161.6; MS (ESI) m/z calcd for C18H25ClN2O 320.2. Found (M + H)+: 321.1.
O-(7-Chloro-4-quinolyl)-6-(N,N-diethylamino)hexanol 15
Employing 0.07 g (0.25 mmol) of O-(7-chloro-4-quinolyl)-1,6-hexanediol in the procedure described above and purification by flash chromatography using CH2Cl2:EtOH (5:1 v/v) containing 0.5% Et3N as the mobile phase gave 0.07 g (0.21 mmol, 83% yield) of a light yellow oil. 1H-NMR (300 MHz, CDCl3) δ = 2.41 (t, J = 7.2 Hz, 6H), 1.35-1.60 (m, 6H), 1.92 (m, 2H), 2.41 (t, J = 7.5 Hz, 2H), 2.51 (q, J = 7.2 Hz, 4H), 4.15 (t, J = 6.9 Hz, 2H), 6.67 (d, J = 5.3 Hz, 1H), 7.41 (dd, J = 2.0 Hz, 9.0 Hz, 1H), 7.99 (d, J = 2.0 Hz, 1H), 8.12 (d, J = 9.0 Hz, 1H), 8.69 (d, J = 5.3 Hz, 1H); 13C-NMR (75 MHz, CDCl3) δ = 11.5, 26.0, 26.9, 27.34, 28.7, 46.8, 52.7, 68.5, 100.8, 119.8, 123.4, 126.3, 127.8, 135.5, 149.6, 152.4, 161.6; GC-MS (CI) m/z calcd for C19H27ClN2O 334.2. Found (M + H)+: 335.3.
Synthesis of S-(7-chloro-4-quinolyl)-2-(N,N-diethylamino)ethanethiol 16.53,58,59
A solution of 1M potassium t-butoxide in t-butyl alcohol (6.0 mL, 6.0 mmol, 1.2 equiv.) was heated to 40 °C and 2-(diethylamino)ethanethiol (0.9 g, 6.0 mmol, 1.2 equiv.) was added dropwise. This mixture was refluxed under nitrogen for 5 minutes. A solution of 4,7-dichloroquinoline (1.0 g, 5.0 mmol, 1 equiv.) in ether was then added dropwise over a period of 10 minutes. The mixture was refluxed for an additional 12 h, cooled to room temperature and then filtered. Excess solvent was removed in vacuo and the yellow residue was purified by flash chromatography using CH2Cl2/MeOH/Et3N (9:0.8:0.2 v/v) as the mobile phase to give a yellow oil (1.3 g, 4.4 mmol, 89% yield). 1H-NMR (CDCl3) δ = 1.05 (t, J = 7.2 Hz, 6H), 2.61 (q, J = 7.2 Hz, 4H), 2.83 (t, J = 6.6 Hz, 2H), 3.19 (t, J = 6.6 Hz, 2H), 7.17 (d, J = 5.1 Hz, 1H), 7.46 (dd, J = 2.1 Hz, 9.9 Hz, 1H), 8.0-8.1 (m, 2H), 8.68 (d, J = 5.1 Hz, 1H); 13C-NMR (75 MHz, CDCl3) δ = 29.9, 36.8, 51.3, 52.6, 116.2, 125.3, 125.3, 127.3, 129.1, 135.8, 148.2, 148.4, 150.4.
7-Chloroquinolyl-4-thiol.60
A solution of 4,7-dichloroquinoline (3.0 g, 15.0 mmol, 1 equiv.) in 100 mL of EtOH was heated to 50 °C and thiourea (1.15 g, 15.0 mmol, 1 equiv.) was added at once. This mixture was shaken vigorously for 3 minutes and then left to cool slowly to room temperature. The white solid was filtered off, dissolved in water and Na2CO3 was added. A yellow–orange precipitate formed which was then filtered off and dissolved in 0.2 M NaOH solution. An insoluble solid, 7,7’-dichloro-4,4’-diquinolylsulfide, was filtered off. The filtrate was acidified with acetic acid to give 2.64 g of yellow crystals (13.5 mmol, 60% yield). 1H-NMR (300 MHz, DMSO-d6) δ = 1.91 (s, 1H), 7.28 (d, J = 6.6 Hz, 1H), 7.48 (dd, J = 2.1 Hz, 9.0 Hz, 1H), 7.70 (d, J = 2.1 Hz, 1H), 7.88 (d, J = 6.6 Hz, 1H), 8.65 (d, J = 8.7 Hz, 1H); 13C-NMR (75 MHz, DMSO-d6) δ = 119.4, 125.4, 126.5, 131.5, 131.8, 135.0, 137.4, 137.7, 193.0.
5. Representative procedure for the synthesis of S-(7-chloro-4-quinolyl)-n-(N,N-diethylamino)alkanethiols
A mixture of 7-chloroquinolyl-4-thiol (0.8 g, 4.1 mmol, 1 equiv.) and KOH (0.11 g, 4.1 mmol, 1 equiv.) in dry CH3CN was stirred at 25° C under inert atmosphere. 1,3-Dibromopropane (0.42 mL, 4.1 mmol, 1 equiv.) was added dropwise and the mixture was stirred at room temperature for 12 h. N,N-Diisopropylethylamine (0.7 mL, 4.1 mmol, 1 equiv.) followed by diethylamine (2.13 mL, 20.5 mmol, 5 equiv.) were added dropwise and the reaction was stirred for an additional 12 h. The reaction mixture was concentrated in vacuo, diluted with water (15.0 mL), and extracted with EtOAc. The combined organic layers were dried over anhydrous MgSO4 and the solvents were removed under reduced pressure to give a light yellow oil. Purification was performed by flash chromatography using EtOAc/hexane/Et3N (7:2.9:0.1 v/v) as the mobile phase to yield a yellow oil (0.8 g, 2.6 mmol, 64% yield).
