

Abstract
Near-infrared light (NIL) promotes a wide range of biological effects including enhancement of energy production, gene expression and prevention of cell death. This is the first report of the in vivo neuroprotective effects of NIL against optic neuropathy induced by mitochondrial complex I inhibition. Subjects were pigmented rats that received single bilateral intravitreal doses of rotenone, a mitochondrial complex I inhibitor, or rotenone plus one of three different doses of NIL. Treatment effects were evaluated at behavioral, structural and neurochemical levels. Rotenone induced a decrease in visual function, as determined by changes in the dark-adapted illuminance sensitivity threshold, escape latency and rate of successful trials in a two-choice visual task, compared with vehicle-treated controls. Behavioral impairment correlated with a decrease in retinal and visual pathway metabolic activity, retinal nerve fiber layer thickness and ganglion cell layer cell density. These changes were prevented by NIL treatments in a dose-dependent manner. Whole-brain cytochrome oxidase and superoxide dismutase activities were also increased in NIL-treated subjects in a dose-dependent manner, suggesting an in vivo transcranial effect of NIL. In whole-brain membrane isolates, NIL prevented the rotenone-induced decrease in cell respiration. The results show that NIL treatment can effectively prevent the neurotoxic effects of rotenone and that it might be used in the treatment of neurodegenerative disorders associated with mitochondrial dysfunction.
Keywords: near-infrared light, cytochrome oxidase, visual function, mitochondrial dysfunction, neuroprotection, rotenone
Introduction
Near-infrared light (NIL) therapy constitutes a novel intervention shown to regulate neuronal function in cell cultures, animal models, and clinical conditions (Eells et al., 2004). NIL treatment exposes a target tissue to a low-power, high-fluency source of directional and monochromatic photon radiation (λ = 630–1000 nm), delivering energy doses that are too low to cause damage, yet high enough to modulate neuronal functions (Sommer et al., 2001). In vitro, photobiomodulation with NIL has shown metabolic-enhancing, antioxidant and anti-apoptotic properties that prevent the neurotoxic effects of mitochondrial inhibitors, such as sodium azide and potassium cyanide (Wong-Riley et al., 2001, 2005; Liang et al., 2006). Such properties support that NIL, or interventions with similar neurobiological effects, may have a role in the treatment of neurodegeneration, a phenomenon that underlies debilitating clinical conditions.
In particular, the detrimental effects of the type of mitochondrial dysfunction implicated in patients with Parkinson's disease (Schapira et al., 1990) and Leber's hereditary optic neuropathy (Brown et al., 2001), has been prevented with NIL treatments in cell cultures treated with rotenone, a mitochondrial complex I inhibitor. Mitochondrial optic neuropathies involve inherited and acquired impairments of mitochondrial function, especially affecting complex I in the respiratory chain (Sadun, 2002). Rotenone is not only a specific complex I inhibitor but also a commonly used pesticide regarded as an environmental mitochondrial toxin (Zhang et al., 2006a). Recently, Liang et al. (2008) demonstrated that NIL treatment prevents not only the decreases in rotenone-induced adenosine triphosphate cellular content and the increases in reactive oxygen species and nitric oxide but also the apoptosis in rat occipital cortical and striatal neuronal cultures (Liang et al., 2008). Eells et al. (2003) described that NIL prevents the morphological and electrophysiological photoreceptor abnormalities induced by reversible inhibition of cytochrome oxidase, but no studies have addressed whether NIL treatment provides neuroprotection against the degenerative effects of other mitochondrial toxins in vivo.
The present report contributes a description of the efficacy of NIL treatment at preventing the toxic effects of rotenone in the retina of living subjects. A rodent model of rotenone-induced retinal degeneration described previously as a screening tool for neuroprotective interventions (Zhang et al., 2002, 2006a,b; Rojas et al., 2008) was used to determine the in vivo neuroprotective effects of three different protocols of NIL treatment. This animal model allows the efficient assessment of neuronal damage at combinatorial levels of organization: visually guided behavior and metabolic activity in the visual pathway are reported as measures of functional effects, whereas a retinal histopathological analysis provides evidence of structural defects. A baseline analysis of visual function was followed by bilateral intravitreal injections of rotenone and delivery of NIL treatments. Postinjection, a second evaluation of visual function was accomplished and eyeballs and brains were analyzed for histopathologic and neurochemical changes. The results showed that NIL treatment prevents retinal neurodegeneration induced by rotenone, in a dose-dependent manner at functional and structural levels. Furthermore, whole-brain-based neurochemical analyses demonstrated that NIL treatment increases both cytochrome oxidase and superoxide dismutase activities, implicating a transcranial effect of NIL treatments on the nervous system.
Materials and Methods
Subjects.
Male Long–Evans rats (n = 42, 40-d-old) were obtained from Harlan. Subjects were handled during 5 d after arrival to allow for habituation. They were maintained in clear plastic cages with food and water ad libitum and subjected to standard light cycles (12 h light/12 h dark). All experimental procedures were approved by the Institutional Animal Care and Use Committee of the University of Texas at Austin. Unless otherwise specified, all chemical reagents were purchased from Sigma-Aldrich.
Behavioral testing.
Visual function was assessed using a descending method of limits in a two-choice visual task apparatus designed to determine changes in the dark-adapted illuminance sensitivity threshold (Hayes and Balkema, 1993; Prusky et al., 2000). Testing in this apparatus consisted of task acquisition, preinjection and postinjection phases (Table 1).
Table 1.
General experimental timeline
| Phase | Period |
|---|---|
| 1. Visual task acquisition | Days 1–9 |
| 2. Prerotenone threshold determination | Days 10–18 |
| 3. Prerotenone NIL treatment | Days 19 and 20 |
| 4. Intravitreal rotenone injection | Day 21 |
| 5. Postrotenone NIL treatment | Days 21–26 |
| 6. Postrotenone threshold determination | Days 29–37 |
| 7. Tissue extraction | Day 38 |
Each phase was divided into working sessions that took place 24 h after the previous one and consisted of five trials in which subjects performed a task. The task consisted of finding an escape platform (12.7 × 12.7 cm) submerged 1–2 cm in a circular tank (radius = 50 cm) containing water at 17−22°C in the shortest possible period of time. Twenty minutes before each session, subjects underwent dark adaptation in the testing room. All sessions were performed in the mornings in a dark room without intramaze visual cues except for the escape platform which received 78.5 lm/m2 of illumination from a white light source located 1.3 m above it. Dispersion of light to the rest of the tank was avoided by connecting the light source to a 1-m-long opaque tube with a diameter of 6 cm. The same place near the tank wall was designated as a starting point whereas the location of the escape platform varied randomly between trials between two defined positions equidistant from the starting point and located at the other end of the tank. With this randomization of the platform position, animals were forced not to learn previous positions of the escape platform but to rely on visual information to find it, thus minimizing errors of habituation and expectation. The two platform positions were separated by a dark barrier dividing the tank into left and right compartments (Fig. 1 A).
Figure 1.

Two-choice visual discrimination task apparatus and near-infrared light administration. A, In each trial, subjects swam from the starting point to the escape platform located in one of the two compartments separated by a dark barrier. The position of the platform was randomized between trials. Entrance to any compartment was considered when the whole body of the subject crossed the imaginary decision line. In the threshold determination phases the illuminance on the platform was gradually decreased, while latencies and rate of successful trials were determined. B, Near-infrared light treatments were delivered to subjects under general anesthesia, via two light-emitting diode arrays located 3.8 cm above their heads.
The water in the tank was stirred and solid surfaces were cleaned with an iodide solution between trials to eliminate olfactory cues. The acquisition phase consisted of nine sessions in which the platform received the maximal illuminance possible in each trial. The trial was stopped if the subject did not find the platform after 60 s. In this case, the subject was guided to the platform with the aid of a plastic stick. The subject remained on the platform for 10 s and was picked up and dried by a researcher. A 30–60 s interval was allowed between trials. For preinjection and postinjection phases, the platform illuminance was systematically reduced 0.5 log units between daily sessions, starting with the maximal illuminance of 78.5 lm/m2 and ending at 2.1 × 10−3 lm/m2. Consequently, the illuminance threshold was determined by progressively decreasing the illuminance level until a behavioral modification was noted. The measured behavioral variables consisted of escape latencies and percentages of successful trials. A successful trial occurred when the first chosen compartment to swim into was the same as the one in which the escape platform was located in that trial and the subject sat on top of the platform. The illuminance level immediately higher to the one producing a significant increase in the escape latency was considered as the dark-adapted illuminance threshold. All sessions were video-recorded and swimming patterns were analyzed. After the preinjection phase, subjects underwent surgery and the postinjection phase started 6 d after surgeries.
Intravitreal injections and tissue processing.
