

Abstract
Cullin7 (CUL7) is a molecular scaffold that organizes an E3 ubiquitin ligase containing the F-box protein Fbw8, Skp1 and the ROC1 RING finger protein. Dysregulation of the CUL7 E3 Ligase has been directly linked to hereditary human diseases as cul7 germline mutations were found in patients with autosomal-recessive 3-M and Yakuts short stature syndromes, which are characterized by profound pre- and postnatal growth retardation. In addition, genetic ablation of CUL7 in mice resulted in intra-uterine growth retardation and perinatal lethality, underscoring its importance for growth regulation. The recent identification of insulin receptor substrate 1, a critical mediator of insulin and insulin-like growth factor-1 signaling, as the proteolytic target of the CUL7 E3 ligase, provided a molecular link between CUL7 and a well-established growth regulatory pathway. This result, coupled with other studies demonstrating interactions between CUL7 and the p53 tumor suppressor protein, as well as the simian virus 40 large T antigen oncoprotein, further implicated CUL7 as a novel player in growth control and suggested pathomechanistic insights into CUL7-linked growth retardation syndromes.
Keywords: cullin7, Fbw8, IRS-1, IGF-1, insulin, ubiquitin, proteasome, growth retardation, 3-M syndrome, yakuts short stature syndrome
The CUL7 E3 Ligase Targets Cyclin D1 and Insulin Receptor Substrate 1 for Ubiquitin-Dependent Degradation
The turnover of intracellular proteins by the Ubiquitin (Ub)-Proteasome System (UPS) is a precisely controlled process that regulates a broad spectrum of fundamental cellular functions, ranging from cell cycle progression to signal transduction1. Central to the UPS is the recognition of a substrate by an E3 Ub ligase, a step pivotal for initiating the ubiquitination reaction that joins the target protein covalently with lysine 48-linked polyubiquitin chains, thereby leading to its degradation by the 26S proteasome.
The cullin-RING complexes constitute the largest group of E3 ligases, which are characterized by two signature components: a cullin (CUL) protein and the RING (for Really Interesting New Gene) finger protein ROC1 (also termed Rbx1 or Hrt1)2. Cullins are molecular scaffolds, capable of integrating both a molecule with substrate-targeting function, and the ROC1 RING domain for tethering an E2 Ub conjugating enzyme. In the prototypic SCF (Skp1·CUL1·F-box protein-ROC1) complex, the CUL1 N-terminus binds to the Skp1·F-box protein substrate-module, whereas the C-terminally located cullin domain anchors ROC1, which recruits Cdc34 and/or Ubc4/5 E2 conjugating enzyme to catalyze the transfer of Ub to the substrate protein.
Cullin7 (CUL7, also known as KIAA0076, p185, p193) is the seventh cullin family member identified to date. It was initially isolated as a cellular protein bound to simian virus 40 (SV40) large T antigen.3,4 As revealed by the subsequent work by Dias et al.5 and Arai et al.,6 CUL7 assembles an SCF-like E3 ligase complex composed of the adapter protein Skp1 (S-phase kinase associated protein 1), ROC1 and the WD40 repeat-containing F-box protein Fbw8 (also named Fbx29, Fbw6 or Fbxw8; see Fig. 1A and Table 1). To date, Fbw8 is the only F-box protein known to bind to CUL7 via Skp1,5,7 underscoring the remarkable selectivity of this cullin protein.
Figure 1.

(A) Composition of the CUL7 E3 Ub ligase complex. The CUL7 protein assembles an SCF-like complex composed of Skp1, Fbw8 and ROC1. While Fbw8 is responsible for substrate protein recognition, ROC1 recruits an Ub-charged E2 Ub-conjugating enzyme for substrate ubiquitination. It remains to be determined how CUL7 binds to the Skp1-Fbw8 heterodimer. (B) Domain organization of the CUL7 protein, as well as localization and relative frequency of CUL7 mutations identified in patients with 3-M (upper half) and Yakuts Short Stature syndrome (lower half). A single mutation (4582insT) of the CUL7 gene, predicted to yield a protein truncated at amino acid position 1553, was found in 43 patients of 37 families with Yakuts short stature syndrome. N8, abbreviation for Nedd8, denotes the CUL7 C-terminal site containing sequence conserved for cullin neddylation, as described previously.75
Table 1.
CUL7 interacting proteins
| CUL7-interacting proteins | Comments | Ref. |
|---|---|---|
| •The CUL7 E3 components | 5, 6 | |
| •Skp1 |
|
|
| •Fbw8 |
|
8, 9 |
| •ROC1 |
|
|
| •Other interacting proteins | ||
| •CUL1 |
|
37 |
| •PARC |
|
23, 76 |
| •Glomulin (Fap68) |
|
6 |
| •p53 |
|
20–22 |
| •SV40 T antigen |
|
3, 4, 27 |
Recently, the CUL7 E3 Ub ligase has been implicated in the proteasomal degradation of two cellular proteins: cyclin D1,8 and insulin receptor substrate 1 (IRS-1).9 Cyclin D1 is an important to S-phase cell cycle progression and is subjected regulator of the G1 to considerable posttranslational regulation (reviewed in ref. 10). The study by Okabe et al.8 demonstrated that Fbw8, the substrate recognition subunit of the CUL7 E3 ligase, mediates the ubiquitination of cyclin D1 in a manner that is dependent upon the phosphorylation of cyclin D1 residue T286 by the ERK2 MAP kinase. Conversion of T286 to alanine or knockdown of Fbw8, CUL1 or CUL7 by RNAi resulted in the stabilization of cyclin D1 and prevented cell cycle progression in a number of different cell types tested. However, it should be noted that Lin et al.11 identified the SCFFBX4-αB-crystallin complex as an E3 ligase for the proteolytic turnover of cyclin D1, and demonstrated the requirement for T286 phosphorylation by glycogen synthase kinase 3β for degradation.
By employing a proteomic approach in search for Fbw8 interacting proteins, Xu et al.9 identified IRS-1 as a proteolytic target of the CUL7 E3 ligase. IRS-1 is a critical component of the signaling pathways downstream of the insulin and insulin-like growth factor 1 (IGF-1) receptor (reviewed in ref. 12). Upon receptor activation, IRS-1 is phosphorylated at multiple tyrosine residues, and then recruits SH2 (Src homology 2)-containing adaptor proteins for the activation of downstream Akt (via PI3K) and RAS/MEK/ERK (via Grb2/SOS) pathways (see Fig. 2). Haruta et al.13 observed that IRS-1 was degraded during prolonged exposure to insulin, in a manner that was sensitive to Wortmannin, a PI3K inhibitor, and to rapamycin, a mammalian target of rapamycin (mTOR) inhibitor. It is now believed that the proteolytic turnover of IRS-1 constitutes a negative feedback loop that restrains the magnitude and/or duration of PI3K activation,14 via a mechanism requiring seryl phosphorylation of IRS-1 by mTOR and its effector kinase S6K (whose activities are stimulated by the PI3K/Akt cascade; see Fig. 2) (reviewed in refs. 14–16).
Figure 2.

