

Abstract
A fully developed understanding of protein glycosylation requires characterization of the modifying oligosaccharides, elucidation of their covalent attachment sites, and determination of the glycan heterogeneity at specific sites. Considering the complexity inherent to protein glycosylation, establishing these features for even a single protein can present an imposing challenge. In order to meet the demands of glycoproteomics, the capability to screen far more complex systems of glycosylated proteins must be developed. Although the proteome wide examination of carbohydrate modification has become an area of keen interest, the intricacy of protein glycosylation has frustrated the progress of large scale, systems oriented research on site-specific protein-glycan relationships. Indeed, the analytical obstacles in this area have been more instrumental in shaping the current glycoproteomic paradigm than have the diverse functional roles and ubiquitous nature of glycans. This report describes the ongoing development and analytically salient features of bead immobilized pronase for glycosylation site footprinting. The present work bears on the ultimate goal of providing analytical tools capable of addressing the diversity of protein glycosylation in a more comprehensive and efficient manner. In particular, this approach has been assessed with respect to reproducibility, sensitivity, and tolerance to sample complexity. The efficiency of pronase immobilization, attainable pronase loading density, and the corresponding effects on glycoprotein digestion rate were also evaluated. In addition to being highly reproducible, the immobilized enzymes retained a high degree of proteolytic activity after repeat usage for up to 6 weeks. This method also afforded a low level of chemical background and provided favorable levels of sensitivity with respect to traditional glycoproteomic strategies. Thus, the application of immobilized pronase shows potential to contribute to the advancement of more comprehensive glycoproteomic research methods that are capable of providing site-specific glycosylation and microheterogeneity information across many proteins.
Keywords: Enzyme immobilization, glycopeptide footprinting, glycoproteomics, pronase, protein glycosylation, site-specific glycosylation analysis
Introduction
The post-translational modification (PTM) of proteins by carbohydrates is becoming increasingly recognized as a vital area of study,1-4 nonetheless the proportion and scale of research devoted to protein glycosylation lags behind efforts aimed at characterizing other PTMs. The study of protein glycosylation has not kept pace with the enormous structural diversity and eclectic functional roles of protein modifying oligosaccharides, but instead has been constrained by the exceedingly challenging nature of protein glycosylation analysis.5-11 While the term “glycoproteomics” has become frequently invoked in the scientific literature, the large scale, systemic examination of protein glycosylation has remained unrealized. Instead, the tremendous complexity of protein glycosylation compounded by the lack of sufficiently developed analytical capabilities have in practice necessitated a certain level of reductionism be applied to the analysis of protein glycosylation. Consequently, “glycoproteomics” has taken on a rather loose and broad definition and can be found to refer to studies of very different scales, depths, objectives, and outcomes. Essentially all such studies can be assigned to one of three categories of reductionist glycoproteomics: proteocentric, glycocentric, or monoproteic.
Proteocentric glycoproteomics generally involves the enrichment of glycan-bearing proteins (e.g., using affinity chromatography or covalent coupling chemistry) followed by standard proteomic analysis. The proteomic determinations may be carried out on the glycoproteins directly, or might be performed after the proteins have been deglycosylated. In the case of direct proteomic analysis, the glycan moieties are often simply disregarded (except for the fact that they are noted to be an impediment to protein identification). When deglycosylation is applied, the released oligosaccharides are usually discarded and the proteomic analysis is aimed towards protein identification and location of former sites of glycosylation. This is most commonly applied to N-linked glycosylation site analysis, wherein enzymatic digestion with peptide N-glycosidase F (PNGase F) results in the conversion of glycosylated asparagine residues into deglycosylated aspartic acid residues. Sites of asparagine to aspartic acid substitution occurring in the context of N-glycosylation consensus sequons are then construed as former sites of oligosaccharide occupancy. While these approaches do allow for the large-scale identification of glycosylated proteins (with or without deglycosylation) and mapping of glycosylation sites (with deglycosylation), all information regarding the general class and composition of the glycans is lost and the identities of oligosaccharides occupying a particular site remain obscure. Given the wide range of possible glycans and the relation of these carbohydrate moieties to an equally large array of metabolic states, the loss of this information severely limits the biological conclusions that may be drawn from such experiments. Proteocentric glycoproteomics has perhaps been most prominently typified by the hydrazide capture method of Aebersold and coworkers12 and the various modifications and applications thereof;13-20 however, many other works can be found to fall into this category.21-25 One such article provides a proteocentric definition of glycoproteomics: “…the identification of specific glycosylation sites on glycoproteins.”22
Glycocentric glycoproteomics is essentially the complement of proteocentric strategies, and is often perceived as falling within the realm of “glycomics” – a more generic term which not only applies to protein linked glycans, but to other glycoconjugates and free oligosaccharides as well. A broad but implicitly glycocentric definition of glycoproteomics can be found in a recent editorial: “…the study of the glycome attached to proteins.”26 In a large scale glycoproteomic study with a glycocentric emphasis, the identities of the glycosylated proteins are usually not of interest, while the classes, compositions, and structures of oligosaccharides released from proteins via PNGase F, β-elimination, or hydrazinolysis are studied in detail.27-33 Again, all information relating the observed glycans to specific proteins or specific sites is lost.
The third major variety of reductionist glycoproteomics is monoproteic in nature. Most commonly, these studies involve proteolytic digestion of a given protein and analysis of the resulting glycopeptides, with the glycans remaining covalently attached to their respective peptide moieties throughout the analysis. Studies of this sort are characterized by more detailed treatment of protein glycosylation and are usually performed with site-specific rigor, thus allowing for association of specific glycans with particular amino acid residues. This level of depth is achieved at the expense of breadth, as work of this type is usually directed at either a single protein or a very simple mixture of proteins.34-44
Taken together, these differently focused approaches to glycoproteomics delineate the present state of the art. Current technologies allow identification of many glycoproteins; characterization of the glycans occurring on many proteins; or more detailed studies of site-specific glycosylation and microheterogeneity for single or very few proteins. Ideally, a truly systems-oriented glycoproteomic analysis would encompass all of these aspects in single experiment or workflow; however, at present this is not feasible. Nonetheless, there are a number of technologies that have potential to surpass the need for reductionist approaches to glycoproteomics. One approach with significant promise for achieving more comprehensive glycoproteomics entails nonspecific enzymatic proteolysis of glycosylated proteins and analysis of the resulting glycopeptides by mass spectrometry (MS). As previously demonstrated by this laboratory45-47 and others,48-52 a highly effective agent for this purpose is pronase - a mixture of proteases exhibiting several different endoprotease and exoprotease activities that in concert are able to hydrolyze virtually any peptide bond. This method of preparing glycopeptides from glycoproteins has several advantages over the use of tryptic digestion. Because glycan modifications sterically interfere with protease action, the peptide bonds adjacent to sites of glycosylation are hydrolyzed at a significantly reduced rate relative to those of nonglycosylated polypeptide regions. This results in a final glycopeptide preparation that is free of interfering nonglycosylated peptides and instead contains only glycopeptides and free amino acids, with a small proportion of dipeptides. This provides a sample with far less complexity than products of tryptic digestion, which contain a mixture of glycosylated and nonglycosylated peptides. Hence, tryptic digestion for glycopeptide preparation further complicates an already difficult analysis and introduces the necessity to fractionate glycosylated and nonglycosylated peptides. Moreover, the common problem of missed tryptic cleavages in the vicinity of glycosylation sites tends to produce glycopeptides with rather large peptide moieties, thus impeding glycopeptide compositional determination and often precluding site-specific glycosylation assignment.