Anesthesia was induced for 3 min with 3% isoflurane (Baxter) and maintained at 1.5% using an E-Z anesthesia vaporizer system (Euthanex). Under a stereomicroscope, a sclerotomy was done in the superior temporal quadrant of each eye to give access to the vitreous body and a 30 ga dental microinjection needle (Monoject; Sherwood Medical Company) was inserted into the vitreous body in a rostro-caudal direction to avoid damaging the lens. The needle was connected by polyethylene tubing (internal diameter = 0.38 mm) to a 10 μl Hamilton microsyringe. The injections were delivered over 6 min using a microinjection pump (CMA Microdialysis) (3 μl, final volume) and the injection needle was left in place for an additional 20 s period to allow for diffusion away from the needle tip and to avoid any efflux of fluid through the injection site. The edges of the injection point were gripped with forceps while the microsyringe was slowly withdrawn. Subjects were divided into control and experimental groups.
In the control group (n = 7), the vitreous body in both eyes of each subject was injected with the vehicle dimethylsulfoxide (DMSO). Subjects in the experimental group (n = 23) received bilateral intravitreal injections of 200 μg/kg rotenone in DMSO. Sixteen days after the rotenone injection, the subjects were decapitated, and the eyeballs and brains were rapidly removed and frozen in isopentane (−40°C). The eyeballs were transferred to cubic aluminum foil containers (2 mm3) filled with Shandon M1 embedding matrix (Thermo Electron Corporation) and stored at −40°C. Forty-micrometer-thick sagittal eyeball and brain coronal sections were obtained at −18°C using a Leica CM300 cryostat (Leica Microsystems) and were mounted on glass slides to create three adjacent series for histological analysis. Sections were stored at −40°C until further use.
Near-infrared light treatments.
NIL treatments were delivered via two R30–123 narrow-angle light-emitting diode arrays (radius = 4.4 cm) (LEDtronics) located 3.8 cm above the subjects' heads (Fig. 1 B). All NIL treatment sessions were given at peak λ = 633 nm, with power density of 2 mW/cm2, during 30 min for an energy density dose of 3.6 J/cm2. This single-session energy density was based on previous studies showing beneficial NIL effects in in vitro and in vivo models of neurodegeneration induced by mitochondrial dysfunction (Eells et al., 2003; Liang et al., 2008). The average power measured at the corneal surface level (approximate area of 49 mm2) at a distance of 3.8 cm was 274 μW, so the estimated energy delivered to this area was 493 μJ in each 30 min session. NIL doses were calculated based on measurements of average power done with a Newport 1830C power meter (Newport). Subjects were anesthetized with 1.5% isoflurane and kept on a thermal bed at 36.5°C during the totality of each one of the NIL treatment sessions. Three different protocols of NIL treatment were assessed in subjects receiving bilateral rotenone intravitreal injections (i.e., the experimental group) for their effectiveness to prevent rotenone-induced retinotoxicity (Table 2).
Table 2.
Near-infrared doses and schedules
| Protocol | Day |
Dose (J/cm2) |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 19 | 20 | 21 a | 22 | 23 | 24 | 25 | 26 | Per day | Total | |
| NIL 1 | • | • | • | 3.6 | 10.8 | |||||
| NIL 2 | • | • | • | • | • | • | 3.6 | 21.6 | ||
| NIL 3 | • | • | • | • | • | • | 3.6 | 21.6 | ||
aIntravitreal injections of rotenone were done on this day.
Protocol NIL 1 (n = 7) consisted of a total dose of 10.8 J/cm2 fractionated in three sessions of NIL treatment each one occurring at 10 min and 24 and 48 h after rotenone injections. Protocol NIL 2 (n = 5) consisted of six sessions of NIL treatment occurring at 10 min and 24, 48, 72, 96, and 120 h after rotenone injections, for a total dose of 21.6 J/cm2. Protocol NIL 3 (n = 5) also consisted of a total dose of 21.6 J/cm2, similar to protocol NIL 2, but in this case treatment sessions occurred at 48 and 24 h before the rotenone injections and continued at 10 min and 24, 48 and 72 h after the injections. These dose fractionation schedules were implemented based on studies reporting that multiple NIL treatment sessions over days are more beneficial than the administration of a single treatment (Brondon et al., 2005). Six subjects in the experimental group received bilateral rotenone injections but no NIL treatment (i.e., rotenone group). Seven subjects in the control group were also further subdivided into radiated (receiving a protocol similar to NIL 1) (n = 3), and nonradiated groups (n = 4). No differences in behavioral or morphological parameters were observed between these two control subgroups so they were combined for additional between-group comparisons. Regardless of whether they received rotenone or DMSO injections or whether they were treated with NIL or not, all subjects were anesthetized with 1.5% isoflurane during similar periods of time at the same intervals, with the exception of an additional group of rotenone-treated subjects (n = 5) that were not given anesthesia.
NADH dehydrogenase histochemistry.
Retinal sections were stained for NADH dehydrogenase activity following a procedure based on the reduction of tetrazolium salts to formazan that allows the identification of retinal layers, including the retinal nerve fiber layer (RNFL) (Jung et al., 2002; Zhang et al., 2002). Fresh-frozen sections were prefixed in a solution containing 0.5% glutaraldehyde/10% sucrose in phosphate buffer (PB) for 7 min at room temperature, followed by 10% sucrose in PB baths (3×, 5 min each). The sections were then incubated at 37°C for 20 min with gentle agitation in PB containing 1 mg/ml NADH, 1.3 mg/ml nitroblue tetrazolium, and 1 m sodium azide. Sections were rinsed in PB, fixed in 10% paraformaldehyde for 30 min at room temperature, dehydrated in 30–100% ethanol series, cleared in xylene and coverslipped with Permount.
Nissl staining.
Frozen retinal sections were delipidized in a series of 95% ethanol, 70% ethanol, distilled water, and 0.1 m sodium acetate buffer at pH 4 (2.5 min each). Sections were stained in 0.1% cresyl violet in sodium acetate buffer at 45°C for 4 min, differentiated in 70% and 95% ethanol and dehydrated in 95% and 100% ethanol. Slides were cleared in xylene and coverslipped with Permount.
Cytochrome oxidase histochemistry.
Retinal sections, brain sections and sections of brain homogenate paste standards (10, 20, 40, 60, and 80 μm thick) were stained with a cytochrome oxidase histochemistry procedure previously described (Gonzalez-Lima and Cada, 1994, 1998; Gonzalez-Lima and Jones, 1994). Briefly, frozen sections were prefixed in 0.5% glutaraldehyde/10% sucrose in PB, pH 7.6, for 5 min at 4°C, followed by three baths of 10% sucrose in PB (5 min each, allowing for gradual increases in temperature). The sections were incubated for 10 min in a solution containing 50 mm Tris, 1.1 mm cobalt chloride, 10% sucrose and 0.5% DMSO. After a 5 min rinse in PB at room temperature, the sections were stained for 60 min at 37°C in a PB solution containing 1.3 mm 3,3′-diaminobenzidine tetrahydrochloride, 75 mg/L cytochrome c, 20 mg/L catalase, 5% sucrose and 0.25% DMSO. Sections were fixed in 4% formalin in PB at room temperature for 30 min, dehydrated in series of 30–100% ethanol series, cleared in xylene (two times, 5 min each) and coverslipped with Permount.
Retinal layer thickness.
Estimates of retinal nerve fiber layer-plus-ganglion cell layer (RNFL + GCL) and inner plexiform layer (IPL) thickness were obtained by systematic analyses of NADH dehydrogenase activity-stained sections, based on previous reports (Zhang et al., 2002; Rojas et al., 2008). The eyeball sections selected for thickness estimates were those with the largest diameter by 2× microscopic inspection, preferentially those in which the largest diameter of the lens, the pupil and the optic nerve were identified. To maximize the efficiency of the estimate while avoiding the effect of retinal thickness by position in the medial-lateral axis of the eye, data were obtained from systematic samples 100–200 μm adjacent to the optic pole within each eyeball section. Retinal images at 20× were analyzed using a microscopic image-processing system including an Olympus BX40 microscope (Olympus America), a CCD camera (Javelin Electronics), a Targa-M8 digitizer and JAVA imaging software (Jandel Scientific). An isotropic uniform random system of test lines was superimposed on captured images and distances between the intercepts of each line with the inner and outer margins of the layer of interest were calculated. An unbiased estimate of thickness was obtained as T ret = Σlength of intercept/no. of intercepts (Gundersen et al., 1978).
Retinal GCL cell density.
Cell density (no. of cells/volume unit) in the GCL was estimated from Nissl-stained sections using the optical dissector stereological method (Harding et al., 1994). The imaging setting consisted of a Labophot-2 binocular bright-field microscope (Nikon Corporation), connected to a DVC-1312C-FW scan camera and DVCView 3.3 imaging software (DVC Company) and a Microcode II digital readout (Boeckeler Instruments). Data were obtained from the retinal optic pole, following a sampling system similar to that used for thickness measurements. Cells were identified at 50× through the thickness of a section and were counted when they first came into focus (except those on top of the sections) within an unbiased counting frame. One sample was obtained from each of three sections per retina. GCL cell density was estimated as Nvcells=ΣQ−/Σv(frame), where ΣQ− is the sum of cell counts per sample, and Σv(frame) is the area of the unbiased counting frame (0.0061 mm2, adjusted for 140× magnification) multiplied by the length of the analyzed region (d). This length was calculated as d = (no. of analyzed sections − 1) × no. of section series (3) × section thickness (40 μm).