Role of IRS-1 in insulin and IGF-1 signaling. Upon ligand binding to the receptor, IRS-1 is recruited to the receptor and phosphorylated on tyrosine residues, which serve as docking sites for adaptor proteins of the PI3K/Akt pathway or Ras-Erk MAPK pathway. Akt signaling is restrained by a negative feedback loop via mTOR and its effector serine/threonine kinase S6K. Phosphorylation of multiple serine residues on IRS-1 by mTOR/S6K may create a phosphodegron required for CUL7 E3 ligase-mediated ubiquitination and proteasomal degradation.
The study by Xu et al.9 demonstrated that Fbw8 binds to IRS-1 and promotes its ubiquitination and proteasomal degradation, and that inactivation/deletion of Fbw8 and CUL7, respectively, accumulated IRS-1. Moreover, Fbw8-induced degradation of IRS-1 was dependent upon mTOR activity and may be mediated by multiple mTOR/S6K target serine residues on IRS-1. Thus, the CUL7 E3 appears to be responsible for mediating mTOR-dependent degradation of IRS-1, thereby functioning as a critical component of the mTOR/IRS-1 negative feedback loop (reviewed in ref. 17), which fine-tunes the PI3K activity in accordance with the magnitude and duration of mTOR/S6K activities.
In support of this, embryonic fibroblasts of CUL7−/− mice were found to accumulate IRS-1 and exhibit increased activation of IRS-1 downstream pathways Akt and MEK/ERK.9 However, despite the over-activation of these pro-mitogenic signaling pathways, CUL7−/− mouse embryonic fibroblasts (MEFs) grew poorly with an increased population of cells arrested in G1-phase. Furthermore, the CUL7−/− MEFs exhibited upregulation of distinct tumor suppressors that included p16 and hypo-phosphorylated retinoblastoma protein (pRb), a large flat morphology and high levels of β-galactosidase activity, all of which are characteristic features of cells undergoing senescence.
It should be noted that previous studies have implicated a role for the SOCS-containing E3 ligases in IRS-1 degradation,18 raising the intriguing possibility that multiple E3 Ub ligases may participate in the proteolytic regulation of IRS-1 in response to cellular and environmental cues.
CUL7 may be Associated with Multiple Non-Proteolytic Functions
In comparison with canonical cullin family members (CUL1-5), CUL7 exhibits several atypical features. Composed of 1698 amino acids in humans, CUL7 is of substantially large size. In addition to the highly conserved cullin domain, it contains two distinct motifs: a DOC domain (similar to the DOC1 of the APC/C),19 and a CPH domain (conserved domain in CUL7, PARC and HERC2 proteins)20 (Fig. 1B). Finally, CUL7 appears to be present only in higher eukaryotes (vertebrates), suggesting a late phylogenetic origin.5 Intriguingly, CUL7 was found to mediate interactions with several proteins in a manner that is independent of Fbw8 and that does not affect the stability of the associated proteins (see Table 1), suggesting that the CUL7 E3 ligase may exert both proteolytic (Fbw8-related) and non-proteolytic effects.
NMR studies revealed a direct binding of CUL7 to p53 and showed that the evolutionarily conserved CUL7 CPH domain is the predominant p53 binding site.20 On p53, the interaction surface was mapped to the tetramerization domain. Given that the oligomerization state of p53 affects both its transcriptional activity and subcellular localization, it was proposed that CUL7 might control p53 function by binding preferentially to the active, tetrameric forms of p53.20 At present there is no experimental evidence that the CUL7 E3 ligase participates in the proteolytic degradation of p53: under conditions in which MDM2 promoted the formation of high molecular weight species of p53-Ub in vitro, CUL7 supported only the mono- and di-ubiquitination of p53.21,22 In addition, no accumulation of p53 protein was detected in cells depleted of CUL7 by RNAi,22 or in the CUL7−/− MEFs.23 However, one study reported the upregulation of p53 protein level in SHEP N-Myc cells depleted of CUL7.24 Interestingly, experimental evidence suggests that the CUL7-p53 interaction may contribute to transcriptional regulation: while CUL7 appears to repress p53 in a luciferase reporter assay,22 p53 seems to be required for upregulation of CUL7 at both mRNA and protein levels after DNA damage induced by etoposide.21
It remains an open question whether CUL7 contributes to p53-dependent apoptosis. Initially, Tsai et al.4 identified a putative BH3 domain in the C-terminus of CUL7 (p193), suggesting that CUL7 might belong to the BH3-only family of pro-apoptotic proteins. It was shown that forced expression of CUL7 in NIH-3T3 cells promoted apoptosis in a manner that was dependent on the integrity of the BH3 domain. Moreover, expression of SV40 large T antigen or Bcl-xL, an antagonist of BH3-only proteins, prevented apoptosis.4 However, given the proximity of this putative BH3 motif with the C-terminally located cullin domain, future work is required to determine whether CUL7’s cullin domain plays a role in apoptosis. Notably, expression of a CUL7 C-terminal truncation mutant with presumptive dominant interfering activity (designated CUL7 1152 stop) was found to confer resistance to MG132- and etoposide-induced apoptosis in U2OS cells, independent of CUL7’s interaction with p53 or PARC.25 However, a recent report by Kim et al.,24 identified CUL7 in a functional screen for inhibitors of Myc-induced apoptosis, showing that expression of CUL7 prevented both c-Myc and N-Myc mediated apoptosis and promoted the transformation of neuroblastoma SHEP cells in a p53-dependent manner. Further studies are thus needed to dissect the relative roles of CUL7 as a regulator of apoptosis in various cell types and tissues.
It has been more than a decade since the CUL7 protein was reported to bind to SV40 T antigen.3,4 SV40 is a member of the polyomaviridae family of DNA viruses capable of inducing tumors in rodents. Owing to the ability of T antigen to transform and immortalize mammalian cells in culture, studies using this oncoprotein yield important insights into the mechanisms of transformation. For instance, it is well documented that T antigen interacts and inhibits both p53 and pRb family proteins, thereby disabling critical cellular tumor suppressive mechanisms (reviewed in ref. 26). DeCaprio and colleagues demonstrated that the association between T antigen and CUL7 is a requirement for SV40 transformation.27 This observation is in agreement with an earlier study that demonstrated that co-expression of both dominant interfering CUL7 1152 stop and dominant interfering p53 were required for E1A-mediated transformation of embryonic stem cell-derived cardiac myocytes28 (unlike T antigen, the E1A viral oncoprotein lacks CUL7 and p53 binding activity). Interestingly, transgenic mice expressing CUL7 1152 stop in the myocardium exhibited enhanced cardiomyocyte proliferation following myocardial infarction,29 which was sufficient to block adverse post-infarction ventricular remodeling. These results are consistent with a CUL7-mediated role in cell cycle progression, although the precise mechanistic underpinnings remain unclear.
T antigen deletion analyses mapped the CUL7 interaction region to a N-terminal motif spanning residues 69 to 83. Moreover, the transformation potential of the T antigenΔ69–83 mutant was significantly reduced.27 However, this mutant could transform cells depleted of CUL7, suggesting that T antigen might neutralize a function of CUL7 that protects against cellular transformation.27 A recent study by Zhao et al.30 suggested that the CUL7-T antigen interaction may be required for the degradation of the Mre11-Rad50-Nbs1 complex, which plays a critical role in DNA damage response pathways. It is possible that T antigen may recruit the CUL7 E3 ligase to mediate degradation of cellular proteins for viral propagation. It remains to be investigated whether other non-proteolytic partners of CUL7 (Table 1) influence the CUL7 proteolytic function by regulating the interactions between the E3 and targets, and/or the catalytic efficiency of ubiquitination.