S-(7-Chloro-4-quinolyl)-3-(N,N-diethylamino)propanethiol 17.58
1H-NMR (300 MHz, CDCl3) δ = 1.11 (t, J = 7.1 Hz, 6H), 1.95-2.07 (m, 2H), 2.55-2.76 (m, 6H), 3.18 (t, J = 6.9 Hz, 2H), 7.17 (d, J = 5.1 Hz, 1H), 7.51 (dd, J = 2.1 Hz, 9.0 Hz, 1H), 8.05-8.09 (m, 2H), 8.72 (d, J = 5.1 Hz, 1H); 13C-NMR (75 MHz, CDCl3) δ = 11.6, 25.8, 29.2, 47.2, 51.7, 116.3, 125.2, 127.4, 129.1, 135.8, 148.1, 148.2, 150.5.
S-(7-Chloro-4-quinolyl)-4-(N,N-diethylamino)butanethiol 18.58
Employing 0.78 g (4.0 mmol) of 7-chloroquinolyl-4-thiol and 0.5 ml (4.0 mmol) of 1,4-dibromobutane in the procedure described above and purification by flash chromatography using EtOAc/hexane/Et3N (7:2.9:0.1 v/v) as the mobile phase gave a yellow oil (0.91 g, 2.8 mmol, 69% yield). 1H-NMR (300 MHz, CDCl3) δ = 1.04 (t, J = 7.0 Hz, 6H), 1.62-1.76 (m, 2H), 1.78-1.92 (m, 2H), 2.45-2.60 (m, 6H), 3.14 (t, J = 7.4 Hz, 2H), 7.20 (d, J = 5.1 Hz, 1H), 7.50 (dd, J = 2.3 Hz, 8.9 Hz, 1H), 8.02-8.10 (m, 2H), 8.71 (d, J = 5.1 Hz, 1H); 13C-NMR (75 MHz, CDCl3) δ = 11.9, 26.8, 26.9, 31.3, 47.0, 52.4, 116.2, 125.2, 127.3, 129.1, 135.8, 148.2, 148.5, 150.4.
S-(7-Chloro-4-quinolyl)-5-(N,N-diethylamino)pentanethiol 19
Employing 0.78 g (4.0 mmol) of 7-chloroquinolyl-4-thiol and 0.55 ml (4.0 mmol) of 1,5-dibromopentane in the procedure described above and purification by flash chromatography using EtOAc/hexane/Et3N (7:2.9:0.1 v/v) as the mobile phase gave a yellow oil (0.81 g, 2.4 mmol, 59% yield). 1H-NMR (300 MHz, CDCl3) δ = 1.13 (t, J = 7.2 Hz, 6H), 1.50-1.70 (m, 4H), 1.80-2.0 (m, 2H), 2.50-2.80 (m, 6H), 3.14 (t, J = 7.2 Hz, 2H), 7.18 (d, J = 4.8 Hz, 1H), 7.51 (dd, J = 1.5 Hz, 9.0 Hz, 1H), 8.00-8.10 (m, 2H), 8.72 (d, J = 4.8 Hz, 1H); 13C-NMR (75 MHz, CDCl3) δ = 11.7, 26.8, 27.3, 28,4, 31.4, 47.1, 52.9, 116.2, 125.3, 127.4, 129.1, 135.8, 148.3, 148.5, 150.5; MS (ESI) m/z calcd for C18H25ClN2S 336.1. Found (M + H)+: 337.1.
S-(7-Chloro-4-quinolyl)-6-(N,N-diethylamino)hexanethiol 20
Employing 0.78 g (4.0 mmol) of 7-chloroquinolyl-4-thiol and 0.6 ml (4.0 mmol) of 1,6-dibromohexane in the procedure described above and purification by flash chromatography using EtOAc/hexane/Et3N (7:2.9:0.1 v/v) as the mobile phase gave a yellow oil (1.02 g, 2.9 mmol, 71% yield). 1H-NMR (300 MHz, CDCl3) δ = 1.11 (t, J = 7.2 Hz, 6H), 1.30-1.50 (m, 2H), 1.50-1.70 (m, 4H), 1.75-1.95 (m, 2H), 2.54 (t, J = 7.4 Hz, 2H), 2.66 (q, J = 7.2 Hz, 4H), 3.12 (t, J = 7.4 Hz, 2H), 7.18 (d, J = 4.8 Hz, 1H), 7.51 (dd, J = 2.0 Hz, 8.9 Hz, 1H), 8.04-8.12 (m, 2H), 8.72 (d, J = 4.8 Hz, 1H); 13C-NMR (75 MHz, CDCl3) δ = 13.3, 28.4, 29.6, 30.7, 31.4, 33.7, 49.4, 55.0, 118.6, 127.7, 129.7, 131.4, 138.2, 150.6, 150.8, 152.9; MS (ESI) m/z calcd for C19H27ClN2S 350.2. Found (M + H)+: 351.2.
1,7-Bis(diethylamido)heptan-4-one
To a solution of 4-ketopimelic acid (0.2 g, 1.2 mmol) in CH3CN was added diisopropylamine (0.5 mL, 2.9 mmol, 2.4 equiv.), PyBop (1.19 g, 2.3 mmol, 1.9 equiv.) and N,N-diisopropylethylamine (0.5 mL, 3.2 mmol, 2.7 equiv.). The reaction was refluxed at 80° C for 48 h. The solvents were removed in vacuo and the residue was dissolved in CH2Cl2 and washed with 2M HCl and water. The organic layer was dried over anhydrous MgSO4 and evaporated under reduced pressure to give 0.31 g (1.1 mmol, 98% yield) of a brown oil. 1H-NMR (300 MHz, CDCl3) δ = 1.07 (t, J = 7.2 Hz, 6H), 1.15 (t, J = 7.2 Hz, 6H), 2.56 (t, J = 6.6 Hz, 4H), 2.82 (t, J = 6.6 Hz, 4H), 3.25-3.44 (m, 8H); 13C-NMR (75 MHz, CDCl3) δ = 13.2, 14.3, 27.1, 37.7, 40.4, 42.0, 171.0, 211.5.