Visual pathway and whole-brain metabolic activity.
Cytochrome oxidase activity was quantified by optical densitometry as detailed by Gonzalez-Lima and Cada (1994). Optical density values for each region of interest were converted to units of cytochrome oxidase activity using calibration curves based on standards of tissue thickness and spectrophotometrically determined cytochrome oxidase activity. Stained sections were placed on a DC-powered light box, digitized and analyzed using a CCD camera (Javelin Electronics), a Targa-M8 digitizer and JAVA imaging software (Jandel Scientific). Digitized images were corrected for slide and light box artifacts by means of background subtraction. For each area of interest, optical density was averaged bilaterally across three retinal or brain sections per subject. Areas of interest included the following: (1) superficial gray matter of the superior colliculus (SupG), (2) dorsal lateral geniculate nucleus (LGN), (3) primary visual cortex (V1) and (4) lateral peristriate cortex (V2). These areas were delineated with reference to AP bregma levels of −4.16 to −7.04 of a standard rat brain atlas (Paxinos and Watson, 1997). For estimation of whole-brain cytochrome oxidase activity, all regions of interest plus images of six entire coronal brain sections/subject within AP bregma levels from −4.16 to −7.04 were captured and analyzed for average optical density intensity. Cytochrome oxidase activity was reported as μmol/min/g of wet tissue.
Superoxide dismutase activity.
Superoxide dismutase (SOD) activity was measured following a procedure based on the ability of SOD to inhibit the spontaneous oxidation of adrenaline to adrenochrome (Misra and Fridovich, 1972). Mitochondrial extraction was done following a modification of the procedure described by Seaton et al. (1996). The forebrains of subjects in the control and experimental groups were diluted 12.5% w/v in ice-cold buffer containing 320 mm sucrose, 1 mm EDTA and 10 mm Tris at pH 7.4 and homogenized in a 7 ml Dounce glass homogenizer (40–50 strokes per brain). The homogenates were further diluted 1:11 with ice-cold buffer and centrifuged at 320 × g for 10 min at 4°C. The supernatant was centrifuged again at 10,000 × g for 30 min. The supernatants and pellets were used to measure cytosolic and mitochondrial SOD activities, respectively. SOD activity controls were determined in a sample mixture containing 0.1 mm EDTA in 50 mm sodium carbonate buffer, pH 10.2. The oxidation of adrenaline bitartrate (0.3 mm) was followed spectrophotometrically at 480 nm and 30°C for 3 min. In separate measurements, aliquots (10 μl) of cytosolic or mitochondrial preparations were added to the sample mixture to produce between 40 and 60% inhibition. Results were expressed as peak units of enzyme activity per gram of wet tissue (1 unit = 50% inhibition of adrenochrome formation, relative to control). All samples were assayed in quadruplicate.
In vitro whole-brain cell respiration.
Cell respiration in brain tissue was measured as a change in partial pressure of oxygen, using dynamic fluorescence quenching as described previously (Zhang et al., 2006a). Aliquots of a pool of whole-brain homogenates from naive Long–Evans rats (n = 7, male, 300–350 g) were solubilized in ice-cold buffer (0.32 m sucrose, 1 mm EDTA, 8.4 mm Trizma HCl, 1.6 mm Trizma base, pH 7), to get 25% w/v tissue solutions. The tissue was diluted 1:80 in sample medium (0.5% deoxycholate in ice-cold buffer, 4 ml of total volume) and vortexed briefly several times during a 10 min incubation period at room temperature, in which cell membrane solubilization took place. Samples with or without 10 μm rotenone were incubated for 10 min on a heating plate at a constant temperature of 37°C, in the dark with 0.1 or 1 J/cm2 NIL. The light-emitting diode arrays were located 3.8 cm from the samples. The change in temperature during this 10 min incubation period with or without NIL radiation was negligible (<0.01°C/min). Cell respiration measurements were done in 15 ml biochemical oxygen demand bottles (Kimble), by means of a fiber optic fluorometric oxygen sensor system (Ocean Optics). The fluorometric probe was immersed into the sample through the capillary pipette dropping stopper, which was sealed with Parafilm. Measurements took place during 4 min, at 37°C and a sample integration time of 120 ms. Cell respiration recordings were always done immediately after the 10 min incubation period, in the dark, and at least in quadruplicate. Results are expressed as percent changes in oxygen concentration compared with baseline, per unit of time.
Statistical analysis.
Repeated measures ANOVA followed by simple effects, simple contrasts, and Dunnett's post hoc correction were used to determine within-group and between-group differences in mean illuminance sensitivity thresholds, escape latencies and successful trials. Individual illuminance thresholds were determined through a series of two-tailed t tests for within-subject escape latencies with a criterion of p < 0.01. Layer thickness, cell density, metabolic activity, SOD activity and cell respiration differences were determined using Dunnett-corrected one-way ANOVA. Within-group, brain-behavior and structure-behavior relationships were determined using group-corrected partial Pearson product–moment correlations. All statistical analyses were conducted using SPSS 11.5 for Windows. A p value of <0.05 was considered significant.
Results
Behavioral effects
After rotenone injection, subjects exposed to NIL treatment preserved their prelesion illuminance sensitivity threshold, a measure based on individual minute changes of visual function, whereas subjects in the rotenone group showed a 72.7% decrease in their illuminance sensitivity threshold, compared with control (p < 0.01) (Fig. 2 A). Further analyses of the psychometric curves for escape latency and successful trials revealed between-group behavioral differences of NIL treatment in a dose-related manner. Subjects treated with the high-dose NIL protocols (NIL 2 and NIL 3) displayed overall escape latencies across different illuminance levels that were not different from control and prelesion conditions (p = 0.76 and p = 0.27, respectively). In contrast, subjects in both the rotenone and the 10.8 J/cm2 NIL (protocol NIL 1) groups showed mean escape latencies that were significantly increased compared with control and prelesion conditions (p < 0.001 and p < 0.05, respectively) (Fig. 2 B). Subjects treated with the high-dose NIL protocols, but not with the low-dose one, also had an overall postrotenone rate of successful trials (73.3%) that was not different from control (92%, p = 0.22), whereas the rotenone alone group performed at chance levels (49.1%), which was different not only from control, but also from prelesion rates (p < 0.01) (Fig. 2 C).
Figure 2.
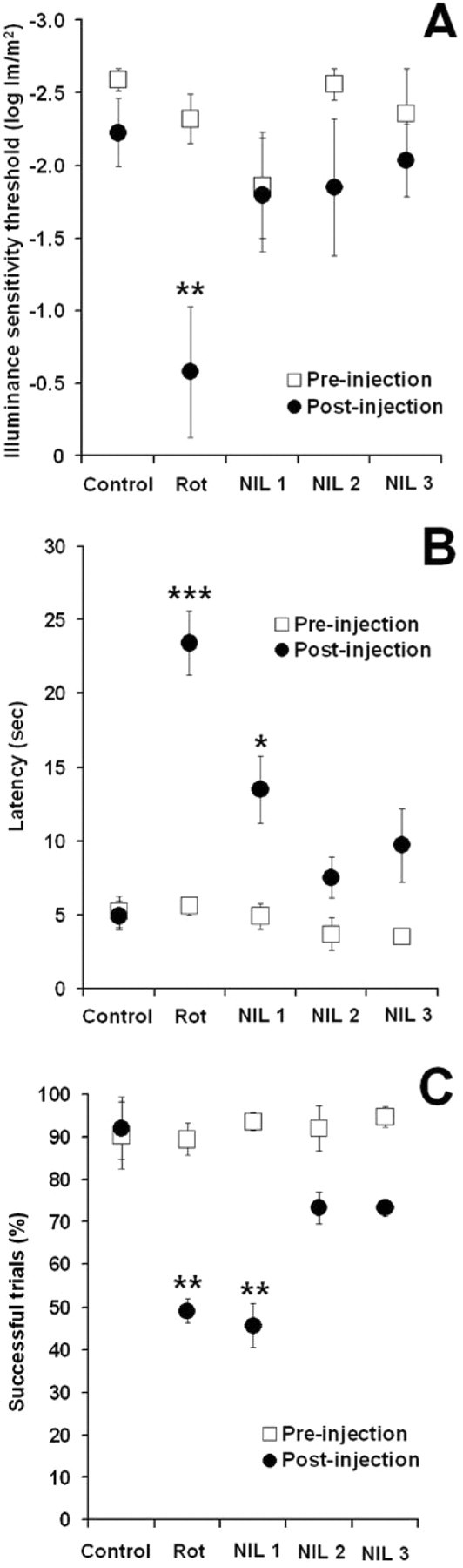
Neuroprotective effects of near-infrared light on visually guided behavior. A, Rotenone (Rot) induced a significant decrease in the illuminance sensitivity threshold but no significant changes postinjection were observed in subjects receiving rotenone plus near-infrared light (effect size = 0.33, power = 0.73). B, Subjects in the Rot group displayed mean postinjection escape latency across illuminance levels that were significantly higher than control and baseline. High NIL doses prevented this change, whereas a low NIL dose was not as effective (effect size = 0.62, power = 0.99). C, Rotenone-treated subjects displayed mean postinjection escape performances across illuminance levels that were not different from chance. This performance was significantly worse than control and baseline. High NIL doses prevented this change, whereas a low NIL dose was not as effective (effect size = 0.41, power = 0.88). NIL 1 = rotenone + 10.8 J/cm2 NIL postinjection, NIL 2 = rotenone + 21.6 J/cm2 postinjection, NIL 3 = rotenone + 10.8 J/cm2 preinjection + 10.8 J/cm2 postinjection. Asterisks indicate a significant difference compared with control. *p < 0.05, **p < 0.01, ***p < 0.001.