CUL7 is a Novel Regulator of Growth
Two recent studies have independently linked mutations of the cul7 gene to hereditary growth retardation syndromes in humans. Cormier-Daire and colleagues identified 25 cul7 germline mutations in patients with 3-M syndrome, an autosomal-recessive disorder characterized by pre- and postnatal growth retardation, facial dysmorphism and skeletal anomalies (Table 2).31 Of these mutations, 19 predict premature termination of translation, with a majority implicated for loss of the functional cullin domain (see Fig. 1B). More recently, Maksimova et al.32 identified 43 patients from 37 Yakuts families, a geographically isolated ethnic group in Russia, with a short stature syndrome similar to 3-M syndrome (Table 2). A novel mutation in the cul7 gene, 4582insT, was found in all these families, and is predicted to produce a truncated protein terminating at amino acid 1553 (see Fig. 1B).
Table 2.
Human diseases linked to dysregulation of CUL7, or IGF-1 and downstream signaling pathways
| Diseases linked to: | Disease names | Gene(s) mutated | Clinical features | Molecular basis | Refs. |
|---|---|---|---|---|---|
| CUL7 E3 | 3-M Yakuts short stature | CUL7 |
|
|
31, 32 |
| IGF-1/IGF-1R | IGF-1 |
|
Homozygous mutation converting valine 44 to methionine in IGF-1, reducing IGF-1R binding by 90-fold | 38 | |
| IGF-1R | Pre- and postnatal growth retardation | Four families with heterozygote IGF-1R mutations, which decrease the receptor function | 39–41 | ||
| RAS/RAF/MEK | Leopard | SHP2 | Common symptoms: | Common molecular culprits: | 60 |
| Noonan | KRAS | •short stature | aberrant activation of the RAS-RAF-ERK pathway | ||
| Cardo-facio-cutanous (CFC) | BRAF | •facial abnormalities | |||
| neurofibromatosis (NF1) | MEK1/2 | •heart defects | |||
| Costello | HRAS | •skin abnormalities | |||
| neurofibromin | •mental retardation | ||||
| TSC-mTOR | Hamartoma: | PTEN | Common symptoms: | Common molecular culprits: | 71 |
| Cowden | LKB | •benign tumor | caused by mutations in tumor-suppressor | ||
| Peutz-Jeghers | TSC | •tumor tissue with | genes that negatively regulate mTOR | ||
| Tuberous sclerosis | disorganized architecture |
Abbreviations: IGF-1R = IGF-1 receptor.
In line with the human hereditary syndromes, targeted disruption of the cul7 gene in mice resulted in severe intrauterine growth retardation (IUGR) with significantly smaller fetuses at later gestational stages and placenta anomalies (see Table 3). Interestingly, disruption of other cullin family members resulted in early embryonic lethality (<E7.5),33–36 while CUL7 knockout mice develop anomalies in later gestational stages (>E12.5). Deletion of the fbw8 gene in mice yielded a similar phenotype as CUL7−/− (see Table 3).7,37 However, while CUL7−/− mice succumb neonataly due to respiratory distress, disruption of the fbw8 gene resulted in a less severe phenotype with abnormalities mainly restricted to the placenta and growth. Approximately 30% of the homozygous Fbw8−/− offspring reached adulthood, albeit displaying body sizes smaller than their wild-type littermates throughout postnatal development. Clearly, these findings establish overlapping function of CUL7 and Fbw8 in growth control, suggesting that CUL7 requires Fbw8 for executing its major growth-regulatory activity. However, the more severe phenotype of the CUL7−/− mice implicates that CUL7 may possess functions that are independent of Fbw8. Of note, impaired proliferation kinetics were observed with the CUL7−/− and Fbw8−/− MEFs, suggesting that both histopathological and cell autonomous effects contribute to the pathogenesis of the CUL7-associated growth retardation syndromes.
Table 3.
Mouse models of CUL7 and Fbw8, as well as genes of function in the IGF-1/IRS1 pathways
| Gene | Model | Phenotype/observations | Ref. | |
|---|---|---|---|---|
| CUL7 | KO |
|
|
6 |
| Placenta: | ||||
|
•smaller materal vessel area in the labyrinth layer | |||
| Mouse embryonic fibroblasts: reduced proliferation rate in culture | ||||
| FBW8 | KO | • fetal growth retardation in later gestational stages (>E12.5)• 70% of Fbw8−/− offspring died at birth of unknown cause, 30% survived but remain smaller throughout adulthood | •no hemorrhages | 7, 37 |
| Placenta: | ||||
|
•smaller maternal vessel area in the labyrinth layer | |||
| Mouse embryonic fibroblasts: reduced proliferation rate in cell culture | ||||
| IGF-1R | KO |
|
45 | |
| IGF-1 | KO |
|
43–45 | |
| IRS-1 | KO | •growth retardation of the fetus in later gestational stages (>E15.5), remain 50 60% smaller throughout adulthood. | 46–47 | |
| •no organ abnormalities, fertile | •insulin resistant | |||
| IGFBP-1 | TG |
|
•fasting hyperglycemia | 63 |
| IGFBP-2 | TG |
|
•fasting hypoglycemia | 64 |
Abbreviations: KO = knockout; TG = transgenic mouse model; IGF-1R = IGF-1 receptor.
despite mice deleted of IGF-1 or IGF-1R revealed no significant changes in placental development, emerging evidence points to a critical role of the IGF system in this process throughout gestation (reviewed in ref. 77). Placenta weight was positively correlated with cord blood level of IGF-1 and -2,78,79 and human placenta explant studies demonstrated a key role of IGF-1 and -2 in promoting cytotrophoblast proliferation and differentiation to synzytiotrophblast cells.80
cul7 is located on human chromosome 6p21.1. In humans, CUL7 mRNA is expressed in various fetal and adult tissues, with highest transcript levels found in fetal kidney and placenta, as well as adult skeletal muscle, heart and pancreas.31 High levels of CUL7 mRNA were found in mouse testes.4 It was revealed that transcript levels of Fbw8 were most abundant in mouse placenta and skeletal muscle, especially of the abdominal walls, diaphragm and intercostal space.7 It remains to be investigated whether and how the expression profile of CUL7 and Fbw8 is correlated with the activity of this E3 ligase.
Possible Pathomechanisms for CUL7-Linked Growth Retardation Syndromes
How might the loss of CUL7 function contribute to growth retardation? Given that genetic disruption of either CUL7 or Fbw8 in mice profoundly impaired placental and embryonic development, as well as proliferation kinetics in embryonic fibroblasts, and given that short stature is the predominant clinical feature of patients with 3-M and Yakuts syndromes, it is tempting to speculate that diminished proteolytic function of the CUL7 E3 ligase is the principal pathogenic mechanism. In support of this notion, biochemical characterization of a subset of 3-M derived CUL7 mutations in vitro provided direct evidence for a reduced ubiquitination activity.31 The finding that IRS-1 is a proteolytic target of the CUL7 E3 is particularly intriguing, as altered activity of the IGF-1 pathway in patients with loss-of-function mutations in the Igf-138 or Igf-1 receptor39–41 gene has also been linked to severe growth retardation defects (Table 2, reviewed in ref. 42). Mouse knockout studies are in agreement with the prominent role for IGF-1 signaling in growth (see Table 3). IGF-1 and IGF-1 receptor (IGF-1R) knockout mice exhibited birth weights only 60% and 45% of the wild type animals, respectively.43–45 Similar to the CUL7−/− mice, IGF-1R knockout mice died soon after birth of respiratory failure.45 Moreover, ablation of the IRS-1 gene resulted in small, insulin-resistant mice.46,47 In addition, Cho et al.,48 showed that mice deficient in Akt1, an isoform of Akt downstream of IGF-1/PI3K (Fig. 2), exhibited both pre- and postnatal growth impairment, with significantly reduced body size.