1,7-Bis(diethylamino)heptan-4-ol
1,7-Bis(diethylamido)heptan-4-one (0.1 g, 0.35 mmol) and lithium aluminum hydride (2.1 ml of a 1M solution in THF, 2.1 mmol, 6 equiv.) in 3 mL of anhydrous toluene were refluxed at 110° C for 48 h. The reaction was quenched with 4M NaOH and extracted with CH2Cl2. The combined organic layers were dried over anhydrous MgSO4 and evaporated under reduced pressure to afford 0.08 g (0.31 mmol, 85% yield) of a brown oil. 1H-NMR (300 MHz, CDCl3) δ = 1.01 (t, J = 7.2 Hz, 12H), 1.32-1.44 (m, 2H), 1.51-1.64 (m, 6H), 2.36-2.65 (m, 12H), 3.51-3.60 (m, 1H); 13C-NMR (75 MHz, CDCl3) δ = 11.3, 24.3, 37.0, 46.6, 53.5, 71.4.
7-Chloro-4-(1’,7’-bis(diethylamino)-4’-heptoxy)quinoline 22
A mixture of 4,7-dichloroquinoline (0.23 g, 1.2 mmol, 3 equiv.), 1,7-bis(diethylamino)heptan-4-ol (0.1 g, 0.39 mmol, 1 equiv.), and a 1.0 M solution of t-BuOK in t-BuOH (0.78 mL, 0.78 mmol, 2 equiv.) was heated under inert atmosphere to 120° C for 72 h with good stirring in a closed vessel. Saturated NaHCO3 was added to the cooled reaction mixture, which was extracted with CH2Cl2, dried over anhydrous MgSO4, and concentrated in vacuo. Purification by flash chromatography using CH2Cl2:EtOH:Et3N (2:1:0.02, v/v) as the mobile phase gave a yellow oil (0.09 g, 0.21 mmol, 54% yield, NMR yield >95%). 1H-NMR (300 MHz, CDCl3) δ = 0.96 (t, J = 7.1 Hz, 12H), 1.37-1.59 (m, 4H), 1.56-1.86 (m, 4H), 2.42-2.54 (overlapping t and q, 12H), 4.60 (sep, J = 5.7 Hz, 1H), 6.77 (d, J = 5.4 Hz, 1H), 7.40 (dd, J = 1.9 Hz, 8.8 Hz, 1H), 7.98 (d, J = 8.8 Hz, 1H), 8.12 (d, J = 1.9 Hz, 1H), 8.69 (d, J = 5.4 Hz, 1H); 13C-NMR (75 MHz, CDCl3) δ = 11.6, 23.0, 47.0, 52.8, 78.6, 101.9, 120.7, 123.9, 126.6, 128.2, 135.9, 150.3, 152.7, 161.3; MS (ESI) m/z calcd for C24H38ClN3O 419.3. Found (M + H)+: 420.2.
1,9-Bis(diethylamido)nonan-5-one
To a mixture of 5-oxoazelaic acid (2.5 g, 12.4 mmol, 1 equiv.) and PyBop (15.4 g, 29.7 mmol, 2.4 equiv.) in anhydrous CH3CN (18.0 mL) under inert atmosphere was added diethylamine (5.11 mL, 49.9 mmol, 4 equiv.) and N,N-diisopropylethylamine (6.0 ml, 34.2 mmol, 2.8 equiv.). The reaction proceeded with good stirring at 35° C for 64 h and then solvents were removed in vacuo. The residue was dissolved in CH2Cl2, washed with a 2M HCl to remove N,N-diisopropylethylamine, dried over anhydrous MgSO4, and concentrated in vacuo to produce a yellow oil (2.39 g, 7.7 mmol, 62% yield). 1H-NMR (300 MHz, CDCl3) δ = 1.08 (t, J = 7.1 Hz, 6H), 1.16 (t, J = 7.2 Hz, 6H), 1.65-1.90 (m, 4H), 2.33 (t, J = 7.5 Hz, 4H), 2.50 (t, J = 7.1 Hz, 4H), 3.10-3.30 (m, 8H); 13C-NMR (75 MHz, CDCl3) δ = 14.9, 14.1, 19.3, 31.8, 40.0, 41.6, 41.9, 171.6, 210.5.
1,9-Bis(diethylamino)nonan-5-ol
1,9-Bis(diethylamido)nonan-5-one (0.1 g, 0.32 mmol) and lithium aluminum hydride in 1M THF (2.1 ml, 2.1 mmol, 6.6 equiv.) were dissolved in 3 mL of anhydrous toluene and refluxed at 110° C for 48 h. The reaction was quenched with 4M NaOH and extracted with CH2Cl2. The combined organic layers were dried over anhydrous MgSO4 and evaporated under reduced pressure to 0.08 g (0.29 mmol, 90% yield) of a brown oil. 1H-NMR (300 MHz, CDCl3) δ = 1.03 (t, J = 6.9 Hz, 12H), 1.32-1.55 (m, 12H), 2.41 (t, J = 6.6 Hz, 4H), 2.55 (q, J = 6.9 Hz, 8H), 3.51-3.61 (m, 1H); 13C-NMR (75 MHz, CDCl3) δ = 11.6, 23.3, 26.9, 37.5, 46.9, 53.0, 71.2.
7-Chloro-4-(1’,9’-bis(diethylamino)-5’-nonoxy)quinoline 23
A mixture of 4,7-dichloroquinoline (0.21 g, 1.05 mmol, 3 equiv.), 1,9-bis(diethylamino)nonan-5-ol (0.1 g, 0.35 mmol, 1 equiv.), and a 1.0 M solution of t-BuOK in t-BuOH (0.70 mL, 0.7 mmol, 2 equiv.) was heated under inert atmosphere to 120° C for 72 h in a closed vessel. Saturated NaHCO3 solution was added to the cooled reaction mixture, which was extracted with CH2Cl2, dried over anhydrous MgSO4, and concentrated in vacuo. Purification by flash chromatography using CH2Cl2:EtOH:Et3N (2:1:0.02, v/v) as the mobile phase gave 0.06 g (0.12 mmol, 35% yield, NMR yield >95%) of a yellow oil. 1H-NMR (300 MHz, CDCl3) δ = 0.98 (t, J = 7.2 Hz, 12H), 1.43-1.95 (m, 12H), 2.40-2.59 (m, 8H), 4.55-4.70 (m, 1H), 6.69 (d, J = 5.3 Hz, 1H), 7.41 (dd, J = 1.8 Hz, 9.8 Hz, 1H), 8.00 (d, J = 1.8 Hz, 1H), 8.15 (d, J = 9.8 Hz, 1H), 8.70 (d, J = 5.3 Hz, 1H); 13C-NMR (75 MHz, CDCl3) δ = 11.9, 23.6, 27.4, 33.8, 47.1, 53.0, 78.9, 101.7, 120.7, 123.9, 126.5, 128.1, 135.9, 150.3, 152.7, 161.5; GC-MS (CI) m/z calcd for C26H42ClN3O 447.3. Found (M + H)+: 448.2.