Structural effects
It was hypothesized that the behavioral effects described above were due to the direct neuroprotective effect of NIL treatments on the retina. Since the intravitreal injection of rotenone has previously been shown to decrease layer thickness and cell density, we assessed the effects of NIL treatments on these two histological variables. Microscopic examination visibly showed a neuroprotective effect of NIL on the retinal layers associated with ganglion cells, including the layer with the axons of ganglion cells (retinal nerve fiber layer), the layer with the somata of ganglion cells (ganglion cell layer) and the layer with the dendrites of ganglion cells (inner plexiform layer). Interestingly, the structural effects of NIL treatments also followed a dose–response pattern similar to that observed in the behavioral experiments, with NIL 2 and 3 protecting against the histopathological effects of rotenone (Fig. 3).
Figure 3.

Neuroprotective effects of near-infrared light on retinal structure. Images of mid-sagittal eyeball sections from the optic pole show profound between-group differences in retinal anatomy. Retinal layers were clearly identified in control subjects. A dark purple staining was found in zones with heavy formazan deposits, which represent retinal regions with high NADH dehydrogenase activity. Layer thinning was a histopathological feature of rotenone-induced neurotoxicity. This was more drastic in the innermost layers including the RNFL, GCL, IPL and inner nuclear layer (INL). Whereas structural defects of similar severity where observed in the group treated with the NIL 1 protocol, protocols NIL 2 and NIL 3 where effective at preventing the retinotoxic effects of rotenone. NIL 1 = rotenone + 10.8 J/cm2 NIL postinjection, NIL 2 = rotenone + 21.6 J/cm2 postinjection, NIL 3 = rotenone + 10.8 J/cm2 preinjection + 10.8 J/cm2 postinjection. OPL = outer plexiform layer, ONL = outer nuclear layer, PR = photoreceptor layer. Light microscopy, 10×.
Morphometric analysis (Fig. 4) showed that rotenone produced a 44.4% decrease in the mean RNFL + GCL thickness (p < 0.01), compared with control. However, 21.6 J/cm2 NIL after rotenone injection (protocol NIL 2) limited the RNFL + GCL thickness reduction to only 12.4%, which was not significantly different from control (p = 0.69). A similar protective effect was observed with protocol NIL 3 (10.8 J/cm2 NIL preinjection plus 10.8 J/cm2 NIL postinjection), which produced a RNFL + GCL thickness reduction of 24.5% (p = 0.15). Retinas exposed to 10.8 J/cm2 NIL postinjection (protocol NIL 1) showed a RNFL+GCL thickness reduction of 37.7% (p < 0.01), indicating that low-dose NIL was not as effective as the high-dose NIL protocols at preventing this neurodegenerative effect of rotenone.
Figure 4.

Effects of near-infrared light on retinal layer thickness. Rotenone induced a significant decrease in the RNFL + GCL and IPL thickness. NIL treatments prevented degeneration in the RNFL+GCL in a dose–response manner. Rot, Rotenone; NIL 1 = rotenone + 10.8 J/cm2 NIL postinjection; NIL 2 = rotenone + 21.6 J/cm2 postinjection; NIL 3 = rotenone + 10.8 J/cm2 preinjection + 10.8 J/cm2 postinjection. Significant difference compared with control at *p < 0.05; significant difference compared with control at **p < 0.01.
A dose–response effect was also observed in the IPL thickness. Rotenone produced a 28.8% decrease in the mean IPL thickness (p < 0.05) and subjects treated with protocols NIL 2 and NIL 3 showed thickness reductions of only 25.1% and 23.7% compared with control (p = 0.11 and p = 0.14, respectively). In contrast, protocol NIL-1-treated subjects showed a 50.5% reduction in thickness compared with control (p < 0.001), which revealed a detrimental structural effect that parallels an absence of beneficial effect at the functional level, with this same dose. Changes in IPL thickness were minimal in NIL-2- and NIL-3-treated groups, but they were proportional with changes in RNFL + GCL thickness as demonstrated by a high correlation between these two measures (r = 0.74, p < 0.001). Similarly, lower IPL thickness values correlated with longer escape latencies in the behavioral task (r = −0.56, p < 0.01).
Rotenone produced a 39.8% reduction in the GCL cell density (p < 0.001), compared with control (17.5 ± 1 vs 8.8 ± 1 cells × 103/mm3). GCL cell densities were similarly different from control in all rotenone plus NIL-treated groups, but GCL cell densities were positively correlated with RNFL + GCL thickness (r = 0.62, p < 0.001). This result not only shows a consistency of treatment effects in two different morphologic variables, but implicates a neuroprotective effect of NIL that spares neural tissue from rotenone-induced neurotoxicity. Furthermore, shorter escape latencies in the behavioral task negatively correlated with both RNFL+GCL thickness (r = −0.71, p < 0.001) and GCL cell density (r = −0.57, p < 0.01) (Fig. 5).
Figure 5.
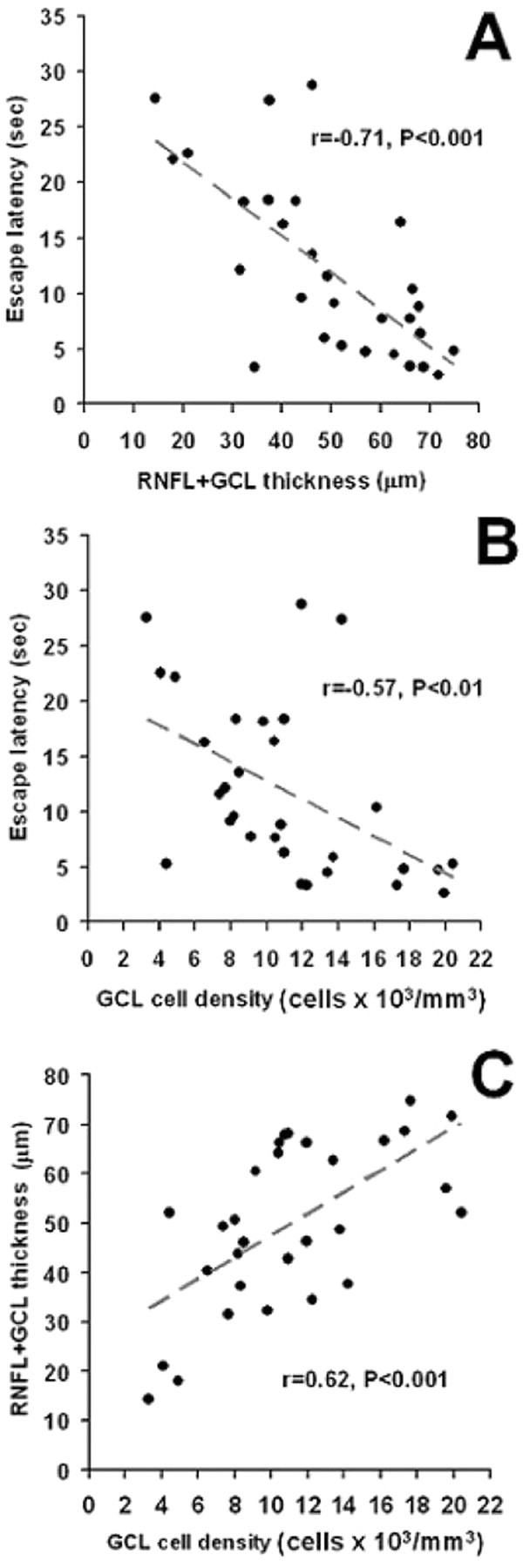
Structure–function correlations. Better-preserved RNFL + GCL thickness (A) and GCL cell densities (B) correlated with shorter mean postinjection escape latencies in the behavioral apparatus. RNFL + GCL thickness was also correlated with GCL cell density (C). These results implicate a functionally relevant neuroprotective effect of NIL that spares neural tissue from rotenone-induced neurotoxicity.