One possible pathogenic mechanism for the CUL7-linked growth retardation comes from studies with the CUL7−/− MEFs. It was observed that while the CUL7−/− MEFs exhibited high levels of IRS-1 and concomitantly, enhanced PI3K/Akt and RAS-MAPK pathways, these cells grew poorly and displayed typical features of oncogene-induced senescence9 (OIS). It was, therefore, proposed that accumulation of IRS-1 due to a dysfunctional CUL7 E3 ligase, triggers OIS, which in turn might contribute to the growth retardation phenotype observed in patients with 3-M/Yakuts syndromes.
OIS is a tumor suppressive program that is initiated upon sustained oncogenic signaling to prevent malignant transformation (reviewed in refs. 49–51). Melanocytic nevi (moles) are a well-characterized example of OIS in cancer biology. Nevi are common benign skin tumors, 80% of which harbor the identical B-Raf (V600E) mutation, which is present in the majority of malignant melanomas. It was revealed that nevi displayed OIS phenotypes, thereby suggesting a role for OIS in preventing melanocytic nevi to progress into a malignant state.52–55 Of particular interest, several components in IRS-1 downstream signaling pathways proved capable of inducing OIS: gain-of-function mutation of Ras (V12),56 B-Raf (V600E)55 and MEK,57 as well as constitutive activation of the PI3K/Akt pathway through depletion of the negative regulator Phosphatase and Tensin homolog (PTEN),58 or transgenic overexpression of Akt,59 respectively. Interestingly, several hereditary short stature syndromes such as Noonan-, Costello- or LEOPARD syndrome are linked to gain-of-function mutations of the Ras-Erk MAPK pathway (Table 2; reviewed in ref. 60). Of note, previous studies have linked premature senescence to Werner syndrome, which is associated with short stature as a main clinical feature (reviewed in ref. 61). Future investigations are required to determine whether high levels of IRS-1 are sufficient to initiate OIS, and whether senescence is a contributing factor to the pathogenesis of growth retardation observed in 3-M/Yakats dwarfism syndromes.
A second hypothetical pathomechanism is based on the increased expression of IGF-1 binding proteins (IGFBP) found in the CUL7−/− and Fbw8−/− MEFs.7 It is well established that the vast majority of IGF-1 molecules in the extracellular compartment are complexed with IGFBP, and that IGFBP-bound IGF-1 exhibits altered activity (reviewed in ref. 62). Several lines of evidence link IGFBPs to the pathogenesis of IUGR: transgenic overexpression of IGFBP-1 and -2 were sufficient to cause fetal growth restriction in transgenic mice (see Table 3).63–65 Moreover, newborns with IUGR were found to have high IGFBP-1 levels that negatively correlated with IGF-1 availability and fetal growth.66 Pathways downstream of IRS-1 such as PI3K/Akt and Erk were reported to increase the expression and secretion of IGFBPs, thereby possibly constituting a negative feedback loop on IGF-1 signaling.67–69 Conceivably, aberrant accumulation of IRS-1 in the CUL7−/− MEFs might trigger the upregulation of IGFBPs, leading to the specific inhibition of IGF-1 receptor signaling. However, IUGR is a complex clinical condition that can result from multiple maternal, fetal and placental dysfunctions (reviewed in ref. 70). It remains to be determined whether the CUL7 E3 ligase (either directly or indirectly) targets additional factors in the IGF-1 signaling network, and/or in other growth-regulatory pathways, thereby further contributing to the pathogenesis of IUGR.
Is CUL7 a Tumor Suppressor or an Oncogene?
Paradoxically, recent studies have associated CUL7 with these two apparently opposing activities. By using a SV40 T antigen model, DeCaprio and co-workers have identified a potential tumor suppressive role for CUL7 in viral transformation.27 In addition, the mTOR/IRS-1 negative feedback loop was linked to the inhibition of malignancy in cells where mTOR is hyper-activated such as hamartoma syndromes. Intriguingly these syndromes are typically benign, rather than malignant (reviewed in ref. 71; see Table 2), and it was proposed that the mTOR/IRS-1 negative feedback loop plays a critical role in restraining PI3K activity, thereby halting the progression to malignancy.72–74 Such a tumor suppression activity might be mediated, at least in part, by the CUL7-mediated targeted degradation of IRS-1. On the contrary, by employing a Myc-induced apoptosis system, Penn and colleagues revealed a growth-promoting function of CUL7, and presented in silico evidence for upregulated CUL7 mRNA level in non-small cell lung carcinoma.24 While further investigations are required to resolve the “tumor suppressor or oncogene” conundrum, it would not be surprising if CUL7 proved to play both growth promoting and suppressive roles in a context-dependent manner.
Concluding Remarks
As discussed above, the CUL7 E3 ligase is implicated in multiple biological functions, including pre- and postnatal growth, cellular senescence, cell cycle regulation, apoptosis and transformation by SV40 T antigen (see Fig. 3). Presumably, execution of these biological functions requires both proteolytic and non-proteolytic activities of the CUL7 E3 ligase.
Figure 3.
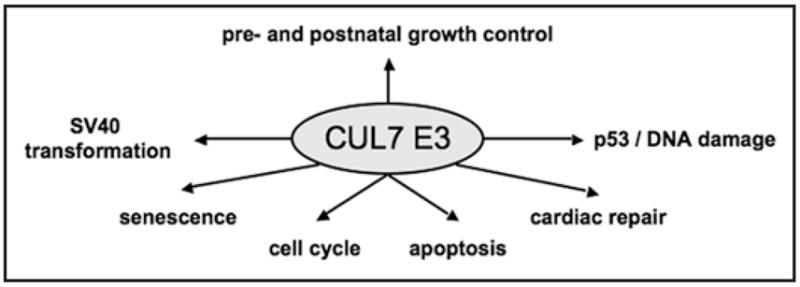
Biological functions of the CUL7 E3 Ub ligase. As described in the text, the CUL7 E3 is associated with multiple biological functions. Studies on 3-M/Yakuts short stature syndromes31–32 as well as with mouse knockouts of CUL7/Fbw8,6,7,37 revealed a prominent role for this E3 in growth control. Moreover, CUL7−/− mouse fibroblasts exhibit senescence phenotype.9 Using the SV40 T antigen model system, it was observed that the CUL7 interaction with T antigen is required for vial transformation.27 Given its ability to target cyclin D1 for degradation,8 the CUL7 E3 may have a role in cell cycle control. In addition, CUL7 was shown to bind to p53.20–25 The CUL7 E3 appears to be able to regulate apoptosis in both p53-dependent24 or independent4 manner. Recent studies have also implicated a role for DNA damage in regulating the p53-CUL7 interactions.21 Finally, CUL7-mediated cell cycle effects have been implicated in cardiac repair.28,29
How does CUL7 function as a unique molecular scaffold by interacting with Skp1-Fbw8 selectively, and by mediating interactions with multiple proteins of both cellular and viral origin (Table 1)? Is it possible that CUL7 “evolves” from CUL1 to assemble an E3 ligase, specifically contributing to complex growth regulatory pathways, such as IGF-1 signaling, demanded by higher organisms? Insights into these questions require structural resolution of the CUL7 E3 ligase and comparison with the previously resolved structures of canonical cullin-based E3s. Structure-based studies will also be critical to reveal the molecular details that govern the interactions between the CUL7 E3 and targets, or its modulator(s), thereby producing information crucial for development of therapeutic and pharmaceutical agents, amenable for both function studies and treatment of human diseases.