A key refinement to the nonspecific proteolysis approach has been recently introduced by this laboratory46 and Temporini et al.51 - the immobilization the pronase proteases. Covalent linkage of the enzymes confers the added benefits of reduced digestion time, prevention of interfering enzyme autolysis products, and facile removal of the enzymes from the digest. This renders the digest amenable to direct analysis by MS using electrospray ionization (ESI) following simple dilution, provided the digestion is performed in an ESI compatible buffer. Although nonspecific proteolysis has thus far served primarily as a platform for monoproteic analysis, it has not escaped our notice that these characteristics are highly desirable from the standpoint of developing a robust, high throughput platform for comprehensive glycoproteomics. As a step towards this objective, the analytical performance of bead immobilized pronase for protein glycosylation profiling is optimized and described. The method has been assessed in terms of enzyme immobilization efficiency, bead coupling capacity, proteolytic activity, digestion reproducibility, and minimum amount of glycoprotein required for characterization. These issues are examined in the context of increasingly complex glycoprotein systems, including mixtures of nonglycosylated, multiply O-glycosylated, and multiply N-glycosylated proteins.
Experimental Section
Enzyme Immobilization
Pronase E (PE) proteases were covalently linked to cyanogen bromide (CNBr) activated sepharose 4B (S4B) beads of 40-165 μm diameter using well established coupling chemistry.53 The PE and S4B beads were both acquired from Sigma (St. Louis, MO). The enzyme immobilization was carried out using an abbreviated version of the protocol reported by Clowers et al.46 and Seipert et al.47, 54 For this expedited coupling procedure, 15, 50, 100, or 150 mg portions of lyophilized S4B beads were weighed into screw cap microfuge tubes. The actual mass of S4B beads dispensed to each tube was recorded to the nearest 0.1 mg. The beads were reconstituted and washed in approximately 1 mL of 1 mM HCl with gentle agitation for 30 min. Each suspension was then centrifuged, and the supernatants were drawn off and discarded. All bead preparations were next washed three times with 500 μL aliquots of 100 mM total phosphate buffer (pH 7.4). After the final washing was removed, the beads were treated with PE solution and fresh phosphate buffer to a total volume of 600 μL. A solution of either 10 mg/mL or 20 mg/mL PE in deionized water was used, depending on the desired mass of PE to be delivered. Either 1 mg, 5 mg, or 10 mg PE was introduced to each aliquot of beads for immobilization. The beads were gently agitated at room temperature for 2 h to allow for enzyme coupling. The supernatants from the coupling reactions were then collected and stored at −20 °C for subsequent assay of residual, non-coupled PE. Remaining CNBr activated sites were then blocked by addition of 500 μL of 1 M aqueous ethanolamine (pH 9.0) and were agitated at room temperature for 2 h. After removing the blocking reaction supernatant, all beads were washed three times with 500 μL portions of 100 mM NH4OAc (pH 7.4). Coupled and washed PE/S4B beads were stored at 4 °C in 500 μL of 100 mM NH4OAc with 0.05% w/w NaN3 (pH 7.4). It should be noted that the coupling procedure was found to be scalable to tenfold greater quantities with no noticeable loss of effectiveness.
Determination of Residual Free Pronase
All coupling supernatants were analyzed for total protein content using a standard Bradford assay. Each assay was performed at a 3.1 mL scale by combining 100 μL coupling supernatant with 3 mL Bradford reagent (Sigma). After allowing 10 min for development, absorbance was measured at 596 nm using a Hewlett-Packard 8452 ultraviolet-visible diode array spectrophotometer (Santa Clara, CA). The concentration of protein was calculated according to a standard curve derived from analysis of bovine serum albumin (BSA) standards prepared in the same manner. A 1.0 mg/mL quantitative BSA standard solution (Sigma) and several dilutions thereof in phosphate buffer served to span the concentration range of 100–1000 μg/mL. Any coupling supernatants exceeding the standard curve range were diluted as necessary in phosphate buffer and reanalyzed. Once the residual PE concentration was determined for each coupling supernatant, the amount of successfully immobilized PE was calculated by difference.
Glycoprotein Digestion
Several model glycoproteins were obtained from commercial sources. Bovine ribonuclease B (RNase B; SwissProt accession P61823), bovine κ-casein (κ-CN; SwissProt accession P02668), chicken egg albumin (CEA; SwissProt accession P01012), and human apo-transferrin (HAT; SwissProt accession P02787) were obtained from Sigma. Bovine fetuin (BF; SwissProt accession P12763) was purchased from CalBiochem (San Diego, CA). Before glycoprotein digestion, each PE/S4B bead preparation was washed three times with 500 μL portions of ammonium acetate buffer. Glycoproteins were dissolved in the ammonium acetate buffer at an initial concentration of 10 μg/μL and in some cases were further diluted to 10-100 ng/μL depending on the desired amount for digestion. A stock mixture of RNase B, κ-CN, BF, and BSA (Sigma) was also prepared in ammonium acetate buffer at a concentration of 1 μg/μL in each protein. Additional ammonium acetate buffer and glycoprotein solution (total volume, 250-500 μL) were added to PE/S4B bead preparations and incubated at 37°C with gentle agitation for varying intervals (ranging from 30 min to 24 h). Digestion blanks containing no glycoprotein were prepared and incubated in the same manner. Supernatants were sampled following centrifugation and were then stored at −20°C until the time of analysis. PE/S4B beads were washed three times with ammonium acetate buffer and stored at 4°C in ammonium acetate / sodium azide buffer for later use.