7-Chloro-4-(1’,3’-bis(diethylamino)-2’-propoxy)quinoline 21
A mixture containing 4,7-dichloroquinoline (0.29 g, 1.48 mmol, 3 equiv.), 1,3-bis(diethylamino)propan-2-ol (0.1 g, 0.49 mmol, 1 equiv.), and t-BuOK in t-BuOH (1.0 mL, 1.0 mmol, 2 equiv.) was heated under inert atmosphere to 70° C for 36 h in a closed vessel. The reaction mixture was allowed to cool to room temperature and saturated NaHCO3 was added. The mixture was then extracted with CH2Cl2, dried over anhydrous MgSO4, and concentrated in vacuo. Purification by flash chromatography using hexane:EtOH:Et3N (2:1:0.01, v/v) as the mobile phase gave a yellow oil (0.16 g, 0.44 mmol, 90% yield). 1H NMR (CDCl3, ppm): 1H-NMR (300 MHz, CDCl3) δ = 1.03 (t, J = 6.9 Hz, 12H), 2.45-2.73 (m, 8H), 2.74-2.92 (m, 4H), 4.65-4.83 (m, 1H), 6.92 (d, J = 5.1 Hz), 7.42 (dd, J = 2.1 Hz, J = 9.0 Hz, 1H), 8.05 (d, J = 2.1 Hz, 1H), 8.13 (d, J = 9.0 Hz, 1H), 8.72 (d, J = 5.1 Hz); 13C-NMR (75 MHz, CDCl3) δ = 12.3, 48.2, 77.9, 102.2, 120.8, 123.9, 126.5, 128.1, 135.8, 150.3, 152.7, 161.5; MS (ESI) m/z calcd for C20H30ClN3O 363.2. Found (M + H)+: 364.2.
II. Cell culture and antimalarial activity measurements
Drug activity was assessed and IC50 were quantified essentially as described 22,23. Drugs were diluted using complete media under sterile conditions and plated in a 96 well plate format. Sorbitol synchronized cultures62 were utilized with >95% of the parasites at the ring stage. Cultures were diluted to give a working stock of 0.5% parasitemia and 2% hematocrit (final hematocrit 1% & 0.5% parasitemia). The plates were incubated for 72 h at 37°C. After 72 h, 50 μL of 10x SYBR green I dye was added to each well, and the plate was incubated for 1 h at 37°C. Fluorescence was measured at 530 nm (490 nm excitation) using a spectra geminiEM plate reader. Data analysis was performed using sigma plot 9.0 software after downloading data in Excel format. For each assay, each drug dilution was analyzed in triplicate, and the results from at least two separate assays are averaged in each case (S.D. < 10 % in each case). All drugs were tested against two chloroquine sensitive, and two chloroquine resistant strains of P. falciparum (GCO3, HB3 and FCB, Dd2, respectively).
III. Heme Affinity Measurements
To measure affinity for monomeric heme, 1.2 mM stock solutions of hemin (sodium salt from Sigma-Aldrich) were prepared in DMSO and stored as 100 mL aliquots. 4.8 μM working solutions were prepared in 40% DMSO / phosphate buffer (fresh stocks prepared daily). Stock solutions of CQ and CQ analogues (diphosphate or dihydrochloride salts) were prepared in 40% DMSO / phosphate buffer and used for the titration experiments (all drugs dilutions were preparedin the same buffer).
1.5 mL cuvettes containing freshly prepared samples of 4.8 μM heme were titrated with increasing concentrations (0 – 50 μM) of drug. Following each addition, the sample was mixed and heme absorbance then recorded at 402 nm. Control experiments were performed by titrating 4.8 μM heme with similar volumes of solvent and no drug present. To measure affinity for μ-oxo dimeric heme, the procedure was similar except hemin was first converted to dimeric form in mild alkaline solution, followed by titration to pH 7.5 using Hepes buffer. The absorbance peak analysis was done using Microsoft excel and Ka were extracted from the ΔAbs402 plots via Scatchard analysis using Microsoft Excel and Sigma plot 9.0.1 software. For each compound, Ka reported are the average of three separate determinations.
IV. Quantification of in vitro Hz Formation Inhibition
A stock solution of 5 mM hemin (Fluka) in 0.1M NaOH was prepared and stored as small aliquots at -20°C. Fresh aliquots were thawed daily to room temperature before use. Lecithin stock solutions were prepared by dissolving in distilled water to 10 mg/ml and similarly stored. 0.5 M propionate was used to buffer experiments in the pH range 5.2 - 5.6.
The assay mixture (1 mL volumes) contained: 200 μL lecithin solution (2 μg/ml final) 20 μl hematin (100 μM final concentration) 20 μl 0.1M HCl (Y) μl propionate buffer (X) μL drug (dependant on the concentration required 0-1000 μM). The addition sequence involved first adding the lecithin followed by heme, HCl, propionate buffer and finally the drug. Each sample was prepared in triplicate. Following addition of the reagents the samples were incubated at 37°C with constant shaking for 18 h. After 18 h the assay was stopped by spinning the samples at 13,200 rpm for 10 minutes followed by carefully aspirating off the supernatant. The pellet was then resuspended in 50 mM bicarbonate buffer pH 9.0 (1 mL) and gently shaken at room temperature for 30 minutes to dissolve uncrystallized heme. The samples were then centrifuged as above and the supernatant removed. Following two additional bicarbonate washes the final pellet (Hz) was dried at 65°C for ~ 1 h. The samples were then dissolved in 0.1M NaOH to solubilize ß-hematin to free heme and ß-hematin formed was then quantified via heme absorbance at 402 nm. Calibration curves were prepared by titrating increasing amounts of heme in the same solvent vs. absorbance at 402 nm.