Finally, it remains to be explored whether CUL7 has a role in insulin signaling, which potentially impacts insulin resistance and therefore, diabetes. Clearly, given its critical role in growth and association with multiple cellular growth-regulatory pathways, the CUL7 E3 ligase has emerged as an exciting new branch of Ub research.
Acknowledgments
A.S. was supported by a research fellowship from the German Research Foundation. Studies carried out in the Pan and Field laboratories were supported by Public Health Service grants CA095634 and GM61051 (to Z-Q.P), and HL085098 (to L.J.F.), respectively. We thank Drs. Ze’ev Ronai, Serge Fuchs, Valerie Cormier-Daire, and Susanne Mühlich, as well as Jordan Kovacev and Hua Yan for critical reading of the manuscript. We extend our appreciation to Dr. Jerard Hurwitz for helpful discussions. We apologize to colleagues whose work could not be cited due to space limitations.
Abbreviations
- CUL7
cullin7
- Ub
ubiquitin
- SCF
Skp1·CUL1· F-box protein
- IRS-1
insulin receptor substrate 1, growth factor 1
- IGF-1R
IGF-1 receptor
- UPS
Ub-proteasome system
- IUGR
intrauterine growth retardation
- OIS
oncogene induced senescence
- SV40
simian virus 40
- mTOR
mammalian target of rapamycin
- MEF
mouse embryonic fibroblast
- IGFBP
IGF-1 binding protein
References
- 1.Hershko A, Ciechanover A. The ubiquitin system. Annu Rev Biochem. 1998;67:425–79. doi: 10.1146/annurev.biochem.67.1.425. [DOI] [PubMed] [Google Scholar]
- 2.Petroski MD, Deshaies RJ. Function and regulation of cullin-RING ubiquitin ligases. Nat Rev Mol Cell Biol. 2005;6:9–20. doi: 10.1038/nrm1547. [DOI] [PubMed] [Google Scholar]
- 3.Kohrman DC, Imperiale MJ. Simian virus 40 large T antigen stably complexes with a 185-kilodalton host protein. J Virol. 1992;66:1752–60. doi: 10.1128/jvi.66.3.1752-1760.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tsai SC, Pasumarthi KB, Pajak L, Franklin M, Patton B, Wang H, Henzel WJ, Stults JT, Field LJ. Simian virus 40 large T antigen binds a novel Bcl-2 homology domain 3-containing proapoptosis protein in the cytoplasm. J Biol Chem. 2000;275:3239–46. doi: 10.1074/jbc.275.5.3239. [DOI] [PubMed] [Google Scholar]
- 5.Dias DC, Dolios G, Wang R, Pan ZQ. CUL7: A DOC domain-containing cullin selectively binds Skp1. Fbx29 to form an SCF-like complex. Proc Natl Acad Sci USA. 2002;99:16601–6. doi: 10.1073/pnas.252646399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Arai T, Kasper JS, Skaar JR, Ali SH, Takahashi C, DeCaprio JA. Targeted disruption of p185/Cul7 gene results in abnormal vascular morphogenesis. Proc Natl Acad Sci USA. 2003;100:9855–60. doi: 10.1073/pnas.1733908100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tsutsumi T, Kuwabara H, Arai T, Xiao Y, Decaprio JA. Disruption of the Fbxw8 gene results in pre- and postnatal growth retardation in mice. Mol Cell Biol. 2008;28:743–51. doi: 10.1128/MCB.01665-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Okabe H, Lee SH, Phuchareon J, Albertson DG, McCormick F, Tetsu O. A critical role for FBXW8 and MAPK in cyclin D1 degradation and cancer cell proliferation. PLoS ONE. 2006;1:128. doi: 10.1371/journal.pone.0000128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xu X, Sarikas A, Dias-Santagata DC, Dolios G, Lafontant PJ, Tsai SC, Zhu W, Nakajima H, Nakajima HO, Field LJ, Wang R, Pan ZQ. The CUL7 E3 ubiquitin ligase targets insulin receptor substrate 1 for ubiquitin-dependent degradation. Mol Cell. 2008;30:403–14. doi: 10.1016/j.molcel.2008.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Alao JP. The regulation of cyclin D1 degradation: roles in cancer development and the potential for therapeutic invention. Mol Cancer. 2007;6:24. doi: 10.1186/1476-4598-6-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lin DI, Barbash O, Kumar KG, Weber JD, Harper JW, Klein-Szanto AJ, Rustgi A, Fuchs SY, Diehl JA. Phosphorylation-dependent ubiquitination of cyclin D1 by the SCF(FBX4-alphaB crystallin) complex. Mol Cell. 2006;24:355–66. doi: 10.1016/j.molcel.2006.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.White MF. IRS proteins and the common path to diabetes. Am J Physiol Endocrinol Metab. 2002;283:413–22. doi: 10.1152/ajpendo.00514.2001. [DOI] [PubMed] [Google Scholar]
- 13.Haruta T, Uno T, Kawahara J, Takano A, Egawa K, Sharma PM, Olefsky JM, Kobayashi M. A rapamycin-sensitive pathway downregulates insulin signaling via phosphorylation and proteasomal degradation of insulin receptor substrate-1. Mol Endocrinol. 2000;14:783–94. doi: 10.1210/mend.14.6.0446. [DOI] [PubMed] [Google Scholar]
- 14.Harrington LS, Findlay GM, Lamb RF. Restraining PI3K: mTOR signalling goes back to the membrane. Trends Biochem Sci. 2005;30:35–42. doi: 10.1016/j.tibs.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 15.Fisher TL, White MF. Signaling pathways: the benefits of good communication. Curr Biol. 2004;14:1005–7. doi: 10.1016/j.cub.2004.11.024. [DOI] [PubMed] [Google Scholar]
- 16.Shah OJ, Hunter T. Tuberous sclerosis and insulin resistance. Unlikely bedfellows reveal a TORrid affair. Cell Cycle. 2005;4:46–51. doi: 10.4161/cc.4.1.1343. [DOI] [PubMed] [Google Scholar]
- 17.Mieulet V, Lamb RF. Shooting the messenger: CULLIN’ insulin signaling with Fbw8. Dev Cell. 2008;14:816–7. doi: 10.1016/j.devcel.2008.05.010. [DOI] [PubMed] [Google Scholar]
- 18.Rui L, Yuan M, Frantz D, Shoelson S, White MF. SOCS-1 and SOCS-3 block insulin signaling by ubiquitin-mediated degradation of IRS1 and IRS2. J Biol Chem. 2002;277:42394–8. doi: 10.1074/jbc.C200444200. [DOI] [PubMed] [Google Scholar]