Sample Preparation and Mass Spectrometry
Prior to MS analysis, digestion supernatants were either diluted or concentrated depending on the quantity of glycoprotein digested. Dilutions were typically at least 10 fold, and were done to result in a final solution composition of 50% aqueous acetonitrile (ACN) / 0.1% formic acid (FA). For low concentration glycoprotein digests, the supernatants were reduced to dryness in a vacuum centrifuge and reconstituted in 25-50 μL deionized water. To compensate for the increased concentration of buffer salts, the digests were then purified by solid-phase extraction using 10 μL Glygen NuTips loaded with graphitic carbon (Columbia, MD). The desalted glycopeptides were eluted from the tips in 10-20 μL 50% ACN / 0.1% FA. For nanoflow electrospray ionization (nESI), glycopeptide solutions were delivered using a syringe pump through a 25 μm inner diameter (ID) fused silica capillary tubing to a New Objective Picoview nESI source (Woburn, MA) at a rate of 100-200 nL/min. Spray voltages of 1600-2200 V were applied to the sample flow path via a liquid junction device. Nanospray emitters fashioned from 75 μm ID fused silica capillary tubing were pulled in-house using a butane torch and were thoroughly cleaned with methanol before use. MS analysis was conducted using a 9.4 T IonSpec QFT FTICR-MS instrument (Lake Forest, CA) fitted with a Waters Z-spray source interface (Manchester, UK). Nanosprayed ions were externally accumulated in a source region hexapole, then transferred through an RF-only quadrupole ion guide to the ICR cell for high resolution, accurate mass analysis. Mass spectra were externally calibrated using a series of maltooligosaccharides as previously described.55
Data Reduction
Calibrated mass spectra were thresholded, deconvoluted, and deisotoped using the IonSpec PeakHunter software package. The resulting peak lists were exported for further analysis using GP Finder, an in-house software package programmed and implemented in IGOR Pro (Version 6, WaveMetrics, Lake Oswego, OR). As described previously,46 GP Finder combines known protein sequence data with monosaccharide composition constraints to search experimental peak lists for potential glycopeptide masses. Monosaccharide composition settings were stringently set to search for specific classes of glycans and minimize the incidence of nonsensical matches. No glycopeptide assignments were made for potential matches having mass errors in excess of 10 ppm. Furthermore, no glycopeptide assignments were made in the absence of complementary glycan compositions occurring on the same peptide tag (which was expected due to the natural site heterogeneity of protein glycosylation), or complementary peptide tags occurring with the same glycan attached (which was expected due to the nonspecific nature of pronase digestion).
Results and Discussion
Reproducibility and Long-Term Stability
Two useful characteristics of pronase coupled beads are their stability during storage and their reusability. These characteristics enable digestions to be carried out in a number of different formats (i.e., cartridge, spin column, 96 well plate, or flow through enzyme reactor) with multiple uses and following several weeks of storage. This furnishes the possibility of high throughput and very low cost glycoprotein digestion. Furthermore, the protease functionalized beads need not be prepared immediately before use, but can be coupled in advance for on demand glycoprotein digestion. In order to assess the extent to which immobilized pronase could be reliably stored and reused, 1 mg digests of RNase B and κ-CN were performed periodically over the course of six weeks using 1 mg PE / 150 mg S4B beads. All digestions were incubated for 24 h, and the same bead preparations were used on each occasion. Portions of each digest were diluted 10 fold prior to nESI-FTICR-MS analysis. As demonstrated in Figure 1, both the RNase B and κ-CN digests remained highly reproducible for six weeks, with all but the most minor components being faithfully replicated at each time point. All of the major peaks corresponded to well characterized RNase B and κ-CN glycopeptides which have been described in detail elsewhere.46 In the case of RNase B, a 16.5 kDa glycoprotein which carries high mannose type N-glycans at a single site of glycosylation,56 the observed glycopeptides corresponded to glycans composed of N-acetylglucosamine (GlcNAc) and mannose (Man) residues (GlcNAc2 Man5 through GlcNAc2 Man9) attached to dipeptides as well as single asparagine residues. For κ-CN, a 21.2 kDa glycoprotein which harbors core 1 type O-glycans at several C-terminal domain sites,57, 58 the observed glycopeptides contained O-glycans composed of N-acetylgalactosamine (GalNAc), galactose (Gal), and N-acetylneuraminic acid (NeuAc) residues (GalNAc1 Gal1, GalNAc1 Gal1 NeuAc1, and GalNAc1 Gal1 NeuAc2) linked primarily to tetra- and pentapeptides. After 10 weekly cycles of digestion and storage, the activity of the pronase beads was found to be slightly diminished. Notably, no additional signals appeared in the glycoprotein digests over the course of six weeks, indicating a lack of any detectable protease autolysis. Autolytic peptides were not detected in supernatants from blank incubations conducted for up to 24 h using pronase coupled beads one week and 10 weeks post conjugation (data not shown). All blank supernatants were prepared in the same manner as the glycoprotein digests.
Figure 1.
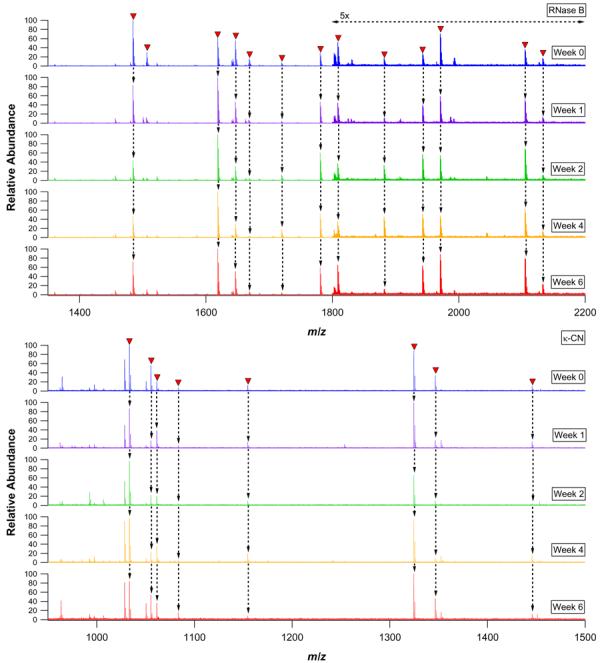
nESI-FTICR mass spectra of 24 h RNase B (upper panel) and κ-CN (lower panel) pronase bead digests performed using the same bead preparations at several intervals over the course of six weeks. Glycopeptide signals are labeled with closed triangles and traced with arrows to the spectra below.