V. pKa Determinations
SPARC pKa calculator is an online tool developed at the University of Georgia by S.W. Karickhoff, L.A.Carreira and S.H. Hilal. Experimental pKa were determined using an Accumet AB15 pH meter and a calomel electrode. 10 mM solutions of the drugs (as dibasic salts) were made in distilled H2O and titrated at room temperature (23.0 ± 2.0°C) using 0.1 M NaOH. Titration plots were generated and pKa’s extracted via inflection points from the second derivative plots; (Δ2pH/ΔV2) vs. V, where V represents the volume of the titrant added and ΔV is the volume increment.
VI. Inversion Recovery and Distance Geometry Calculations
Relaxation rates of individual protons were converted into distances16 to the paramagnetic Fe center at one face of the μ-oxo dimer by applying the Solomon-Bloembergen equation:
| (1) |
where S is the total electron spin, r is the distance between the proton and the paramagnetic Fe, and γN, ge, μ0, and μb are constants. Measurements of magnetic susceptibility for the samples used in the relaxation experiments indicate that the μ-oxo dimer has an effective spin state of ½ per Fe. Thus, S=1/2 is used in equation 1. The effective correlation time (τc) is defined via the relation 1/τ(effective)) 1/τ(rotation) + 1/τ(exchange) + 1/τ(electron relaxation). Since the electron relaxation time (7×10-12 s) is the shortest among these time periods, it is essentially the effective correlation time. The factor 0.4 comes from simplifying the spectral density functions using (2π ×500 MHz and 2π ×329 GHz for the proton and electron angular frequencies, respectively). Using the distances derived from equation 1 as restraints, distance geometry/simulated annealing protocol is employed to solve the drug – μ-oxo dimer structures. The noncovalent complex is dynamic and the NMR spectrum is an average between free and complexed drug molecules. The distances 1/r6 are also time-averaged and since shorter distances are weighed more in this type of averaging, the r values obtained from the relaxation rates are used as minima in the distance geometry calculations (Further details are available from reference 16).
Supplementary Material
1H and 13C-NMR spectra of all compounds. HPLC chromatograms for compounds 1-23. This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
We thank the NIH (Grant RO1AI060792) for financial support.
Footnotes
Supported by the NIH: RO1AI060792
Abbreviations: CQ; chloroquine, CQS(R); chloroquine sensitive (resistant), DV; digestive vacuole, Hz; hemozoin, MQ; mefloquine, QN; quinine, t-BUOK; potassium tert butoxide, VAR; vacuolar accumulation ratio
References
- 1.Kumar KA, Sano G, Boscardin S, Nussenzweig RS, Nussenzweig MC, Zavala F, Nussenzweig V. The circumsporozoite protein is an immunodominant protective antigen in irradiated sporozoites. Nature. 2006;444(7121):937–940. doi: 10.1038/nature05361. [DOI] [PubMed] [Google Scholar]
- 2.Hoffman SL. Malaria: a protective paradox. Nature. 2006;444(7121):824–827. doi: 10.1038/nature05409. [DOI] [PubMed] [Google Scholar]
- 3.Vennerstrom L, Arbe-Barnes S, Brun R, Charman SA, Chiu FC, Chollet J, Dong Y, Dorn A, Hunziker D, Matile H, McIntosh K, Padmanilayam M, Santo Tomas J, Scheurer C, Scorneaux B, Tang Y, Urwyler H, Wittlin S, Charman WN. Identification of an antimalarial synthetic trioxolane drug development candidate. Nature. 2004;430(7002):900–904. doi: 10.1038/nature02779. [DOI] [PubMed] [Google Scholar]
- 4.Tang Y, Dong Y, Wittlin S, Charman SA, Chollet J, Chiu FC, Charman WN, Matile H, Urwyler H, Dorn A, Bajpai S, Wang X, Padmanilayam M, Karle JM, Brun R, Vennerstrom JL. Weak base dispiro-1,2,4-trioxolanes: potent antimalarial ozonides. Bioorg Med Chem Lett. 2007;17:1260–1265. doi: 10.1016/j.bmcl.2006.12.007. [DOI] [PubMed] [Google Scholar]
- 5.Dong Y, Tang Y, Chollet J, Matile H, Wittlin S, Charman SA, Charman WN, Tomas JS, Scheurer C, Snyder C, Scorneaux B, Bajpai S, Alexander SA, Wang X, Padmanilayam M, Cheruku SR, Brun R, Vennerstrom JL. Effect of functional group polarity on the antimalarial activity of spiro and dispiro-1,2,4-trioxolanes. Bioorg Med Chem. 2006;14:6368–6382. doi: 10.1016/j.bmc.2006.05.041. [DOI] [PubMed] [Google Scholar]
- 6.Posner GH, Paik IH, Chang W, Borstnik K, Sinishtaj S, Rosenthal AS, Shapiro TA. Malaria-Infected Mice Are Cured by a Single Dose of Novel Artemisinin Derivatives. J Med Chem. 2007;50:2516–2519. doi: 10.1021/jm070149m. [DOI] [PubMed] [Google Scholar]
- 7.Paik IH, Xie S, Shapiro TA, Labonte T, Narducci Sarjeant AA, Baege AC, Posner GH. Second generation, orally active, antimalarial, artemisinin-derived trioxane dimers with high stability, efficacy, and anticancer activity. J Med Chem. 2006;49:2731–2734. doi: 10.1021/jm058288w. [DOI] [PubMed] [Google Scholar]