Metabolic effects
Cytochrome oxidase histochemistry-based analysis of the metabolic activity in the visual pathway was used as an ancillary measure of the functional effects of NIL treatment, especially searching for signs of functional deafferentation after rotenone administration. For example, in the superior colliculus, the main recipient of retinal ganglion cell axons in the rat, metabolic activity was clearly affected by the intravitreal rotenone injections. Mean cytochrome oxidase activity was 21.8% lower in rotenone-treated subjects, compared with control. NIL treatments prevented this effect in a dose–response manner (Fig. 6).
Figure 6.

Central metabolic effects of rotenone-induced visual deafferentation. A, The metabolic activity in the superior colliculus, a main recipient of afferent visual information in the rat, was severely affected by the intravitreal rotenone injections. Mean cytochrome oxidase activity was 21.8% lower in rotenone-treated subjects, compared with control. NIL treatments prevented this effect in a dose–response manner. The black bar represents the group mean. B, Tectal images of brain coronal sections stained with the cytochrome oxidase histochemical technique show a less intense staining in the superficial gray matter of the superior colliculus (SupG) in a rotenone-treated subject (region between white arrows). This contrasts with the intense staining observed in control and NIL-treated subjects. These changes took place 16 d after rotenone injection. IntG, Intermediate gray matter of the superior colliculus; Rot, rotenone; NIL 1 = rotenone + 10.8 J/cm2 NIL postinjection; NIL 2 = rotenone + 21.6 J/cm2 postinjection; NIL 3 = rotenone + 10.8 J/cm2 preinjection + 10.8 J/cm2 postinjection.
In addition, being cytochrome oxidase activity a maker of increases as well as decreases in neuronal metabolic activity, it was expected that it would reveal not only between-group differences due to rotenone neurotoxicity but also signs of neural activation correlating with both functional and structural changes due to NIL treatments. The results supported these predictions. First, the local metabolic activity in the retina showed a significant 32.4% decrease after rotenone, compared with control (p < 0.01). The metabolic activity was also lower in the rest of the treatment groups, compared with control (Fig. 7), but significantly correlated with both behavioral and structural variables. In other words, although NIL treatment did not increase the cytochrome oxidase activity in the retina, compared with the rotenone-treated group, subjects with higher retinal metabolic activity displayed shorter escape latency (r = −0.54, p < 0.01) and a higher percentage of successful trials in the behavioral task (r = 0.44, p < 0.05). Similarly, a higher retinal metabolic activity positively correlated with intact retinal structure, including RNFL plus GCL thickness (r = 0.60, p < 0.001), IPL thickness (r = 0.57, p < 0.01) and GCL cell density (r = 0.61, p < 0.001).
Figure 7.

Effect of near-infrared light treatment on retinal and visual pathway metabolic activity. Rotenone significantly decrease the metabolic activity in the retina, superficial gray matter of the superior colliculus (SupG), dorsal lateral geniculate nucleus (LGN), primary visual cortex (V1) and lateral peristriate cortex (V2). NIL treatments prevented this effect in a dose–response manner. In thalamic and cortical regions, the metabolic activity of high-dose NIL-treated subjects reached values higher than control. Rot, Rotenone; NIL 1 = rotenone + 10.8 J/cm2 NIL postinjection; NIL 2 = rotenone + 21.6 J/cm2 postinjection; NIL 3 = rotenone + 10.8 J/cm2 preinjection + 10.8 J/cm2 postinjection. Lower than control at *p < 0.05; lower than control at **p < 0.01; lower than control at ***p < 0.001. Higher than control at + p < 0.05; higher than control at ++ p < 0.01; higher than control at +++ p < 0.001.
Figure 7 also shows that between-group effects of NIL treatments on brain metabolic activity were evident trans-synaptically in central visual pathway relays at midbrain, thalamic and cortical levels. The functional deafferentation induced by the retinotoxic effects of rotenone were evidenced as a significant decrease in the metabolic activity in each one of the analyzed neuroanatomical regions compared with control. Also, the effects of NIL treatment appeared in a dose–response manner. Visual pathway metabolic activity in subjects treated with the low-dose NIL was significantly lower than control and similar to the rotenone group, whereas high-dose NIL prevented the decrease in metabolic activity observed in the rotenone group. Remarkably, protocols NIL 2 and NIL 3 not only prevented the local decreases in metabolic activity in visual structures, but actually increased their activity to levels higher than control. These metabolic increases relative to control were more reliable in higher brain regions such as LGN, V1 and especially V2, where an increase of 26.1% was observed with protocol NIL 3.
Transcranial NIL neurochemical effects
The results of regional brain metabolic activity measures suggested the possibility that some of these effects were mediated not only by the afferent influences of retinal inputs, but by a direct transcranial effect of near-infrared radiation on the entire brain. To test this hypothesis, whole-brain measures of cytochrome oxidase activity were obtained from the histochemically stained brain sections of subjects treated with the different NIL doses. Compared with subjects not exposed to NIL (the control and rotenone alone groups), a dose-dependent increase in whole-brain cytochrome oxidase activity of 14.8% (p = 0.08), 56.7% (p < 0.001) and 66.6% (p < 0.001) was observed in subjects treated with protocols NIL 1, NIL 2 and NIL 3, respectively (Fig. 8).
Figure 8.

Transcranial effects of near-infrared light on whole-brain metabolic capacity. A dose–response effect was observed, with the protocol including prerotenone treatment sessions producing the highest increase in cytochrome oxidase activity. NIL 1 = rotenone + 10.8 J/cm2 NIL postinjection; NIL 2 = rotenone + 21.6 J/cm2 postinjection; NIL 3 = rotenone + 10.8 J/cm2 preinjection + 10.8 J/cm2 postinjection. Significant difference compared with control at *p < 0.01; significant difference compared with control at **p < 0.001.
Further evidence of a dose–response transcranial effect of NIL treatments was obtained analyzing the whole brain cytosolic and mitochondrial SOD activity of subjects in each treatment group. While protocols NIL 1 and NIL 2 had no significant positive effect due to high variance, protocol NIL 3 induced a 51.5% increase in cytosolic SOD activity, relative to subjects not exposed to NIL (the control and rotenone alone groups) (Fig. 9).
Figure 9.

Transcranial effects of near-infrared light on whole-brain superoxide dismutase activity. Whereas subjects treated with the low-dose (NIL 1) protocol tended to show lower SOD activity levels, subjects treated with the high-dose protocols (NIL 2 and NIL 3) increased SOD activity compared with control. NIL 1 = rotenone + 10.8 J/cm2 NIL postinjection; NIL 2 = rotenone + 21.6 J/cm2 postinjection; NIL 3 = rotenone + 10.8 J/cm2 preinjection + 10.8 J/cm2 postinjection. Significantly lower than control at + p < 0.05; significantly higher than control at *p < 0.01.
Anesthesia and in vitro studies
A series of control studies were conducted to verify that NIL effects were not due to anesthesia effects or photodegradation of rotenone, but rather to direct NIL stimulation of mitochondrial respiration. First, the use of general anesthesia with isoflurane to immobilize the subjects while they received NIL radiation raised the concern that the anesthetic state induced by isoflurane would be the real cause of neuroprotection, instead of NIL treatments. However, subjects in the rotenone group were also anesthetized with isoflurane for the same period of time as subjects receiving NIL treatment; the mean illuminance sensitivity threshold and behavioral performance in this rotenone group were not improved compared with a rotenone-treated group not exposed to isoflurane (p = 0.17 and p = 0.79). Also, subjects in the rotenone and the rotenone-plus-isoflurane groups showed no differences in other measures such as retinal layer thickness (p = 0.42), GCL cell density (p = 0.49) and retinal metabolic activity (p = 0.20).
Second, there was the concern that NIL may photodegrade rotenone. But compared with rotenone solutions incubated in the dark, rotenone solutions radiated with NIL showed no decrease in their pro-oxidant properties, as measured by its ability to induce lipid peroxidation in brain homogenates [3 ± 0.1 vs 3.2 μmol/L thiobarbituric acid-reactive substances (TBARS), p = 0.8; 10 μm rotenone and 10 μm rotenone plus 3.6 J/cm2, respectively]. Levels of lipid peroxidation in brain homogenates treated with these two solutions were higher than that of a control (1.5 ± 0.1 TBARS μmol/L, p < 0.001).
Finally, we verified that NIL directly stimulates mitochondrial respiration in vitro. Cytochrome oxidase catalyzes the synthesis of water in mitochondria, the main oxygen-consuming reaction in neurons, and thus constitutes the rate-limiting step for cell respiration. Whole-brain homogenates were radiated with NIL to test the hypothesis that in vitro treatment would directly prevent the decrease in the rate of oxygen consumption induced by rotenone. Rotenone was expected to block the electron flow through the respiratory chain components and inhibit oxygen consumption. Whereas rotenone alone induced nearly a 75% decrease in the cell respiration rate (p < 0.05), in the presence of rotenone, even a small dose of NIL treatment (0.1 J/cm2) increased cell respiration rates to levels similar to control (Fig. 10).