Minimum Protein Quantity Required for Digestion
While pronase digestion offers a number of advantages for protein glycosylation profiling, one potential limitation lies in reduced sensitivity towards each glycosylation site as compared to specific digestion. Because glycosylation is by nature a heterogeneous modification, the glycopeptides derived from specific proteolysis are expected to be represented by a single peptide moiety linked to several different glycans. Thus, for a particular glycosylation site having five glycoforms, the observance of five glycopeptides is anticipated. By contrast, when glycoproteins are nonspecifically digested, each glycosylation site is represented by a population of glycopeptides that results from a convolution of glycan and peptide heterogeneities. For example, a site of glycosylation having five glycoforms and represented in a digest by dipeptides and tripeptides (as can occur in pronase digests) could be divided into as many as 25 distinct glycopeptides. Although the peptide tag heterogeneity confers additional confidence when assigning sites of glycan modification, the dual heterogeneity compounds the division of a given glycosite into a greater number of distinct glycopeptides, each with reduced molar contribution. On the balance, pronase digestion eliminates nonglycosylated peptides that would otherwise cause signal suppression of glycosylated peptides in tryptic preparations. This ostensible improvement in glycopeptide ionization efficiency should somewhat offset any stoichiometric disadvantage.
To determine the lowest quantity of glycoprotein required for glycoprofiling, decreasingly concentrated solutions of RNase B were digested for 24 h using 1 mg PE / 150 mg S4B bead preparations. Figure 2 provides mass spectra of glycopeptides derived from as little as 100 ng (∼7 pmol) RNase B. Following concentration and desalting, and accounting for sample infusion rate and MS scan time (10-20 s/scan), each spectrum represents only about 3.5-7.0% consumption of the total digest. Thus, the spectrum for the 100 ng (7 pmol) digest was acquired based on the consumption of 500 fmol RNase B digest. The 10 μg digest yielded 16 glycopeptides, all of which have been previously observed and described in detail.46 At the 1 μg level, nine glycopeptides were observed, and at the 100 ng level, only six glycopeptides could still be detected. In each digest, the observed glycopeptides were accounted for as primarily dipeptides linked to the series of five high mannose type N-oligosaccharides.
Figure 2.

nESI-FTICR mass spectra of 10 μg, 1 μg, and 100 ng RNase B digests. Glycopeptide signals are labeled with closed triangles. Each trace represents the summation of 10 MS scans.
Model Glycoproteins with Multiple Potential Glycosylation Sites
To further explore the efficacy of bead immobilized pronase for site specific glycoprofiling, several proteins with increasingly complex glycosylation were subjected to pronase bead digestion and direct nESI-FTICR-MS analysis. These included: CEA, a 42.9 kDa glycoprotein with two potential N-glycosylation sites and a known population of high mannose and hybrid type N-glycans;59-61 HAT, a 77.1 kDa glycoprotein having two consensus N-glycosylation sequons and carrying sialylated complex type N-linked oligosaccharides;62-65 and BF, a 38.4 kDa glycoprotein containing three potential sites of N-glycan attachment, an array of sialylated complex type N-glycans, and several known and suspected sites of core 1 and core 2 type O-glycan modification.66-69
The pronase bead digests of these glycoproteins are presented in Figure 3. The glycopeptides observed in the CEA digest could all be assigned to dipeptides and tripeptides with high mannose and hybrid type N-oligosaccharides linked to 293N (Supplementary Table 1). As expected based on previous studies, no evidence of glycosylation was found at the second potential N-linked site, 312N.
Figure 3.
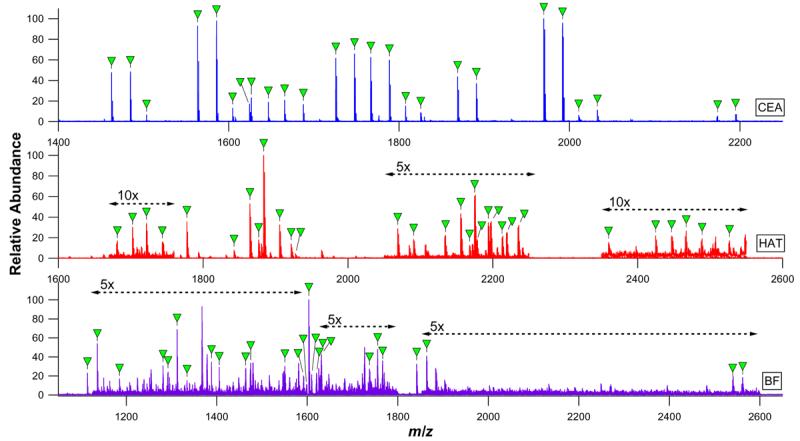
nESI-FTICR mass spectra of CEA, HAT, and BF digests. Glycopeptide signals are labeled with closed triangles and correspond to the glycopeptide compositions listed in Supplementary Tables 1 through 3.
The HAT glycopeptide preparation was found to contain a mixture of glycosylated asparagine residues, dipeptides, and tripeptides, with the corresponding glycan moieties being assigned to nonsialylated, monosialylated, and disialylated complex type N-linked glycans (Supplementary Table 2). While the peptide sequence tags remaining attached to these glycopeptides were small, their compositions were sufficient to allow confirmation of HAT glycosylation at both potential sites: 432N and 430N.
As anticipated, glycopeptides derived from BF were of both the O-linked and the N-linked classes (Supplementary Table 3). Monosialylated and disialylated O-glycans were very clearly associated with 271S, but were ambiguously associated with 280T and 282S due to the close proximity of the two sites and the lack of any observed peptide tags encompassing only one of the sites. Glycopeptides carrying glycans simultaneously at both sites were not observed, although glycosylation at either or both sites in different glycoforms of the protein cannot be ruled out based on this result alone. O-glycosylation at 341S, a suspected glycosylation site, was not observed. Notably, the O-linked glycopeptides were found to have peptide footprints comprised of five to seven amino acid residues, while the N-linked glycopeptides observed in the same digest were exclusively linked to tripeptides. This finding is consistent with results for κ-CN O-glycopeptides, which in this work and previous work46 were found to be degraded to tetrapeptide tags at the smallest. This result is in contrast to N-linked glycopeptides, which have been repeatedly shown to be reducible to tripeptides, dipeptides, and single asparagine residues. The occurrence of the two behaviors within the same digest as observed here is particularly striking, and suggests that the locally reduced pronase susceptibility conferred by N-linked and O-linked glycans is in some way distinct. The N-linked complement of BF glycopeptides was mapped to 156N, 176N, both of which were found to be occupied by biantennary and triantennary complex type oligosaccharides decorated by two to four sialic acid residues.