- 19.Grossberger R, Gieffers C, Zachariae W, Podtelejnikov AV, Schleiffer A, Nasmyth K, Mann M, Peters JM. Characterization of the DOC1/APC10 subunit of the yeast and the human anaphase-promoting complex. J Biol Chem. 1999;274:14500–7. doi: 10.1074/jbc.274.20.14500. [DOI] [PubMed] [Google Scholar]
- 20.Kaustov L, Lukin J, Lemak A, Duan S, Ho M, Doherty R, Penn LZ, Arrowsmith CH. The conserved CPH domains of Cul7 and PARC are protein-protein interaction modules that bind the tetramerization domain of p53. J Biol Chem. 2007;282:11300–7. doi: 10.1074/jbc.M611297200. [DOI] [PubMed] [Google Scholar]
- 21.Jung P, Verdoodt B, Bailey A, Yates JR, 3rd, Menssen A, Hermeking H. Induction of cullin 7 by DNA damage attenuates p53 function. Proc Natl Acad Sci USA. 2007;104:11388–93. doi: 10.1073/pnas.0609467104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Andrews P, He YJ, Xiong Y. Cytoplasmic localized ubiquitin ligase cullin 7 binds to p53 and promotes cell growth by antagonizing p53 function. Oncogene. 2006;25:4534–48. doi: 10.1038/sj.onc.1209490. [DOI] [PubMed] [Google Scholar]
- 23.Skaar JR, Arai T, DeCaprio JA. Dimerization of CUL7 and PARC is not required for all CUL7 functions and mouse development. Mol Cell Biol. 2005;25:5579–89. doi: 10.1128/MCB.25.13.5579-5589.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim SS, Shago M, Kaustov L, Boutros PC, Clendening JW, Sheng Y, Trentin GA, Barsyte-Lovejoy D, Mao DY, Kay R, Jurisica I, Arrowsmith CH, Penn LZ. CUL7 is a novel anti-apoptotic oncogene. Cancer Res. 2007;67:9616–22. doi: 10.1158/0008-5472.CAN-07-0644. [DOI] [PubMed] [Google Scholar]
- 25.Dowell JD, Tsai SC, Dias-Santagata DC, Nakajima H, Wang Z, Zhu W, Field LJ. Expression of a mutant p193/CUL7 molecule confers resistance to MG132- and etoposide-induced apoptosis independent of p53 or Parc binding. Biochim Biophys Acta. 2007;1773:358–66. doi: 10.1016/j.bbamcr.2006.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hahn WC, Dessain SK, Brooks MW, King JE, Elenbaas B, Sabatini DM, DeCaprio JA, Weinberg RA. Enumeration of the simian virus 40 early region elements necessary for human cell transformation. Mol Cell Biol. 2002;22:2111–23. doi: 10.1128/MCB.22.7.2111-2123.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kasper JS, Kuwabara H, Arai T, Ali SH, DeCaprio JA. Simian virus 40 large T antigen’s association with the CUL7 SCF complex contributes to cellular transformation. J Virol. 2005;79:11685–92. doi: 10.1128/JVI.79.18.11685-11692.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pasumarthi KB, Tsai SC, Field LJ. Coexpression of mutant p53 and p193 renders embryonic stem cell-derived cardiomyocytes responsive to the growth-promoting activities of adenoviral E1A. Circ Res. 2001;88:1004–11. doi: 10.1161/hh1001.090878. [DOI] [PubMed] [Google Scholar]
- 29.Nakajima HO, Tsai SC, Field LJ. Expression of mutant p193 and p53 permits cardiomyocyte cell cycle reentry after myocardial infarction in transgenic mice. Circ Res. 2004;94:1606–14. doi: 10.1161/01.RES.0000132279.99249.f4. [DOI] [PubMed] [Google Scholar]
- 30.Zhao X, Madden-Fuentes RJ, Lou BX, Pipas JM, Gerhardt J, Rigell CJ, Fanning E. Ataxia telangiectasia-mutated damage-signaling kinase- and proteasome-dependent destruction of Mre11-Rad50-Nbs1 subunits in Simian virus 40-infected primate cells. J Virol. 2008;82:5316–28. doi: 10.1128/JVI.02677-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Huber C, Dias-Santagata D, Glaser A, O’Sullivan J, Brauner R, Wu K, Xu X, Pearce K, Wang R, Uzielli ML, Dagoneau N, Chemaitilly W, Superti-Furga A, Dos Santos H, Megarbane A, Morin G, Gillessen-Kaesbach G, Hennekam R, Van der Burgt I, Black GC, Clayton PE, Read A, Le Merrer M, Scambler PJ, Munnich A, Pan ZQ, Winter R, Cormier-Daire V. Identification of mutations in CUL7 in 3-M syndrome. Nat Genet. 2005;37:1119–24. doi: 10.1038/ng1628. [DOI] [PubMed] [Google Scholar]
- 32.Maksimova N, Hara K, Miyashia A, Nikolaeva I, Shiga A, Nogovicina A, Sukhomyasova A, Argunov V, Shvedova A, Ikeuchi T, Nishizawa M, Kuwano R, Onodera O. Clinical, molecular and histopathological features of short stature syndrome with novel CUL7 mutation in Yakuts: new population isolate in Asia. J Med Genet. 2007;44:772–8. doi: 10.1136/jmg.2007.051979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li B, Ruiz JC, Chun KT. CUL-4A is critical for early embryonic development. Mol Cell Biol. 2002;22:4997–5005. doi: 10.1128/MCB.22.14.4997-5005.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Singer JD, Gurian-West M, Clurman B, Roberts JM. Cullin-3 targets cyclin E for ubiquitination and controls S phase in mammalian cells. Genes Dev. 1999;13:2375–87. doi: 10.1101/gad.13.18.2375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang Y, Penfold S, Tang X, Hattori N, Riley P, Harper JW, Cross JC, Tyers M. Deletion of the Cul1 gene in mice causes arrest in early embryogenesis and accumulation of cyclin E. Curr Biol. 1999;9:1191–4. doi: 10.1016/S0960-9822(00)80024-X. [DOI] [PubMed] [Google Scholar]
- 36.Dealy MJ, Nguyen KV, Lo J, Gstaiger M, Krek W, Elson D, Arbeit J, Kipreos ET, Johnson RS. Loss of Cul1 results in early embryonic lethality and dysregulation of cyclin E. Nat Genet. 1999;23:245–8. doi: 10.1038/13886. [DOI] [PubMed] [Google Scholar]
- 37.Tsunematsu R, Nishiyama M, Kotoshiba S, Saiga T, Kamura T, Nakayama KI. Fbxw8 is essential for Cul1-Cul7 complex formation and for placental development. Mol Cell Biol. 2006;26:6157–69. doi: 10.1128/MCB.00595-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Walenkamp MJ, Karperien M, Pereira AM, Hilhorst-Hofstee Y, van Doorn J, Chen JW, Mohan S, Denley A, Forbes B, van Duyvenvoorde HA, van Thiel SW, Sluimers CA, Bax JJ, de Laat JA, Breuning MB, Romijn JA, Wit JM. Homozygous and heterozygous expression of a novel insulin-like growth factor-I mutation. J Clin Endocrinol Metab. 2005;90:2855–64. doi: 10.1210/jc.2004-1254. [DOI] [PubMed] [Google Scholar]