Figure 10.

Effect of near-infrared light on cell respiration in vitro. Rotenone decreased the rate of oxygen consumption. Small (0.1 J/cm2) and large (1 J/cm2) pulses of NIL reversed the inhibitory effect of rotenone. Rot, Rotenone (10 μm). Significant difference compared with control at *p < 0.05.
Discussion
This is the first report providing evidence that NIL treatment via light-emitting diodes exerts neuroprotective effects against rotenone-induced neurotoxicity in vivo. These neuroprotective effects were observed at the behavioral, morphologic and neurochemical levels in a dose–response manner. These results add to a large body of compelling evidence suggesting that the biological effects of NIL could be effectively used to treat neurodegenerative conditions, in particular those associated with mitochondrial dysfunction. In vitro NIL effects include the following: (1) increased expression of the antiapoptotic protein Bcl-2 and reduced expression of the proapoptotic protein Bax (Shefer et al., 2002), (2) decreased numbers of apoptotic cells after exposure to the amyloid β protein (Duan et al., 2003), (3) improved function of cortical neurons inactivated by toxins (Wong-Riley et al., 2005; Liang et al., 2006), (4) increased survival and ATP content of striatal neurons after rotenone- and MPP+-induced toxicity and decreased oxidative stress and nitric oxide production (Liang et al., 2008), (5) increased neurite outgrowth (Wollman and Rochkind, 1998), (6) regulation of genes encoding DNA repair proteins, antioxidant enzymes and molecular chaperones (Eells et al., 2004), and (7) increased proliferation of olfactory ensheathing stem cells (Byrnes et al., 2005b), Schwann cells (Van Breugel and Bär, 1993), astrocytes, and oligodendrocytes (Rochkind et al., 1990). In vivo, NIL induces peripheral and central nerve regeneration after trauma (Anders et al., 1993; Byrnes et al., 2005a), reduces neuroinflammation (Byrnes et al., 2005a), and prevents methanol-induced photoreceptor degeneration (Eells et al., 2003). In humans, NIL has been successfully applied in the treatment of neuropathic pain (Iijima et al., 1991), prevention of oral mucositis in immunocompromised patients (Whelan et al., 2002), and in stimulation of wound closure and skin graft healing (Conlan et al., 1996).
An in vivo model of retinal degeneration induced by rotenone was used in this study. Rotenone-treated subjects showed decreased visual function, which correlated with abnormal retinal morphology, and with impoverished retinal and visual pathway metabolic activity. Rotenone is a natural pesticide regarded as a probable environmental neurotoxin (Sherer et al., 2001). Rotenone is a mitochondrial complex I inhibitor that induces oxidative stress, energy depletion and neuronal death, and has been used to model neurodegenerative disorders associated with complex I dysfunction, including Parkinson's disease and optic neuropathy (Betarbet et al., 2000; Zhang et al., 2002; Sherer et al., 2003; Beretta et al., 2006).
NIL treatments were effective at preventing rotenone-induced retinotoxicity in a dose–response manner. A total dose of 21.6 J/cm2 fractionated in six sessions of 3.6 J/cm2 postinjection (protocol NIL 2) was highly effective at preventing the rotenone-induced visual deficits. Rates of successful trials, escape latencies and illuminance sensitivity thresholds in NIL-2-treated subjects were similar to control, and correlated with preservation of the retinal nerve fiber layer and sustained metabolic activation of the central visual pathway. This high dose was similarly effective when part of it was delivered as a pretreatment previous to rotenone injections (protocol NIL 3). In contrast, when only half of the total NIL dose (10.8 J/cm2) was given after the injection (protocol NIL 1), no neuroprotective effects were observed. This supports that prophylactic administration of NIL occurring before the mitochondrial insult contributed to the postrotenone neuroprotective effects, as previously demonstrated in in vitro systems (Wong-Riley et al., 2005). In addition, the effectiveness of fractionated NIL protocols is in agreement with previous studies showing that NIL doses given within the same day are not as effective as those that are given over several days and that stimulatory effects are observed at daily energy densities between 1 × 10−3 J/cm2 and 10 J/cm2 (Brondon et al., 2005; Liang et al., 2008).
Neural metabolic activity was determined by means of quantification of cytochrome oxidase histochemistry, which detects within-brain regional differences in long-term metabolic demands (Wong-Riley, 1989; Gonzalez-Lima and Cada, 1994). As expected, more cytochrome oxidase activity was revealed in the retina and central visual pathway of control animals, in contrast with the decrease observed in rotenone-treated subjects. Rotenone's neurotoxicity was evident as a decrease in retinal and visual pathway cytochrome oxidase activity, relative to control. This is evidence of functional deafferentation and a trans-synaptic metabolic effect of the lesions (Wong-Riley 1979; Wong-Riley and Welt, 1980), which was reversed by NIL treatments.
Remarkably, the whole-brain metabolic activity and not only that of the visual pathway was increased after NIL treatments in a dose–response manner, which suggests a direct effect of NIL treatments on the brain, independent of the afferent visual input. This evidence also suggests that the neuroprotective effect of NIL may be mediated by a buildup of the cytochrome oxidase holoenzyme pool. Previous spectroscopic measures have shown that although the directionality of the electromagnetic radiation produced by light-emitting diode arrays is lower than that of laser sources, photons at wavelengths between 630 and 800 nm are able to travel ∼23 cm even in layers of tissues with relatively low transparencies such as skin, connective tissue, muscle, bone and spinal cord (Chance et al., 1988a,b; Byrnes et al., 2005a). Further neurochemical analyses demonstrated that brains exposed to NIL treatments showed an increase not only in cytochrome oxidase activity but also in SOD activity. The increase in SOD was observed in a dose–response manner, with higher NIL protocols being more effective than the low-dose protocol. This effect was observed in forebrain samples, thus being independent of retinal inputs, which supports the notion of a transcranial biological effect of NIL. Finally, the increases in SOD activity were seen in cytosolic fractions, which suggest the involvement of extramitochondrial messengers or effectors mediating responses to NIL. These data are consistent with previous studies suggesting that the mechanism of action of NIL treatment includes regulation of gene expression (Eells et al., 2004; Byrnes et al., 2005b; Liang et al., 2006) and suggest that genes involved in neuronal bioenergetics (e.g., cytochrome oxidase) and antioxidant defenses (e.g., SOD) may play a role in the intrinsic mechanisms of survival against mitochondrial insults.
However, what mechanisms allow upregulation of enzyme genes relevant to neuroprotection after NIL treatments? Direct catalytic enhancement of cytochrome oxidase activity, alteration of metabolic pathways and actions of alternate photoacceptors are possible mechanisms that merit further investigation. Cytochrome oxidase is regarded as the primary photoacceptor of light in the red to near-infrared region of the visible spectrum (Yamanaka et al., 1988; Pastore et al., 2000). In neural tissue, it is the most abundant metalloprotein and wavelength peaks in its absorption spectrum (670 and 830 nm) highly correlate with its peaks in catalytic activity and with ATP content in vitro (Eells et al., 2004). The in vitro study provides evidence of a direct stimulatory effect of NIL on mitochondrial respiration independently of in vivo enzyme buildup. In vitro, brief pulses of NIL produced a respiratory enhancement that counteracted the inhibitory effect of rotenone, suggesting a direct stimulation of the catalytic activity of cytochrome oxidase and acceleration of the electron flow. This interpretation agrees with previous observations demonstrating that low-level laser radiation increases cytochrome c oxidation in the presence of cytochrome oxidase (Pastore et al., 2000). However, the respiratory enhancing effects demonstrated here were not observed in control conditions, but only in systems in which rotenone was added. This suggests that only a partially inhibited respiratory chain is permissible to direct NIL-induced increases in electron flow at the cytochrome oxidase levels, but not an otherwise uncoupled system in which electron flow occurs at the highest possible rate (Kadenbach, 2003).
NIL has also been shown to induce transient biochemical changes that impact a variety of metabolic pathways and redox reactions. For instance, NIL is able to modify the interactions between cytochrome oxidase and nitric oxide and to promote the generation of free radicals (Karu, 1999; Karu et al., 2005). Although cytochrome oxidase is regarded as a major mediator of NIL effects, other proteins may also have a role, whether directly acting as photoacceptors or being part of cellular cascades triggering cell survival. Of particular interest are other heme-containing proteins implicated in intrinsic mechanisms of neuroprotection and cell death. An example is neuroglobin, which is predominantly expressed in the brain and shows upregulation in response to oxygen deprivation, and protects neurons from ischemic and hypoxic death (Khan et al., 2008; Wang et al., 2008). Neuroglobin is expressed in the retina and reduces cytosolic cytochrome c thus being implicated in responses to NIL as both a photoacceptor and intracellular effector (Ostojić et al., 2006; Fago et al., 2008). The role of this and other proteins in NIL-tissue interactions together with cytochrome oxidase's role should be further investigated.
