

Cytochrome P450 refers to a group of enzymes that catalyze the monooxygenation of various organic molecules in the following reaction:
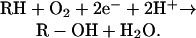 |
P450s are best known for their role in drug metabolism and detoxification. Humans have 57 functional P450 genes and 46 pseudogenes (http://drnelson.utmem.edu/human.genecount.html), and several of these, located in liver microsomes, are induced by a variety of foreign organic molecules. Hydroxylation of these substances by P450s renders these relatively insoluble organic compounds more soluble for easier elimination. In addition, P450s participate in the metabolism of sex hormones, vitamin D, and bile acids in mammals (1, 2); ecdysones in insects (3); and terpenes in plants (4). P450s also play an important role in microorganisms in the use of various organic compounds as carbon sources and in the production of important natural products such as antibiotics.
P450s range in size from 40 to 50 kDa and contain a single heme group. Oxygen binds to the heme iron where the enzyme catalyzes cleavage of the oxygen O—O bond, leaving behind an iron-linked oxygen atom that provides a potent oxidant. The substrate is held precisely in place such that the correct carbon atom is close to the iron-linked O atom for regio- and stereoselective hydroxylation. In general, those P450s involved in the production of important intermediates such as in steroid metabolism are highly specific. However, many of the drug-metabolizing P450s are not specific and are capable of hydroxylating a variety of diverse and unrelated compounds. Several P450 crystal structures now are known, and it was not too surprising, based on sequence alignments, that the overall fold is maintained in all P450s (Fig. 1). A particularly challenging problem, however, is to understand how P450s adapt to accommodate different substrates given the restriction that the same fold is maintained in all P450s. The first P450 structure (5) presented a puzzle as to how the substrate enters the active site, because the substrate-binding pocket was found to be inaccessible to the outside world. The prevailing view developed since then is that the F and G helices and the loop connecting these helices (Fig. 1) are flexible and undergo an open/close motion, which allows substrates to enter and products to leave. The new P450 2B4 structure by Scott et al. (6), published in this issue of PNAS, provides a dramatic example of the range of motions available to P450s. P450 2B4 is especially important. Formerly known as P450 LM2 (liver microsome 2), P450 2B4 was the first microsomal P450 to be purified and well characterized by Coon et al. (7) and since then has served as a paradigm for detailed biophysical and biochemical investigations. Hence, a good deal is known about this P450, and, with the structure now in hand, many loose ends can to be tied together.
Fig. 1.

Schematic diagram of P450 BM-3 in the partially open conformation. The B′, F, G, and I helices are labeled. The F and G helices and the F/G loop are highlighted in magenta. These helices slide over the surface of the I helix, which leads to an open/close motion of the access channel leading to the active site.
P450 2B4, like other membrane-bound P450s, presents a special challenge for crystallization. Scott et al. (6) followed the pioneering work from Eric Johnson's laboratory (8) on engineering the P450 to be both soluble and monodisperse, which led to the first mammalian P450 structure, P450 2C5 (9). This involved removal of the N-terminal region that helps to anchor the P450 to the microsomal membrane and mutating key residues on the surface that further improves both solubility and homogeneity. One difference in the P450 2B4 work is that no mutations in the F/G helix region were required. A fortuitous and totally unexpected finding in the new P450 2B4 structure is how two P450 2B4 molecules form a dimer. Molecules related by crystallographic symmetry form a tight dimer, which, in itself, is not so unusual. However, a His residue between the F and G helices in molecule A penetrates into the active site of the crystallographically related molecule B, where it forms a His—Fe bond. The interaction is symmetric, because the His of molecule B also coordinates the iron of molecule A, requiring that the active site adopt a conformation involving the F and G helices far more open than has been observed in previous structures (Fig. 2). One could argue that such an open conformation is an artifact of crystallization. However, Scott et al. (6) show that the same dimer very likely forms in solution. Moreover, forces that hold protein molecules together in a crystal lattice are weak, so the protein cannot adopt a conformation in the crystal that is not energetically accessible in solution. In effect, the P450 2B4 structure has been ”trapped” in a wide-open conformation and nicely illustrates the rather wide range of motions available to P450s. Before the P450 2B4 structure, the most that was known about motion in P450s derived from the bacterial fatty acid monooxygenase P450 BM-3 (10, 11) and, more recently, P450 2CD (12). The substrate-bound and -free structures of P450 BM-3 show that the F and G helices slide as a unit over the L helix, which leads to the open/close motion. It might be anticipated that P450 2B4 will experience the same type of motion.
Fig. 2.