- 8.Jambou R, Legrand E, Niang M, Khim N, Lim P, Volney B, Ekala MT, Bouchier C, Esterre P, Fandeur T, Mercereau-Puijalon O. Resistance of Plasmodium falciparum field isolates to in-vitro artemether and point mutations of the SERCA-type PfATPase6. Lancet. 2005;366(9501):1960–1963. doi: 10.1016/S0140-6736(05)67787-2. [DOI] [PubMed] [Google Scholar]
- 9.De D, Krogstad FM, Byers LD, Krogstad DJ. Structure-activity relationships for antiplasmodial activity among 7-substituted 4-aminoquinolines. J Med Chem. 1998;41:4918–4926. doi: 10.1021/jm980146x. [DOI] [PubMed] [Google Scholar]
- 10.Delarue S, Girault S, Maes L, Debreu-Fontaine MA, Labaeid M, Grellier P, Sergheraert C. Synthesis and in vitro and in vivo antimalarial activity of new 4-anilinoquinolines. J Med Chem. 2001;44:2827–2833. doi: 10.1021/jm010842o. [DOI] [PubMed] [Google Scholar]
- 11.Stocks PA, Raynes KJ, Bray PG, Park BK, O’Neill PM, Ward SA. Novel short chain chloroquine analogues retain activity against chloroquine resistant K1 Plasmodium falciparum. J Med Chem. 2002;45:4975–4983. doi: 10.1021/jm0108707. [DOI] [PubMed] [Google Scholar]
- 12.O’Neill PM, Willock DJ, Hawley SR, Bray PG, Storr RC, Ward SA, Park BK. Synthesis, antimalarial activity, and molecular modeling of tebuquine analogues. J Med Chem. 1997;40:437–448. doi: 10.1021/jm960370r. [DOI] [PubMed] [Google Scholar]
- 13.Madrid PB, Liou AP, DeRisi JL, Guy RK. Incorporation of an intramolecular hydrogen-bonding motif in the side chain of 4-aminoquinolines enhances activity against drug-resistant P. falciparum. J Med Chem. 2006;49:4535–4543. doi: 10.1021/jm0600951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ursos LM, Roepe PD. Chloroquine resistance in the malarial parasite, Plasmodium falciparum. Med Res Rev. 2002;22:465–491. doi: 10.1002/med.10016. [DOI] [PubMed] [Google Scholar]
- 15.Fidock DA, Nomura T, Talley AK, Cooper RA, Dzekunov SM, Ferdig MT, Ursos LM, Sidhu AB, Naude B, Deitsch KW, Su XZ, Wootton JC, Roepe PD, Wellems TE. Mutations in the P. falciparum digestive vacuole transmembrane protein PfCRT and evidence for their role in chloroquine resistance. Mol Cell. 2000;6:861–871. doi: 10.1016/s1097-2765(05)00077-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Leed A, DuBay K, Ursos LM, Sears D, de Dios AC, Roepe PD. Solution structures of antimalarial drug-heme complexes. Biochemistry. 2002;41:10245–10255. doi: 10.1021/bi020195i. [DOI] [PubMed] [Google Scholar]
- 17.Chong CR, Sullivan DJ., Jr Inhibition of heme crystal growth by antimalarials and other compounds: implications for drug discovery. Biochem Pharmacol. 2003;66:2201–2212. doi: 10.1016/j.bcp.2003.08.009. [DOI] [PubMed] [Google Scholar]
- 18.de Dios AC, Casabianca LB, Kosar A, Roepe PD. Structure of the amodiaquine-FPIX μ-oxo dimer solution complex at atomic resolution. Inorg Chem. 2004;43:8078–8084. doi: 10.1021/ic0489948. [DOI] [PubMed] [Google Scholar]
- 19.de Dios AC, Tycko R, Ursos LMB, Roepe PD. NMR Studies of Chloroquine –Ferriprotoporphyrin IX Complex. J Phys Chem A. 2003;107:5821–5825. [Google Scholar]
- 20.Bennett TN, Kosar AD, Ursos LM, Dzekunov S, Singh Sidhu AB, Fidock DA, Roepe PD. Drug resistance-associated PfCRT mutations confer decreased Plasmodium falciparum digestive vacuolar pH. Mol Biochem Parasitol. 2004;133:99–114. doi: 10.1016/j.molbiopara.2003.09.008. [DOI] [PubMed] [Google Scholar]
- 21.Gligorijevic B, Bennett T, McAllister R, Urbach JS, Roepe PD. Spinning disk confocal microscopy of live, intraerythrocytic malarial parasites. 2. Altered vacuolar volume regulation in drug resistant malaria. Biochemistry. 2006;45:12411–12423. doi: 10.1021/bi0610348. [DOI] [PubMed] [Google Scholar]
- 22.Bennett TN, Paguio M, Gligorijevic B, Seudieu C, Kosar AD, Davidson E, Roepe PD. Novel, rapid, and inexpensive cell-based quantification of antimalarial drug efficacy. Antimicrob Agents Chemother. 2004;48:1807–1810. doi: 10.1128/AAC.48.5.1807-1810.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Smilkstein M, Sriwilaijaroen N, Kelly JX, Wilairat P, Riscoe M. Simple and inexpensive fluorescence-based technique for high-throughput antimalarial drug screening. Antimicrob Agents Chemother. 2004;48:1803–1806. doi: 10.1128/AAC.48.5.1803-1806.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Johnson JD, Dennull RA, Gerena L, Lopez-Sanchez M, Roncal NE, Waters NC. Assessment and continued validation of the malaria SYBR green I-based fluorescence assay for use in malaria drug screening. Antimicrob Agents Chemother. 2007;51:1926–1933. doi: 10.1128/AAC.01607-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vippagunta SR, Dorn A, Matile H, Bhattacharjee AK, Karle JM, Ellis WY, Ridley RG, Vennerstrom JL. Structural specificity of chloroquine-hematin binding related to inhibition of hematin polymerization and parasite growth. J Med Chem. 1999;42:4630–4639. doi: 10.1021/jm9902180. [DOI] [PubMed] [Google Scholar]