In summary, the present experiments demonstrate that NIL treatment via light-emitting diodes provides neuroprotection against rotenone-induced retinotoxicity. NIL treatments also appear to have in vivo transcranial neurochemical effects that involve metabolic-enhancing and antioxidant systems. These data suggest that NIL could be used as a noninvasive neuroprotective approach against neurological conditions caused by mitochondrial dysfunction.
Footnotes
This work was supported in part by National Institutes of Health Grant MH65728 (to F.G.-L.) and CONACYT 187413 (to J.C.R.). This work was performed by J.C.R. in partial fulfillment of his requirements for a Ph.D. degree in neuroscience at the University of Texas at Austin. We appreciate the valuable contributions of the Dissertation Committee Members: Drs. Theresa Jones, Timothy Schallert, Andrea Gore, and Henry Rylander. We are also in debt to Dr. Eimeira Padilla and Frank Puga for their help with brain extractions, Megan Webb and Shreya Gandhy for their help with behavioral experiments, Dr. William M. Lackowski, Christian Balderrama, and Michelle Machie for their help with NIL measures and treatments, and Drs. Aleksandra Bruchey and Chris Hayworth for their comments to improve this manuscript.
References
- Anders JJ, Borke RC, Woolery SK, Van de Merwe WP. Low power laser irradiation alters the rate of regeneration of the rat facial nerve. Lasers Surg Med. 1993;13:72–82. doi: 10.1002/lsm.1900130113. [DOI] [PubMed] [Google Scholar]
- Beretta S, Wood JP, Derham B, Sala G, Tremolizzo L, Ferrarese C, Osborne NN. Partial mitochondrial complex I inhibition induces oxidative damage and perturbs glutamate transport in primary retinal cultures. Relevance to Leber hereditary optic neuropathy (LHON) Neurobiol Dis. 2006;24:308–317. doi: 10.1016/j.nbd.2006.07.016. [DOI] [PubMed] [Google Scholar]
- Betarbet R, Sherer TB, MacKenzie G, Garcia-Osuna M, Panov AV, Greenamyre JT. Chronic systemic pesticide exposure reproduces features of Parkinson's disease. Nat Neurosci. 2000;3:1301–1306. doi: 10.1038/81834. [DOI] [PubMed] [Google Scholar]
- Brondon P, Stadler I, Lanzafame RJ. A study of the effects of phototherapy dose interval on photobiomodulation of cell cultures. Lasers Surg Med. 2005;36:409–413. doi: 10.1002/lsm.20183. [DOI] [PubMed] [Google Scholar]
- Brown MD, Zhadanov S, Allen JC, Hosseini S, Newman NJ, Atamonov VV, Mikhailovskaya IE, Sukernik RI, Wallace DC. Novel mtDNA mutations and oxidative phosphorylation dysfunction in Russian LHON families. Hum Genet. 2001;109:33–39. doi: 10.1007/s004390100538. [DOI] [PubMed] [Google Scholar]
- Byrnes KR, Waynant RW, Ilev IK, Wu X, Barna L, Smith K, Heckert R, Gerst H, Anders JJ. Light promotes regeneration and functional recovery and alters the immune response after spinal cord injury. Lasers Surg Med. 2005a;36:171–185. doi: 10.1002/lsm.20143. [DOI] [PubMed] [Google Scholar]
- Byrnes KR, Wu X, Waynant RW, Ilev IK, Anders JJ. Low power laser irradiation alters gene expression of olfactory ensheathing cells in vitro. Lasers Surg Med. 2005b;37:161–171. doi: 10.1002/lsm.20202. [DOI] [PubMed] [Google Scholar]
- Chance B, Nioka S, Kent J, McCully K, Fountain M, Greenfeld R, Holtom G. Time-resolved spectroscopy of hemoglobin and myoglobin in resting and ischemic muscle. Anal Biochem. 1988a;174:698–707. doi: 10.1016/0003-2697(88)90076-0. [DOI] [PubMed] [Google Scholar]
- Chance B, Leigh JS, Miyake H, Smith DS, Nioka S, Greenfeld R, Finander M, Kaufmann K, Levy W, Young M, et al. Comparison of time-resolved and -unresolved measurements of deoxyhemoglobin in brain. Proc Natl Acad Sci U S A. 1988b;85:4971–4975. doi: 10.1073/pnas.85.14.4971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conlan MJ, Rapley JW, Cobb CM. Biostimulation of wound healing by low-energy laser irradiation. A review. J Clin Periodontol. 1996;23:492–496. doi: 10.1111/j.1600-051x.1996.tb00580.x. [DOI] [PubMed] [Google Scholar]
- Duan R, Zhu L, Liu TC, Li Y, Liu J, Jiao J, Xu X, Yao L, Liu S. Light emitting diode irradiation protect against the amyloid beta 25–35 induced apoptosis of PC12 cell in vitro. Lasers Surg Med. 2003;33:199–203. doi: 10.1002/lsm.10216. [DOI] [PubMed] [Google Scholar]
- Eells JT, Henry MM, Summerfelt P, Wong-Riley MT, Buchmann EV, Kane M, Whelan NT, Whelan HT. Therapeutic photobiomodulation for methanol-induced retinal toxicity. Proc Natl Acad Sci U S A. 2003;100:3439–3444. doi: 10.1073/pnas.0534746100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eells JT, Wong-Riley MT, VerHoeve J, Henry M, Buchman EV, Kane MP, Gould LJ, Das R, Jett M, Hodgson BD, Margolis D, Whelan HT. Mitochondrial signal transduction in accelerated wound and retinal healing by near-infrared light therapy. Mitochondrion. 2004;4:559–567. doi: 10.1016/j.mito.2004.07.033. [DOI] [PubMed] [Google Scholar]
- Fago A, Mathews AJ, Brittain T. A role for neuroglobin: resetting the trigger level for apoptosis in neuronal and retinal cells. IUBMB Life. 2008;60:398–401. doi: 10.1002/iub.35. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Lima F, Cada A. Cytochrome oxidase activity in the auditory system of the mouse: a qualitative and quantitative histochemical study. Neuroscience. 1994;63:559–578. doi: 10.1016/0306-4522(94)90550-9. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Lima F, Cada A. Quantitative histochemistry of cytochrome oxidase activity: theory, methods, and regional brain vulnerability. In: Gonzalez-Lima F, editor. Cytochrome oxidase in neuronal metabolism and Alzheimer's disease. New York: Plenum; 1998. pp. 55–90. [Google Scholar]
- Gonzalez-Lima F, Jones D. Quantitative mapping of cytochrome oxidase activity in the central auditory system of the gerbil: a study with calibrated activity standards and metal-intensified histochemistry. Brain Res. 1994;660:34–49. doi: 10.1016/0006-8993(94)90836-2. [DOI] [PubMed] [Google Scholar]
- Gundersen HJ, Jensen TB, Osterby R. Distribution of membrane thickness determined by lineal analysis. J Microsc. 1978;113:27–43. doi: 10.1111/j.1365-2818.1978.tb00091.x. [DOI] [PubMed] [Google Scholar]
- Harding AJ, Halliday GM, Cullen K. Practical considerations for the use of the optical disector in estimating neuronal number. J Neurosci Methods. 1994;51:83–89. doi: 10.1016/0165-0270(94)90028-0. [DOI] [PubMed] [Google Scholar]
- Hayes JM, Balkema GW. Elevated dark-adapted thresholds in hypopigmented mice measured with a water maze screening apparatus. Behav Genet. 1993;23:395–403. doi: 10.1007/BF01067442. [DOI] [PubMed] [Google Scholar]
- Iijima K, Shimoyama N, Shimoyama M, Mizuguchi T. Evaluation of analgesic effect of low-power He:Ne laser on postherpetic neuralgia using VAS and modified McGill pain questionnaire. J Clin Laser Med Surg. 1991;9:121–126. doi: 10.1089/clm.1991.9.121. [DOI] [PubMed] [Google Scholar]
- Jung C, Higgins CM, Xu Z. A quantitative histochemical assay for activities of mitochondrial electron transport chain complexes in mouse spinal cord sections. J Neurosci Methods. 2002;114:165–172. doi: 10.1016/s0165-0270(01)00524-6. [DOI] [PubMed] [Google Scholar]
- Kadenbach B. Intrinsic and extrinsic uncoupling of oxidative phosphorylation. Biochim Biophys Acta. 2003;1604:77–94. doi: 10.1016/s0005-2728(03)00027-6. [DOI] [PubMed] [Google Scholar]
- Karu T. Primary and secondary mechanisms of action of visible to near-IR radiation on cells. J Photochem Photobiol B. 1999;49:1–17. doi: 10.1016/S1011-1344(98)00219-X. [DOI] [PubMed] [Google Scholar]