Spaced filled diagrams of P450 BM-3 and P450 2B4 in the same orientation. The F and G helices are in magenta. The P450 BM-3 model is of the substrate-free structure, which adopts a partially open conformation that allows substrates to enter (10, 17). In P450 2B4, these same elements of structure are positioned very differently, leading to a wide-open active site. This difference illustrates the range of motions possible in P450s while retaining the overall P450-fold.
Such open and close motion, however, does not fully explain how a single P450 can accommodate a variety of different substrates. In addition to closure of the active site, there may also be a more dramatic reshaping of the active site. The best candidates for such changes include the F/G loop and B′ helix. In some P450 structures, the electron density for the F/G loop is visible only in the substrate-bound form (12), suggesting that the F/G loop may be able to shape itself around the substrate. The one clear example of such reshaping is in the thermophilic P450, CYP119. The structure of this P450 has been solved with different types of inhibitors bound to the heme iron (13, 14). A rather dramatic unfolding of the C terminus of the F helix and large movements of the F/G loop were observed, further supporting the view that the F/G loop and associated elements of structure can shape themselves around active site ligands. A P450 involved in steroid metabolism, CYP51, provides yet another example of an open active site (15). In this case, however, the open heme pocket is due more to a break in the I helix that opens up a new access channel to the active site. Although quite speculative, it is intriguing to consider the possibility that once a steroid substrate binds, the I helix ”repairs” itself, resulting in a closed active site. The final example is a chemically modified form of the bacterial P450cam, in which a Cys residue near the active site was modified with a bulky ferrocene. In this case, the B′ helix completely rearranges, resulting in large motions (16). Although not physiologically relevant, the ferrocene work illustrates the flexibility available to a P450 precisely in the region where substrate entry is thought to occur. These various structures of open P450 conformations thus solve the early puzzle of how substrates enter. Large conformational changes due primarily to motions of the F and G helices as well as the F/G loop are responsible. The current P450 2B4 structure very likely represents the extreme end of an open conformation of the drug-metabolizing microsomal P450s. An intriguing aspect of this open conformation is that the connecting segment between the F and G helices in P450 2B4 is ordered and helical. Assuming that the active site closes when a substrate binds, the F/G loop region might move into the active site, which, as in CYP119, may require unfolding of helical segments. We anxiously await a P450 2B4 structure with substrate bound.
See companion article on page 13196.
References
- 1.Hayaishi, O. (1974) Molecular Mechanisms of O2 Activation (Academic, New York).
- 2.Gunsalus, I. C., Pederson, T. C. & Sligar, S. G. (1975) Annu. Rev. Biochem. 44, 377-407. [DOI] [PubMed] [Google Scholar]
- 3.Smith, S. L., Bollenbacher, W. E., Cooper, D. Y., Schleyer, H., Weilgus, J. J. & Gilbert, L. I. (1979) Mol. Cell. Endocrinol. 15, 111-113. [DOI] [PubMed] [Google Scholar]
- 4.Murphy, P. I. & West, C. A. (1969) Arch. Biochem. Biophys. 133, 395-407. [DOI] [PubMed] [Google Scholar]
- 5.Poulos, T. L., Finzel, B. C. & Howard, A. J. (1987) J. Mol. Biol. 195, 687-700. [DOI] [PubMed] [Google Scholar]
- 6.Scott, E. E., He, Y. A., Wester, M. R., White, M. A., Chin, C. C., Halpert, J. R., Johnson, E. F. & Stout, C. D. (2003) Proc. Natl. Acad. Sci. USA 100, 13196-13201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Coon, M. J., van der Hoeven, T. A., Kaschnitz, R. M. & Stroble, H. W. (1973) Ann. N.Y. Acad. Sci. 212, 449-457. [DOI] [PubMed] [Google Scholar]
- 8.Come, J. & Johnson, E. F. (2000) J. Biol. Chem. 275, 2545-2553. [DOI] [PubMed] [Google Scholar]
- 9.Williams, P. A., Cosme, J., Sridhar, V., Johnson, E. E. & McRee, D. E. (2000) Mol. Cell 5, 121-131. [DOI] [PubMed] [Google Scholar]
- 10.Li, H. & Poulos, T. L. (1997) Nat. Struct. Biol. 4, 140-146. [DOI] [PubMed] [Google Scholar]
- 11.Haines, D. C., Tomchick, D. R., Machius, M. & Peterson, J. A. (2001) Biochemistry 40, 13456-13465. [DOI] [PubMed] [Google Scholar]
- 12.Wester, M. R., Johnson, E. F. & Stout, C. D. (2003) Biochemistry 42, 9335-9345. [DOI] [PubMed] [Google Scholar]
- 13.Yano, J. K., Koo, L. S., Schuller, D. J., Li, H., Ortiz de Montellano, P. R. & Poulos, T. L. (2000) J. Biol. Chem. 275, 31086-31092. [DOI] [PubMed] [Google Scholar]
- 14.Park, S. Y., Yamane, K., Adachi, S., Shiro, Y., Maves, S. A., Weiss, K. E. & Sligar, S. G. (2002) J. Inorg. Biochem. 91, 491-501. [DOI] [PubMed] [Google Scholar]
- 15.Podust, L. M., Poulos, T. L. & Waterman, M. R. (2001) Proc. Natl. Acad. Sci. USA 98, 3068-3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.DiGleria, K., Nickerson, D. P., Hill, H. A. O., Wong, L. L. & Fulop, V. (1998) J. Am. Chem. Soc. 120, 46-52. [Google Scholar]
- 17.Ravichandran, K. G., Boddupalli, S. S., Hasermann, C. A., Peterson, J. A. & Deisenhofer, J. (1993) Science 261, 731-736. [DOI] [PubMed] [Google Scholar]