In sum, these results illustrate that nonspecific proteolysis is readily applicable to a wide assortment of glycoproteins having different sizes and types of glycosylation. Another noteworthy difference among these glycoproteins is the varying number of disulfide bridges: one in the case of CEA, six in the case of BF, and 19 in the case of HAT. All of these proteins were efficiently digested despite the lack of any reductive treatment prior to proteolysis.
Pronase Coupling Efficiency and Activity
Considering the downstream objective of comprehensive glycoprofiling of protein mixtures, the need for ever more rapid and efficient proteolysis is evident. Even if some degree of glycoprotein enrichment and fractionation is applied to a mixture prior to proteolysis, a large number of glycosylated proteins are expected to remain against a background of nonglycosylated proteins. A further complication is manifest in the widely varying proteolytic susceptibilities among glycoproteins. To address these complexities, the coupling of varying ratios of PE and S4B was systematically explored with respect to protease immobilization efficiency, protease capacity of the beads, and protease activity.
All experiments described thus far were conducted using an amount of pronase similar to that which would customarily be used in a free enzyme digest. However, with enzyme immobilization comes the opportunity to use far more pronase than would otherwise be possible or even desirable. The limit of enzyme solubility - which in the case of PE is approximately 20 mg/mL - ultimately restricts the amount of protease that can be used in a free enzyme digestion. Moreover, including large amounts of free enzyme would undoubtedly lead to the production of many protease-protease digestion products, thus complicating the digest while leading to protease inactivation. The drawbacks associated with both autolysis and the accompanying enzyme inactivation are avoided through the use of immobilized proteases. Furthermore, the immobilization of the enzymes allows quantities of protease to be used in a given reaction that surpass the quantity of free enzyme that could be dissolved in solution.
The efficiency for coupling of different quantities of PE to varying amounts of S4B beads is shown in Figure 4. This result illustrates the capacity to couple considerably more than the 1 mg PE per 150 mg S4B, as has typically been performed for this procedure. Indeed, the reaction of 10 mg PE with only 15 mg S4B is about 70% efficient, yielding pronase coupled beads that are approximately one third enzyme by mass (0.45 mg coupled PE per mg S4B dry mass). This corresponds to an effective PE concentration of 135 mg/mL, which represents a nearly seven fold increase in PE concentration as compared to the maximum aqueous solubility of PE. While maximizing the amount of immobilized pronase should provide higher proteolytic activity, minimizing the quantity of beads in the digestion reduces the possibility of protein loss through nonspecific interaction with the beads.
Figure 4.
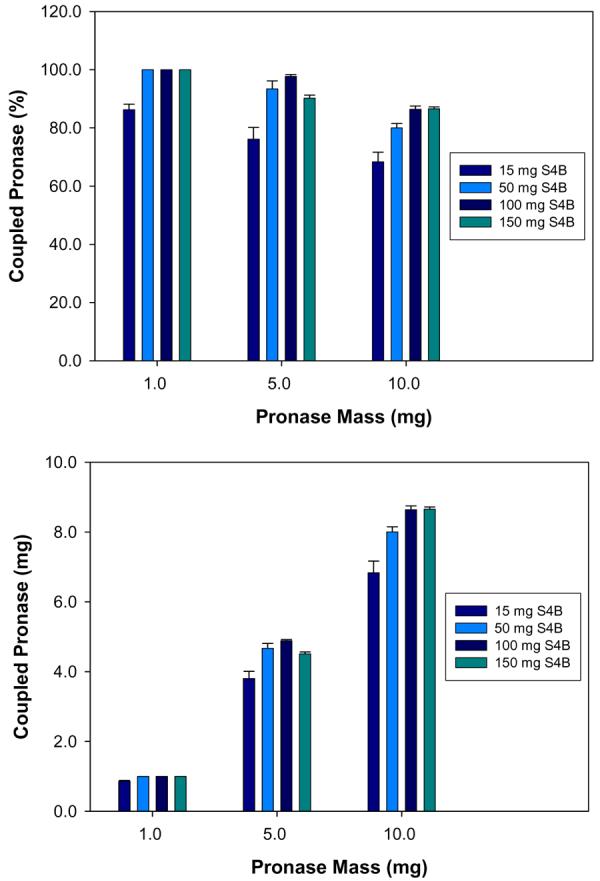
Mean coupling efficiencies of differing masses of PE to varying amounts of S4B beads. Error bars represent the standard deviation of four coupling reactions. The amount of successfully coupled PE is expressed in both relative (upper panel) and absolute (lower panel) terms.
In order to determine whether the apparently increased quantity of coupled pronase would be effective in accelerating the digestion of glycoproteins, RNase B and κ-CN were incubated for abbreviated times with 150 mg portions of S4B that had been coupled to either 1 mg, 5 mg, or 10 mg PE. As seen in Figure 5, only the intact RNase B glycoforms with GlcNAc2 Man5 (at z = 7+ and 6+) and GlcNAc2 Man6 (at z = 7+) were detected when digestion was performed for 30 min with 1 mg PE. When κ-CN was digested for 120 min with 1 mg PE, the major observed species was a multiply charged polypeptide with unknown composition (z = 11 through z = 8). However, when the digestions were performed for the same duration but with 5 mg and 10 mg of coupled PE, well known and previously observed46 glycopeptides appeared, with the concomitant depletion of the intact protein and large polypeptides. Thus, the efficient conjugation of large amounts of pronase to sepharose beads allows for accelerated glycoprotein profiling and should also provide advantages when digesting larger, more complicated glycoproteins or glycoprotein mixtures.
Figure 5.