- Karu TI, Pyatibrat LV, Afanasyeva NI. Cellular effects of low power laser therapy can be mediated by nitric oxide. Lasers Surg Med. 2005;36:307–314. doi: 10.1002/lsm.20148. [DOI] [PubMed] [Google Scholar]
- Khan AA, Mao XO, Banwait S, DerMardirossian CM, Bokoch GM, Jin K, Greenberg DA. Regulation of hypoxic neuronal death signaling by neuroglobin. FASEB J. 2008;22:1737–1747. doi: 10.1096/fj.07-100784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang HL, Whelan HT, Eells JT, Meng H, Buchmann E, Lerch-Gaggl A, Wong-Riley M. Photobiomodulation partially rescues visual cortical neurons from cyanide-induced apoptosis. Neuroscience. 2006;139:639–649. doi: 10.1016/j.neuroscience.2005.12.047. [DOI] [PubMed] [Google Scholar]
- Liang HL, Whelan HT, Eells JT, Wong-Riley MT. Near-infrared light via light-emitting diode treatment is therapeutic against rotenone- and 1-methyl-4-phenylpyridinium ion-induced neurotoxicity. Neuroscience. 2008;153:963–974. doi: 10.1016/j.neuroscience.2008.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misra HP, Fridovich I. The role of superoxide anion in the autoxidation of epinephrine and a simple assay for superoxide dismutase. J Biol Chem. 1972;247:3170–3175. [PubMed] [Google Scholar]
- Ostojić J, Sakaguchi DS, de Lathouder Y, Hargrove MS, Trent JT, 3rd, Kwon YH, Kardon RH, Kuehn MH, Betts DM, Grozdanić S. Neuroglobin and cytoglobin: oxygen-binding proteins in retinal neurons. Invest Ophthalmol Vis Sci. 2006;47:1016–1023. doi: 10.1167/iovs.05-0465. [DOI] [PubMed] [Google Scholar]
- Pastore D, Greco M, Passarella S. Specific helium-neon laser sensitivity of the purified cytochrome c oxidase. Int J Radiat Biol. 2000;76:863–870. doi: 10.1080/09553000050029020. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The rat brain in stereotaxic coordinates. San Diego: Academic; 1997. [DOI] [PubMed] [Google Scholar]
- Prusky GT, West PW, Douglas RM. Behavioral assessment of visual acuity in mice and rats. Vision Res. 2000;40:2201–2209. doi: 10.1016/s0042-6989(00)00081-x. [DOI] [PubMed] [Google Scholar]
- Rochkind S, Vogler I, Barr-Nea L. Spinal cord response to laser treatment of injured peripheral nerve. Spine. 1990;15:6–10. doi: 10.1097/00007632-199001000-00003. [DOI] [PubMed] [Google Scholar]
- Rojas JC, Saavedra JA, Gonzalez-Lima F. Neuroprotective effects of memantine in a mouse model of retinal degeneration induced by rotenone. Brain Res. 2008;1215:208–217. doi: 10.1016/j.brainres.2008.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadun AA. Mitochondrial optic neuropathies. J Neurol Neurosurg Psychiatry. 2002;72:423–425. doi: 10.1136/jnnp.72.4.423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schapira AH, Cooper JM, Dexter D, Clark JB, Jenner P, Marsden CD. Mitochondrial complex I deficiency in Parkinson's disease. J Neurochem. 1990;54:823–827. doi: 10.1111/j.1471-4159.1990.tb02325.x. [DOI] [PubMed] [Google Scholar]
- Seaton TA, Jenner P, Marsden CD. Mitochondrial respiratory enzyme function and superoxide dismutase activity following brain glutathione depletion in the rat. Biochem Pharmacol. 1996;52:1657–1663. doi: 10.1016/s0006-2952(96)00452-2. [DOI] [PubMed] [Google Scholar]
- Shefer G, Partridge TA, Heslop L, Gross JG, Oron U, Halevy O. Low-energy laser irradiation promotes the survival and cell cycle entry of skeletal muscle satellite cells. J Cell Sci. 2002;115:1461–1469. doi: 10.1242/jcs.115.7.1461. [DOI] [PubMed] [Google Scholar]
- Sherer TB, Betarbet R, Greenamyre JT. Pesticides and Parkinson's disease. ScientificWorldJournal. 2001;1:207–208. doi: 10.1100/tsw.2001.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherer TB, Betarbet R, Testa CM, Seo BB, Richardson JR, Kim JH, Miller GW, Yagi T, Matsuno-Yagi A, Greenamyre JT. Mechanism of toxicity in rotenone models of Parkinson's disease. J Neurosci. 2003;23:10756–10764. doi: 10.1523/JNEUROSCI.23-34-10756.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommer AP, Pinheiro AL, Mester AR, Franke RP, Whelan HT. Biostimulatory windows in low-intensity laser activation: lasers, scanners, and NASA's light-emitting diode array system. J Clin Laser Med Surg. 2001;19:29–33. doi: 10.1089/104454701750066910. [DOI] [PubMed] [Google Scholar]
- Van Breugel HH, Bär PR. He-Ne laser irradiation affects proliferation of cultured rat Schwann cells in a dose-dependent manner. J Neurocytol. 1993;22:185–190. doi: 10.1007/BF01246357. [DOI] [PubMed] [Google Scholar]
- Wang X, Liu J, Zhu H, Tejima E, Tsuji K, Murata Y, Atochin DN, Huang PL, Zhang C, Lo EH. Effects of neuroglobin overexpression on acute brain injury and long-term outcomes after focal cerebral ischemia. Stroke. 2008;39:1869–1874. doi: 10.1161/STROKEAHA.107.506022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whelan HT, Connelly JF, Hodgson BD, Barbeau L, Post AC, Bullard G, Buchmann EV, Kane M, Whelan NT, Warwick A, Margolis D. NASA light-emitting diodes for the prevention of oral mucositis in pediatric bone marrow transplant patients. J Clin Laser Med Surg. 2002;20:319–324. doi: 10.1089/104454702320901107. [DOI] [PubMed] [Google Scholar]
- Wollman Y, Rochkind S. In vitro cellular processes sprouting in cortex microexplants of adult rat brains induced by low power laser irradiation. Neurol Res. 1998;20:470–472. doi: 10.1080/01616412.1998.11740550. [DOI] [PubMed] [Google Scholar]
- Wong-Riley M. Changes in the visual system of monocularly sutured or enucleated cats demonstrable with cytochrome oxidase histochemistry. Brain Res. 1979;171:11–28. doi: 10.1016/0006-8993(79)90728-5. [DOI] [PubMed] [Google Scholar]
- Wong-Riley MT. Cytochrome oxidase: an endogenous metabolic marker for neuronal activity. Trends Neurosci. 1989;12:94–101. doi: 10.1016/0166-2236(89)90165-3. [DOI] [PubMed] [Google Scholar]
- Wong-Riley MT, Welt C. Histochemical changes in cytochrome oxidase of cortical barrels after vibrissal removal in neonatal and adult mice. Proc Natl Acad Sci U S A. 1980;77:2333–2337. doi: 10.1073/pnas.77.4.2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong-Riley MT, Bai X, Buchmann E, Whelan HT. Light-emitting diode treatment reverses the effect of TTX on cytochrome oxidase in neurons. Neuroreport. 2001;12:3033–3037. doi: 10.1097/00001756-200110080-00011. [DOI] [PubMed] [Google Scholar]
- Wong-Riley MT, Liang HL, Eells JT, Chance B, Henry MM, Buchmann E, Kane M, Whelan HT. Photobiomodulation directly benefits primary neurons functionally inactivated by toxins: role of cytochrome c oxidase. J Biol Chem. 2005;280:4761–4771. doi: 10.1074/jbc.M409650200. [DOI] [PubMed] [Google Scholar]
- Yamanaka T, Fukumori Y, Numata M, Yamazaki T. The variety of molecular properties of bacterial cytochromes containing heme a. Ann N Y Acad Sci. 1988;550:39–46. doi: 10.1111/j.1749-6632.1988.tb35320.x. [DOI] [PubMed] [Google Scholar]
- Zhang X, Jones D, Gonzalez-Lima F. Mouse model of optic neuropathy caused by mitochondrial complex I dysfunction. Neurosci Lett. 2002;326:97–100. doi: 10.1016/s0304-3940(02)00327-0. [DOI] [PubMed] [Google Scholar]
- Zhang X, Rojas JC, Gonzalez-Lima F. Methylene blue prevents neurodegeneration caused by rotenone in the retina. Neurotox Res. 2006a;9:47–57. doi: 10.1007/BF03033307. [DOI] [PubMed] [Google Scholar]
- Zhang X, Jones D, Gonzalez-Lima F. Neurodegeneration produced by rotenone in the mouse retina: a potential model to investigate environmental pesticide contributions to neurodegenerative diseases. J Toxicol Environ Health A. 2006b;69:1681–1697. doi: 10.1080/15287390600630203. [DOI] [PubMed] [Google Scholar]