- 26.Ridley RG, Hofheinz W, Matile H, Jaquet C, Dorn A, Masciadri R, Jolidon S, Richter WF, Guenzi A, Girometta M-A, Urwyler H, Huber W, Thaithong S, Peters W. 4-Aminoquinoline analogs of chloroquine with shortened side chains retain activity against chloroquine-resistant Plasmodium falciparum. Antimicrob Agents Chemother. 1996;40:1846–1854. doi: 10.1128/aac.40.8.1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hofheinz W, Jaquet C, Jolidon S. Aminochinolin-Derivate mit einer Wirksamkeit gegen Malariaerreger. 94116281.0. European patent application. 1995 June;
- 28.Tarbell DS, Shakespeare N, Claus CJ, Bunnett JF. The synthesis of some 7-chloro-4-(3-alkylaminopropylamino)-quinolines. J Am Chem Soc. 1946;68:1217–1219. doi: 10.1021/ja01211a023. [DOI] [PubMed] [Google Scholar]
- 29.De D, Byers LD, Krogstad DJ. Antimalarials: synthesis of 4-aminoquinolines that circumvent drug resistance in malaria parasites. J Heterocycl Chem. 1997;34:315–320. [Google Scholar]
- 30.Surrey AR, Lesher GY, Mayer JR, Webb WG. Hypotensive agents. 11. The preparation of quaternary salts of some 4-dialkylaminoalkylaminoquinolines. J Am Chem Soc. 1959;81:2894–2897. [Google Scholar]
- 31.Wellems TE, Walker-Jonah A, Panton LJ. Genetic mapping of the chloroquine-resistance locus on Plasmodium falciparum chromosome 7. Proc Natl Acad Sci U S A. 1991;88:3382–3386. doi: 10.1073/pnas.88.8.3382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bennett TN, Patel J, Ferdig MT, Roepe PD. Plasmodium falciparum Na+/H+ exchanger activity and quinine resistance. Mol Biochem Parasitol. 2007;153:48–58. doi: 10.1016/j.molbiopara.2007.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang H, Paguio M, Roepe PD. The antimalarial drug resistance protein Plasmodium falciparum chloroquine resistance transporter binds chloroquine. Biochemistry. 2004;43:8290–8296. doi: 10.1021/bi049137i. [DOI] [PubMed] [Google Scholar]
- 34.Constantinidis I, Satterlee JD. UV-visible and carbon NMR studies of chloroquine binding to urohemin I chloride and uroporphyrin I in aqueous solutions. J Am Chem Soc. 1988;110:4391–4395. [Google Scholar]
- 35.Egan TJ, Mavuso WW, Ross DC, Marques HM. Thermodynamic factors controlling the interaction of quinoline antimalarial drugs with ferriprotoporphyrin IX. J Inorg Biochem. 1997;68:137–145. doi: 10.1016/s0162-0134(97)00086-x. [DOI] [PubMed] [Google Scholar]
- 36.Egan TJ, Ncokazi KK. Effects of solvent composition and ionic strength on the interaction of quinoline antimalarials with ferriprotoporphyrin IX. J Inorg Biochem. 2004;98:144–152. doi: 10.1016/j.jinorgbio.2003.09.007. [DOI] [PubMed] [Google Scholar]
- 37.Cheruku SR, Maiti S, Dorn A, Scorneaux B, Bhattacharjee AK, Ellis WY, Vennerstrom JL. Carbon isosteres of the 4-aminopyridine substructure of chloroquine: effects on pK(a), hematin binding, inhibition of Hz formation, and parasite growth. J Med Chem. 2003;46(14):3166–9. doi: 10.1021/jm030038x. [DOI] [PubMed] [Google Scholar]
- 38.Dorn A, Vippagunta SR, Matile H, Jaquet C, Vennerstrom JL, Ridley RG. An assessment of drug-haematin binding as a mechanism for inhibition of haematin polymerisation by quinoline antimalarials. Biochem Pharmacol. 1998;55:727–736. doi: 10.1016/s0006-2952(97)00510-8. [DOI] [PubMed] [Google Scholar]
- 39.Reeves DC, Liebelt DA, Lakshmanan V, Roepe PD, Fidock DA, Akabas MH. Chloroquine-resistant isoforms of the Plasmodium falciparum chloroquine resistance transporter acidify lysosomal pH in HEK293 cells more than chloroquine-sensitive isoforms. Mol Biochem Parasitol. 2006;150:288–299. doi: 10.1016/j.molbiopara.2006.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Naude B, Brzostowski JA, Kimmel AR, Wellems TE. Dictyostelium discoideum expresses a malaria chloroquine resistance mechanism upon transfection with mutant, but not wild-type, Plasmodium falciparum transporter PfCRT. J Biol Chem. 2005;280:25596–25603. doi: 10.1074/jbc.M503227200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Musonda CC, Gut J, Rosenthal PJ, Yardley V, Carvalho de Souza RC, Chibale K. Application of multicomponent reactions to antimalarial drug discovery. Part 2: New antiplasmodial and antitrypanosomal 4-aminoquinoline γ-and δ-lactams via a catch and release’ protocol. Bioorg Med Chem. 2006;14:5605–5615. doi: 10.1016/j.bmc.2006.04.035. [DOI] [PubMed] [Google Scholar]
- 42.Solomon VR, Puri SK, Srivastava K, Katti SB. Design and synthesis of new antimalarial agents from 4-aminoquinoline. Bioorg Med Chem. 2005;13:2157–2165. doi: 10.1016/j.bmc.2004.12.051. [DOI] [PubMed] [Google Scholar]
- 43.Biot C, Daher W, Ndiaye CM, Melnyk P, Pradines B, Chavain N, Pellet A, Fraisse L, Pelinski L, Jarry C, Brocard J, Khalife J, Forfar-Bares I, Dive D. Probing the Role of the Covalent Linkage of Ferrocene into a Chloroquine Template. J Med Chem. 2006;49:4707–4714. doi: 10.1021/jm060259d. [DOI] [PubMed] [Google Scholar]