- 39.Abuzzahab MJ, Schneider A, Goddard A, Grigorescu F, Lautier C, Keller E, Kiess W, Klammt J, Kratzsch J, Osgood D, Pfaffle R, Raile K, Seidel B, Smith RJ, Chernausek SD. IGF-I receptor mutations resulting in intrauterine and postnatal growth retardation. N Engl J Med. 2003;349:2211–22. doi: 10.1056/NEJMoa010107. [DOI] [PubMed] [Google Scholar]
- 40.Kawashima Y, Kanzaki S, Yang F, Kinoshita T, Hanaki K, Nagaishi J, Ohtsuka Y, Hisatome I, Ninomoya H, Nanba E, Fukushima T, Takahashi S. Mutation at cleavage site of insulin-like growth factor receptor in a short-stature child born with intrauterine growth retardation. J Clin Endocrinol Metab. 2005;90:4679–87. doi: 10.1210/jc.2004-1947. [DOI] [PubMed] [Google Scholar]
- 41.Walenkamp MJ, van der Kamp HJ, Pereira AM, Kant SG, van Duyvenvoorde HA, Kruithof MF, Breuning MH, Romijn JA, Karperien M, Wit JM. A variable degree of intrauterine and postnatal growth retardation in a family with a missense mutation in the insulin-like growth factor I receptor. J Clin Endocrinol Metab. 2006;91:3062–70. doi: 10.1210/jc.2005-1597. [DOI] [PubMed] [Google Scholar]
- 42.Cianfarani S, Geremia C, Puglianiello A, Maiorana A, Germani D. Late effects of disturbed IGF signaling in congenital diseases. Endocr Dev. 2007;11:16–27. doi: 10.1159/000111054. [DOI] [PubMed] [Google Scholar]
- 43.Powell-Braxton L, Hollingshead P, Warburton C, Dowd M, Pitts-Meek S, Dalton D, Gillett N, Stewart TA. IGF-I is required for normal embryonic growth in mice. Genes Dev. 1993;7:2609–17. doi: 10.1101/gad.7.12b.2609. [DOI] [PubMed] [Google Scholar]
- 44.Baker J, Liu JP, Robertson EJ, Efstratiadis A. Role of insulin-like growth factors in embryonic and postnatal growth. Cell. 1993;75:73–82. [PubMed] [Google Scholar]
- 45.Liu JP, Baker J, Perkins AS, Robertson EJ, Efstratiadis A. Mice carrying null mutations of the genes encoding insulin-like growth factor I (Igf-1) and type 1 IGF receptor (Igf1r) Cell. 1993;75:59–72. [PubMed] [Google Scholar]
- 46.Araki E, Lipes MA, Patti ME, Bruning JC, Haag B, 3rd, Johnson RS, Kahn CR. Alternative pathway of insulin signalling in mice with targeted disruption of the IRS-1 gene. Nature. 1994;372:186–90. doi: 10.1038/372186a0. [DOI] [PubMed] [Google Scholar]
- 47.Tamemoto H, Kadowaki T, Tobe K, Yagi T, Sakura H, Hayakawa T, Terauchi Y, Ueki K, Kaburagi Y, Satoh S, et al. Insulin resistance and growth retardation in mice lacking insulin receptor substrate-1. Nature. 1994;372:182–6. doi: 10.1038/372182a0. [DOI] [PubMed] [Google Scholar]
- 48.Cho H, Thorvaldsen JL, Chu Q, Feng F, Birnbaum MJ. Akt1/PKBalpha is required for normal growth but dispensable for maintenance of glucose homeostasis in mice. J Biol Chem. 2001;276:38349–52. doi: 10.1074/jbc.C100462200. [DOI] [PubMed] [Google Scholar]
- 49.Di Micco R, Fumagalli M, d’Adda di Fagagna F. Breaking news: high-speed race ends in arrest—how oncogenes induce senescence. Trends Cell Biol. 2007;17:529–36. doi: 10.1016/j.tcb.2007.07.012. [DOI] [PubMed] [Google Scholar]
- 50.Yaswen P, Campisi J. Oncogene-induced senescence pathways weave an intricate tapestry. Cell. 2007;128:233–4. doi: 10.1016/j.cell.2007.01.005. [DOI] [PubMed] [Google Scholar]
- 51.Mooi WJ, Peeper DS. Oncogene-induced cell senescence--halting on the road to cancer. N Engl J Med. 2006;355:1037–46. doi: 10.1056/NEJMra062285. [DOI] [PubMed] [Google Scholar]
- 52.Chin L, Merlino G, DePinho RA. Malignant melanoma: modern black plague and genetic black box. Genes Dev. 1998;12:3467–81. doi: 10.1101/gad.12.22.3467. [DOI] [PubMed] [Google Scholar]
- 53.Bennett DC. Human melanocyte senescence and melanoma susceptibility genes. Oncogene. 2003;22:3063–9. doi: 10.1038/sj.onc.1206446. [DOI] [PubMed] [Google Scholar]
- 54.Pollock PM, Harper UL, Hansen KS, Yudt LM, Stark M, Robbins CM, Moses TY, Hostetter G, Wagner U, Kakareka J, Salem G, Pohida T, Heenan P, Duray P, Kallioniemi O, Hayward NK, Trent JM, Meltzer PS. High frequency of BRAF mutations in nevi. Nat Genet. 2003;33:19–20. doi: 10.1038/ng1054. [DOI] [PubMed] [Google Scholar]
- 55.Michaloglou C, Vredeveld LC, Soengas MS, Denoyelle C, Kuilman T, van der Horst CM, Majoor DM, Shay JW, Mooi WJ, Peeper DS. BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature. 2005;436:720–4. doi: 10.1038/nature03890. [DOI] [PubMed] [Google Scholar]
- 56.Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 1997;88:593–602. doi: 10.1016/s0092-8674(00)81902-9. [DOI] [PubMed] [Google Scholar]
- 57.Acosta JC, O’Loghlen A, Banito A, Guijarro MV, Augert A, Raguz S, Fumagalli M, Da Costa M, Brown C, Popov N, Takatsu Y, Melamed J, d’Adda di Fagagna F, Bernard D, Hernando E, Gil J. Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell. 2008;133:1006–18. doi: 10.1016/j.cell.2008.03.038. [DOI] [PubMed] [Google Scholar]
- 58.Chen Z, Trotman LC, Shaffer D, Lin HK, Dotan ZA, Niki M, Koutcher JA, Scher HI, Ludwig T, Gerald W, Cordon-Cardo C, Pandolfi PP. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature. 2005;436:725–30. doi: 10.1038/nature03918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Miyauchi H, Minamino T, Tateno K, Kunieda T, Toko H, Komuro I. Akt negatively regulates the in vitro lifespan of human endothelial cells via a p53/p21-dependent pathway. EMBO J. 2004;23:212–20. doi: 10.1038/sj.emboj.7600045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bentires-Alj M, Kontaridis MI, Neel BG. Stops along the RAS pathway in human genetic disease. Nat Med. 2006;12:283–5. doi: 10.1038/nm0306-283. [DOI] [PubMed] [Google Scholar]