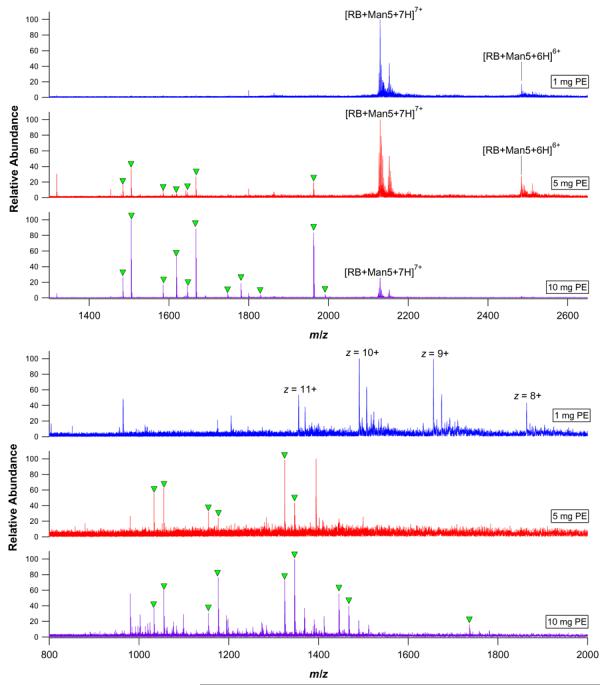
nESI-FTICR mass spectra of immobilized pronase digests of RNase B with 30 minutes of incubation (upper panel) and immobilized pronase digests of κ-CN with 120 minutes of incubation (lower panel). The digests using 1 mg, 5 mg, or 10 mg of PE are shown. Well known glycopeptides are labeled with closed triangles.
Digestion and Analysis of a Glycoprotein Mixture
A cocktail of the three glycoproteins RNase B, κ-CN, and BF was prepared and supplemented with the nonglycosylated protein BSA. The mixture was then digested using 10 mg PE / 150 mg S4B beads. The resulting digests were analyzed with the goal of establishing whether a more complex glycopeptide mixture could be adequately characterized using the current approach. The resulting digest of this mixture was expected to provide high mannose and complex type N-linked glycopeptides in addition to core 1 and core 2 type O-linked glycopeptides. Another question addressed by this experiment was whether a reasonably clean glycopeptide preparation could still be achieved despite the burdening of the mixture with a significant proportion of nonglycosylated protein.
The nESI-FTICR mass spectrum resulting from direct analysis of the unfractionated, unpurified protein mixture digest is provided in Figure 6. A total of 31 putative glycopeptide assignments could be made, with representation of all glycoproteins present in the mixture (Supplementary Table 4). These assignments accounted for most of the major signals (although a small number of peaks remained unaccounted for). Thus, the presence of BSA did not appear to have contributed any appreciable interference or complication of the digest. Most of the assigned glycopeptides were mapped to RNase B, which was not unexpected due to the relatively high mole fraction of this protein in the mixture. Nonetheless, several higher molecular weight glycopeptides mapping to BF were also detected (e.g., m/z 1895.6805 with z = 2, assigned as the [M+H+Na]2+ ion of 153APLNDS158 + HexNAc5 Hex6 NeuAc4). As expected, assignment of the O-linked glycopeptides was subject to greater complication owing to the lack of a consensus O-glycosylation sequon. Specifically, two potential O-linked glycopeptide compositions were found to have two potential assignments. For example, the glycopeptide at m/z 1312.5545 could be assigned either to the [M+Na]+ ion of the κ-CN glycopeptide 182(T)VQVTS(T)188 + HexNAc1 Hex1 NeuAc1 (3.2 ppm mass error), or the [M+H]+ ion of the BF glycopeptide 269(A)PSAVPD(A)276 + HexNAc1 Hex1 NeuAc1 (1.5 ppm mass error). A related signal corresponding to a glycoform having one additional NeuAc residue was also detected at m/z 1603.6481. While in a separate experiment 271S on BF was found to contribute significantly to the observed glycopeptide population (see Figure 3 and Supplementary Table 3), glycosylation on 186T of κ-CN has not been observed here or in previous studies.46, 58 Therefore, assignment of this signal to the κ-CN glycopeptide seems unlikely. While in this case it was possible to draw on other available information to provisionally assign this O-linked BF glycopeptide, this situation underscores the possibility for coincidence of even very accurately measured masses with multiple potential glycopeptide compositions. More comprehensive glycoproteomics will require that the presently described approach be used in concert with additional analytical dimensions (i.e., chromatography and tandem mass spectrometry); nonetheless, these experiments demonstrate that bead immobilized pronase digestion of glycoprotein mixtures is in principle capable of generating representative glycopeptides from a complex mixture of glycoproteins. Further, this can be accomplished with little or no interference by nonglycosylated peptides despite the large amount of proteases used, and despite the presence of a background nonglycosylated protein.
Figure 6.

nESI-FTICR-MS analysis of a 1:1:1:1 (by mass) mixture of RNase B, κ-CN, BF, and the nonglycosylated protein BSA. Glycopeptide signals are labeled with closed triangles and correspond to the glycopeptide compositions listed in Supplementary Table 4.
Conclusions
The development of versatile, efficient, and robust analytical tools remains an important step towards the realization of comprehensive, systems oriented glycoproteomics. Achieving this order of breadth and depth in glycoproteomic research will undoubtedly require the integration of several complementary preparative, analytical, and bioinformatic tools. On the basis of the present findings, nonspecific proteolysis with immobilized pronase can play a pivotal role as a new technology for large scale glycoproteomics. Glycopeptide footprinting using immobilized pronase has been demonstrated to produce high quality glycopeptide preparations with high reproducibility. The immobilization of the proteolytic enzymes has also been found to allow a 100 to 1000 fold reduction in the quantity of glycoprotein required for characterization when compared to the 0.1 mg to 1.0 mg quantities commonly used for conventional pronase digestion.45, 48, 50-52 Furthermore, pronase can be effectively immobilized in an approximately 70-fold greater enzyme to bead mass ratio than previously reported,46 allowing effective pronase concentrations that are approximately seven fold greater than the maximum aqueous solubility of free pronase with commensurate increases in digestion rate. This allows informative glycopeptides to be produced in as little as 30 minutes, or approximately 100 times faster than the up to 48 hour digestions customary with free pronase.45, 48 Importantly, the increased densities of immobilized pronase were achieved without introducing any detectable levels of contaminating autolysis. In concert, these features of immobilized pronase have enabled an initial example of glycopeptide footprinting in a mixture of glycosylated and non-glycosylated proteins with both N-linked and O-linked glycan modifications present. This provides the first evidence that pronase digestion is applicable to studies much more complex than monoproteic. Moreover, the result demonstrates that the method is tolerant of significant background levels of non-glycosylated protein. By virtue of immobilization, pronase digestion becomes amenable to implementation in a number of more convenient and higher throughput formats (i.e., cartridge, spin column, 96 well plate, or flow through enzyme reactor). The benefits afforded by enzyme immobilization add to the basic advantages of nonspecific proteolysis, including the tunability of glycosylated peptide length and the ability to circumvent protein denaturation, reduction, and alkylation steps.