- 44.Musonda CC, Taylor D, Lehman J, Gut J, Rosenthal PJ, Chibale K. Application of multi-component reactions to antimalarial drug discovery. Part 1: Parallel synthesis and antiplasmodial activity of new 4-aminoquinoline Ugi adducts. Bioorg Med Chem Lett. 2004;14:3901–3905. doi: 10.1016/j.bmcl.2004.05.063. [DOI] [PubMed] [Google Scholar]
- 45.Singh C, Malik H, Puri SK. Synthesis and antimalarial activity of a new series of trioxaquines. Bioorg Med Chem. 2004;12:1177–1182. doi: 10.1016/j.bmc.2003.11.021. [DOI] [PubMed] [Google Scholar]
- 46.Chipeleme A, Gut J, Rosenthal PJ, Chibale K. Synthesis and biological evaluation of phenolic Mannich bases of benzaldehyde and (thio)semicarbazone derivatives against the cysteine protease falcipain-2 and a chloroquine resistant strain of Plasmodium falciparum. Bioorg Med Chem. 2007;15:273–282. doi: 10.1016/j.bmc.2006.09.055. [DOI] [PubMed] [Google Scholar]
- 47.Dechy-Cabaret O, Benoit-Vical F, Robert A, Meunier B. Preparation and antimalarial activities of “trioxaquines”, new modular molecules with a trioxane skeleton linked to a 4-aminoquinoline. ChemBioChem. 2000;1:281–283. doi: 10.1002/1439-7633(20001117)1:4<281::AID-CBIC281>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 48.Beagley P, Blackie MAL, Chibale K, Clarkson C, Moss JR, Smith PJ. Synthesis and antimalarial activity in vitro of new ruthenocene-chloroquine analogues. J Chem Soc, Dalton Trans. 2002:4426–4433. [Google Scholar]
- 49.Dechy-Cabaret O, Benoit-Vical F, Loup C, Robert A, Gornitzka H, Bonhoure A, Vial H, Magnaval J-F, Seguela J-P, Meunier B. Synthesis and antimalarial activity of trioxaquine derivatives. Chem Eur J. 2004;10:1625–1636. doi: 10.1002/chem.200305576. [DOI] [PubMed] [Google Scholar]
- 50.Madrid PB, Wilson NT, DeRisi JL, Guy RK. Parallel Synthesis and Antimalarial Screening of a 4-Aminoquinoline Library. J Comb Chem. 2004;6:437–442. doi: 10.1021/cc0340473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chibale K, Ojima I, Haupt H, Geng X, Pera P, Bernacki RJ. Modulation of human mammary cell sensitivity to paclitaxel by new quinoline sulfonamides. Bioorg Med Chem Lett. 2001;11:2457–2460. doi: 10.1016/s0960-894x(01)00462-0. [DOI] [PubMed] [Google Scholar]
- 52.Jin W, Arakawa Y, Yasuzawa H, Taki T, Hashiguchi R, Mitsutani K, Shoga A, Yamaguchi Y, Kurosaki H, Shibata N, Ohta M, Goto M. Comparative Study of the Inhibition of Metallo-β-Lactamases (IMP-1 and VIM-2) by Thiol Compounds That Contain a Hydrophobic Group. Biol Pharm Bull. 2004;27:851–856. doi: 10.1248/bpb.27.851. [DOI] [PubMed] [Google Scholar]
- 53.Gallo S, Atifi S, Mahamoud A, Santelli-Rouvier C, Wolfart K, Molnar J, Barbe J. Synthesis of aza mono, bi and tricyclic compounds. Evaluation of their anti MDR activity. Eur J Med Chem. 2003;38:19–26. doi: 10.1016/s0223-5234(02)01422-8. [DOI] [PubMed] [Google Scholar]
- 54.Madrid PB, Sherrill J, Liou AP, Weisman JL, DeRisi JL, Guy RK. Synthesis of ring-substituted 4-aminoquinolines and evaluation of their antimalarial activities. Bioorg Med Chem Lett. 2005;15:1015–1018. doi: 10.1016/j.bmcl.2004.12.037. [DOI] [PubMed] [Google Scholar]
- 55.Surrey AR, Hammer HF. Some 7-substituted 4-aminoquinoline derivatives. J Am Chem Soc. 1946;68:113–116. doi: 10.1021/ja01205a036. [DOI] [PubMed] [Google Scholar]
- 56.Drake NL, Creech HJ, Garman JA, Haywood ST, Peck RM, Van Hook JO, Walton E. Synthetic antimalarials. The preparation of certain 4-aminoquinolines. J Am Chem Soc. 1946;68:1208–1213. doi: 10.1021/ja01211a021. [DOI] [PubMed] [Google Scholar]
- 57.Cheng J, Zeidan R, Mishra S, Liu A, Pun SH, Kulkarni RP, Jensen GS, Bellocq NC, Davis ME. Structure-Function Correlation of Chloroquine and Analogues as Transgene Expression Enhancers in Nonviral Gene Delivery. J Med Chem. 2006;49:6522–6531. doi: 10.1021/jm060736s. [DOI] [PubMed] [Google Scholar]
- 58.Clinton RO, Suter CM. Some dialkylaminoalkyl sulfides and ethers derived from quinoline and acridine heterocycles. J Am Chem Soc. 1948;70:491–494. doi: 10.1021/ja01182a017. [DOI] [PubMed] [Google Scholar]
- 59.Ghisalberti D, Mahamoud A, Chevalier J, Baitiche M, Martino M, Pages J-M, Barbe J. Chloroquinolines block antibiotic efflux pumps in antibiotic-resistant Enterobacter aerogenes isolates. Int J Antimicrob Agents. 2006;27:565–569. doi: 10.1016/j.ijantimicag.2006.03.010. [DOI] [PubMed] [Google Scholar]
- 60.Surrey AR. Basic esters and amides of 4-quinolylmercaptoacetic acid derivatives. J Am Chem Soc. 1948;70:2190–2193. [Google Scholar]
- 61.Ferrari V, Cutler DJ. Temperature dependence of the acid dissociation constants of chloroquine. J Pharm Sci. 1987;76(7):554–556. doi: 10.1002/jps.2600760714. [DOI] [PubMed] [Google Scholar]
- 62.Lambros C, Vanderberg JP. Synchronization of Plasmodium falciparum erythrocytic stages in culture. The Journal of Parasitology. 1979;65:418–420. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
1H and 13C-NMR spectra of all compounds. HPLC chromatograms for compounds 1-23. This material is available free of charge via the Internet at http://pubs.acs.org.