- 61.Hanada K, Hickson ID. Molecular genetics of RecQ helicase disorders. Cell Mol Life Sci. 2007;64:2306–22. doi: 10.1007/s00018-007-7121-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Firth SM, Baxter RC. Cellular actions of the insulin-like growth factor binding proteins. Endocr Rev. 2002;23:824–54. doi: 10.1210/er.2001-0033. [DOI] [PubMed] [Google Scholar]
- 63.Rajkumar K, Barron D, Lewitt MS, Murphy LJ. Growth retardation and hyperglycemia in insulin-like growth factor binding protein-1 transgenic mice. Endocrinology. 1995;136:4029–34. doi: 10.1210/endo.136.9.7544274. [DOI] [PubMed] [Google Scholar]
- 64.Hoeflich A, Wu M, Mohan S, Foll J, Wanke R, Froehlich T, Arnold GJ, Lahm H, Kolb HJ, Wolf E. Overexpression of insulin-like growth factor-binding protein-2 in transgenic mice reduces postnatal body weight gain. Endocrinology. 1999;140:5488–96. doi: 10.1210/endo.140.12.7169. [DOI] [PubMed] [Google Scholar]
- 65.Watson CS, Bialek P, Anzo M, Khosravi J, Yee SP, Han VK. Elevated circulating insulin-like growth factor binding protein-1 is sufficient to cause fetal growth restriction. Endocrinology. 2006;147:1175–86. doi: 10.1210/en.2005-0606. [DOI] [PubMed] [Google Scholar]
- 66.Klauwer D, Blum WF, Hanitsch S, Rascher W, Lee PD, Kiess W. IGF-I, IGF-II, free IGF-I and IGFBP-1, -2 and -3 levels in venous cord blood: relationship to birthweight, length and gestational age in healthy newborns. Acta Paediatr. 1997;86:826–33. doi: 10.1111/j.1651-2227.1997.tb08605.x. [DOI] [PubMed] [Google Scholar]
- 67.Frost RA, Nystrom GJ, Lang CH. Stimulation of insulin-like growth factor binding protein-1 synthesis by interleukin-1beta: requirement of the mitogen-activated protein kinase pathway. Endocrinology. 2000;141:3156–64. doi: 10.1210/endo.141.9.7641. [DOI] [PubMed] [Google Scholar]
- 68.Martin JL, Baxter RC. Expression of insulin-like growth factor binding protein-2 by MCF-7 breast cancer cells is regulated through the phosphatidylinositol 3-kinase/AKT/mammalian target of rapamycin pathway. Endocrinology. 2007;148:2532–41. doi: 10.1210/en.2006-1335. [DOI] [PubMed] [Google Scholar]
- 69.Mehrian-Shai R, Chen CD, Shi T, Horvath S, Nelson SF, Reichardt JK, Sawyers CL. Insulin growth factor-binding protein 2 is a candidate biomarker for PTEN status and PI3K/Akt pathway activation in glioblastoma and prostate cancer. Proc Natl Acad Sci USA. 2007;104:5563–8. doi: 10.1073/pnas.0609139104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cetin I, Foidart JM, Miozzo M, Raun T, Jansson T, Tsatsaris V, Reik W, Cross J, Hauguel-de-Mouzon S, Illsley N, Kingdom J, Huppertz B. Fetal growth restriction: a workshop report. Placenta. 2004;25:753–7. doi: 10.1016/j.placenta.2004.02.004. [DOI] [PubMed] [Google Scholar]
- 71.Inoki K, Corradetti MN, Guan KL. Dysregulation of the TSC-mTOR pathway in human disease. Nat Genet. 2005;37:19–24. doi: 10.1038/ng1494. [DOI] [PubMed] [Google Scholar]
- 72.Um SH, Frigerio F, Watanabe M, Picard F, Joaquin M, Sticker M, Fumagalli S, Allegrini PR, Kozma SC, Auwerx J, Thomas G. Absence of S6K1 protects against age- and diet-induced obesity while enhancing insulin sensitivity. Nature. 2004;431:200–5. doi: 10.1038/nature02866. [DOI] [PubMed] [Google Scholar]
- 73.Shah OJ, Wang Z, Hunter T. Inappropriate activation of the TSC/Rheb/mTOR/S6K cassette induces IRS1/2 depletion, insulin resistance, and cell survival deficiencies. Curr Biol. 2004;14:1650–6. doi: 10.1016/j.cub.2004.08.026. [DOI] [PubMed] [Google Scholar]
- 74.Harrington LS, Findlay GM, Gray A, Tolkacheva T, Wigfield S, Rebholz H, Barnett J, Leslie NR, Cheng S, Shepherd PR, Gout I, Downes CP, Lamb RF. The TSC1-2 tumor suppressor controls insulin-PI3K signaling via regulation of IRS proteins. J Cell Biol. 2004;166:213–23. doi: 10.1083/jcb.200403069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Pan ZQ, Kentsis A, Dias DC, Yamoah K, Wu K. Nedd8 on cullin: building an expressway to protein destruction. Oncogene. 2004;23:1985–97. doi: 10.1038/sj.onc.1207414. [DOI] [PubMed] [Google Scholar]
- 76.Skaar JR, Florens L, Tsutsumi T, Arai T, Tron A, Swanson SK, Washburn MP, DeCaprio JA. PARC and CUL7 form atypical cullin RING ligase complexes. Cancer Res. 2007;67:2006–14. doi: 10.1158/0008-5472.CAN-06-3241. [DOI] [PubMed] [Google Scholar]
- 77.Forbes K, Westwood M. The IGF axis and placental function a mini review. Horm Res. 2008;69:129–37. doi: 10.1159/000112585. [DOI] [PubMed] [Google Scholar]
- 78.Giudice LC, de Zegher F, Gargosky SE, Dsupin BA, de las Fuentes L, Crystal RA, Hintz RL, Rosenfeld RG. Insulin-like growth factors and their binding proteins in the term and preterm human fetus and neonate with normal and extremes of intrauterine growth. J Clin Endocrinol Metab. 1995;80:1548–55. doi: 10.1210/jcem.80.5.7538146. [DOI] [PubMed] [Google Scholar]
- 79.Ong K, Kratzsch J, Kiess W, Costello M, Scott C, Dunger D. Size at birth and cord blood levels of insulin, insulin-like growth factor I (IGF-I), IGF-II, IGF-binding protein-1 (IGFBP-1), IGFBP-3, and the soluble IGF-II/mannose-6-phosphate receptor in term human infants. The ALSPAC Study Team. Avon Longitudinal Study of Pregnancy and Childhood. J Clin Endocrinol Metab. 2000;85:4266–9. doi: 10.1210/jcem.85.11.6998. [DOI] [PubMed] [Google Scholar]
- 80.Forbes K, Westwood M, Baker PN, Aplin JD. Insulin-like growth factor I and II regulate the life cycle of trophoblast in the developing human placenta. Am J Physiol Cell Physiol. 2008;294:1313–22. doi: 10.1152/ajpcell.00035.2008. [DOI] [PubMed] [Google Scholar]