These advantages notwithstanding, there are two important caveats to the use of pronase for glycopeptide footprinting analysis. First, the natural heterogeneity of glycans at a specific site is compounded by peptide length heterogeneity resulting from nonspecific proteolysis, thus potentially reducing the effective sensitivity of the method. This being said, a digest resulting from several picomoles of glycoprotein can still yield useful information. It should not be overlooked that the peptide tag heterogeneity, while perhaps a disadvantage from the standpoint of sensitivity, is advantageous in that the observation of multiple peptide tags implicating a single site of glycosylation provides additional confidence in glycopeptide assignments. Furthermore, the elimination of non-glycosylated peptides minimizes ion suppression of the glycosylated peptides – a factor that reduces any presumptive stoichiometric disadvantage. The second compromise of nonspecific proteolysis is that no peptide digestion constraints (e.g., tryptic cleavage rules) can be applied when searching for potential glycopeptide matches. Again, this complication is somewhat offset by the richness in information provided by sequential cleavage of amino acid residues about the site of glycan attachment.
When performing glycopeptide footprinting at the monoproteic level, the high mass accuracy and high resolution furnished by FTICR-MS are usually sufficient to allow direct analysis of glycoprotein digests. This sort of analysis should also be well within the capabilities of other high performance mass analyzers. To exhaustively assign glycopeptides from mixtures of greater complexity, the incorporation chromatographic separation will be necessary. In addition, the use of MS/MS to aid in accurate glycopeptide assignment will be essential; with this in mind, considerable effort has been invested in understanding the MS/MS dissociation pathways of pronase derived glycopeptides.47, 54 It should also be acknowledged that the relatively short peptide tags typically produced by nonspecific proteolysis might be of limited use for identifying a glycosylated protein in a mixture; however, it is possible to obtain longer peptide tags by adjusting the digestion time. Thus, it is possible that larger pronase glycopeptides could play a role in confirming protein identity. Another option for identifying the glycoproteins in a complex mixture would be to perform a standard tryptic digestion and protein identification experiment in parallel with immobilized pronase based glycopeptide footprinting. When used in concert with additional appropriate technologies, glycopeptide footprinting with immobilized pronase can eventually surpass the present limitations of conventional reductionist glycoproteomics.
Supplementary Material
Acknowledgments
Funding for this work was provided by the National Institutes of Health (GM49077), the California Dairy Research Foundation (06LEC-01-NH), UC Discovery (05GEB01NHB), and Dairy Management Incorporated.
Footnotes
Supporting Information Available
Supplementary Table 1: N-linked glycopeptides derived from immobilized pronase digestion of CEA. Supplementary Table 2: N-linked glycopeptides derived from immobilized pronase digestion of HAT. Supplementary Table 3: N-linked and O-linked glycopeptides derived from immobilized pronase digestion of BF. Supplementary Table 4: N-linked and O-linked glycopeptides derived from immobilized pronase digestion of a mixture of RNase B, κ-CN, BF, and the nonglycosylated protein BSA.
References
- 1.Varki A. Biological roles of oligosaccharides - all of the theories are correct. Glycobiology. 1993;3:97–130. doi: 10.1093/glycob/3.2.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dwek RA. Glycobiology: toward understanding the function of sugars. Chemical Reviews. 1996;96:683–720. doi: 10.1021/cr940283b. [DOI] [PubMed] [Google Scholar]
- 3.Rudd PM, Dwek RA. Glycosylation: heterogeneity and the 3D structure of proteins. Critical Reviews in Biochemistry and Molecular Biology. 1997;32:1–100. doi: 10.3109/10409239709085144. [DOI] [PubMed] [Google Scholar]
- 4.Spiro RG. Protein glycosylation: nature, distribution, enzymatic formation, and disease implications of glycopeptide bonds. Glycobiology. 2002;12:43r–56r. doi: 10.1093/glycob/12.4.43r. [DOI] [PubMed] [Google Scholar]
- 5.Wuhrer M, Deelder AM, Hokke CH. Protein glycosylation analysis by liquid chromatography-mass spectrometry. Journal of Chromatography B. 2005;825:124–133. doi: 10.1016/j.jchromb.2005.01.030. [DOI] [PubMed] [Google Scholar]
- 6.Morelle W, Canis K, Chirat F, Faid V, Michalski JC. The use of mass spectrometry for the proteomic analysis of glycosylation. Proteomics. 2006;6:3993–4015. doi: 10.1002/pmic.200600129. [DOI] [PubMed] [Google Scholar]
- 7.Morelle W, Michalski JC. Analysis of protein glycosylation by mass spectrometry. Nature Protocols. 2007;2:1585–1602. doi: 10.1038/nprot.2007.227. [DOI] [PubMed] [Google Scholar]
- 8.Morris HR, Chalabi S, Panico M, Sutton-Smith M, Clark GF, Goldberg D, Dell A. Glycoproteomics: past, present and future. International Journal of Mass Spectrometry. 2007;259:16–31. [Google Scholar]
- 9.Wuhrer M, Catalina MI, Deelder AM, Hokke CH. Glycoproteomics based on tandem mass spectrometry of glycopeptides. Journal of Chromatography B. 2007;849:115–128. doi: 10.1016/j.jchromb.2006.09.041. [DOI] [PubMed] [Google Scholar]
- 10.Dalpathado DS, Desaire H. Glycopeptide analysis by mass spectrometry. Analyst. 2008;133:731–738. doi: 10.1039/b713816d. [DOI] [PubMed] [Google Scholar]
- 11.Temporini C, Calleri E, Massolini G, Caccialanza G. Integrated analytical strategies for the study of phosphorylation and glycosylation in proteins. Mass Spectrometry Reviews. 2008;27:207–236. doi: 10.1002/mas.20164. [DOI] [PubMed] [Google Scholar]
- 12.Zhang H, Li XJ, Martin DB, Aebersold R. Identification and quantification of N-linked glycoproteins using hydrazide chemistry, stable isotope labeling and mass spectrometry. Nature Biotechnology. 2003;21:660–666. doi: 10.1038/nbt827. [DOI] [PubMed] [Google Scholar]
- 13.Liu T, Qian WJ, Gritsenko MA, Camp DG, 2nd, Monroe ME, Moore RJ, Smith RD. Human plasma N-glycoproteome analysis by immunoaffinity subtraction, hydrazide chemistry, and mass spectrometry. Journal of Proteome Research. 2005;4:2070–2080. doi: 10.1021/pr0502065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lewandrowski U, Moebius J, Walter U, Sickmann A. Elucidation of N-glycosylation sites on human platelet proteins: a glycoproteomic approach. Molecular and Cellular Proteomics. 2006;5:226–233. doi: 10.1074/mcp.M500324-MCP200. [DOI] [PubMed] [Google Scholar]
- 15.Pan S, Wang Y, Quinn JF, Peskind ER, Waichunas D, Wimberger JT, Jin J, Li JG, Zhu D, Pan C, Zhang J. Identification of glycoproteins in human cerebrospinal fluid with a complementary proteomic approach. Journal of Proteome Research. 2006;5:2769–2779. doi: 10.1021/pr060251s. [DOI] [PubMed] [Google Scholar]
- 16.Ramachandran P, Boontheung P, Xie Y, Sondej M, Wong DT, Loo JA. Identification of N-linked glycoproteins in human saliva by glycoprotein capture and mass spectrometry. Journal of Proteome Research. 2006;5:1493–1503. doi: 10.1021/pr050492k. [DOI] [PubMed] [Google Scholar]
- 17.Bernhard OK, Kapp EA, Simpson RJ. Enhanced analysis of the mouse plasma proteome using cysteine-containing tryptic glycopeptides. Journal of Proteome Research. 2007;6:987–995. doi: 10.1021/pr0604559. [DOI] [PubMed] [Google Scholar]
- 18.Sun B, Ranish JA, Utleg AG, White JT, Yan X, Lin B, Hood L. Shotgun glycopeptide capture approach coupled with mass spectrometry for comprehensive glycoproteomics. Molecular and Cellular Proteomics. 2007;6:141–149. doi: 10.1074/mcp.T600046-MCP200. [DOI] [PubMed] [Google Scholar]
- 19.Tian Y, Zhou Y, Elliott S, Aebersold R, Zhang H. Solid-phase extraction of N-linked glycopeptides. Nature Protocols. 2007;2:334–339. doi: 10.1038/nprot.2007.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhou Y, Aebersold R, Zhang H. Isolation of N-linked glycopeptides from plasma. Analytical Chemistry. 2007;79:5826–5837. doi: 10.1021/ac0623181. [DOI] [PubMed] [Google Scholar]
- 21.Qiu R, Regnier FE. Comparative glycoproteomics of N-linked complex-type glycoforms containing sialic acid in human serum. Analytical Chemistry. 2005;77:7225–7231. doi: 10.1021/ac050554q. [DOI] [PubMed] [Google Scholar]
- 22.Alvarez-Manilla G, Atwood JA, Guo Y, Warren NL, Orlando R, Pierce M. Tools for glycoproteomic analysis: size exclusion chromatography facilitates identification of tryptic glycopeptides with N-linked glycosylation sites. Journal of Proteome Research. 2006;5:701–708. doi: 10.1021/pr050275j. [DOI] [PubMed] [Google Scholar]
- 23.Durham M, Regnier FE. Targeted glycoproteomics: serial lectin affinity chromatography in the selection of O-glycosylation sites on proteins from the human blood proteome. Journal of Chromatography A. 2006;1132:165–173. doi: 10.1016/j.chroma.2006.07.070. [DOI] [PubMed] [Google Scholar]
- 24.Schwientek T, Mandel U, Roth U, Müller S, Hanisch FG. A serial lectin approach to the mucin-type O-glycoproteome of Drosophila melanogaster S2 cells. Proteomics. 2007;7:3264–3277. doi: 10.1002/pmic.200600793. [DOI] [PubMed] [Google Scholar]
- 25.Xu Z, Zhou X, Lu H, Wu N, Zhao H, Zhang L, Zhang W, Liang YL, Wang L, Liu Y, Yang P, Zha X. Comparative glycoproteomics based on lectins affinity capture of N-linked glycoproteins from human Chang liver cells and MHCC97-H cells. Proteomics. 2007;7:2358–2370. doi: 10.1002/pmic.200600041. [DOI] [PubMed] [Google Scholar]
- 26.Packer N. Introducing Glycoproteomics, a new section of Proteomics. Proteomics. 2006;6:6121–6123. [Google Scholar]
- 27.Sagi D, Kienz P, Denecke J, Marquardt T, Peter-Katalinic J. Glycoproteomics of N-glycosylation by in-gel deglycosylation and matrix-assisted laser desorption/ionisation-time of flight mass spectrometry mapping: application to congenital disorders of glycosylation. Proteomics. 2005;5:2689–2701. doi: 10.1002/pmic.200401312. [DOI] [PubMed] [Google Scholar]
- 28.Hato M, Nakagawa H, Kurogochi M, Akama TO, Marth JD, Fukuda MN, Nishimura S-I. Unusual N-glycan structures in alpha-mannosidase II/IIx double null embryos identified by a systematic glycomics approach based on two-dimensional LC mapping and matrix-dependent selective fragmentation method in MALDI-TOF/TOF mass spectrometry. Molecular and Cellular Proteomics. 2006;5:2146–2157. doi: 10.1074/mcp.M600213-MCP200. [DOI] [PubMed] [Google Scholar]
- 29.Liu X, McNally DJ, Nothaft H, Szymanski CM, Brisson JR, Li J. Mass spectrometry-based glycomics strategy for exploring N-linked glycosylation in eukaryotes and bacteria. Analytical Chemistry. 2006;78:6081–6087. doi: 10.1021/ac060516m. [DOI] [PubMed] [Google Scholar]
- 30.Costello CE, Contado-Miller JM, Cipollo JF. A glycomics platform for the analysis of permethylated oligosaccharide alditols. Journal of the American Society for Mass Spectrometry. 2007;18:1799–1812. doi: 10.1016/j.jasms.2007.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jang-Lee J, Curwen RS, Ashton PD, Tissot B, Mathieson W, Panico M, Dell A, Wilson RA, Haslam SM. Glycomics analysis of schistosoma mansoni egg and cercarial secretions. Molecular and Cellular Proteomics. 2007;6:1485–1499. doi: 10.1074/mcp.M700004-MCP200. [DOI] [PubMed] [Google Scholar]
- 32.Llop E, Gallego RG, Belalcazar V, Gerwig GJ, Kamerling JP, Segura J, Pascual JA. Evaluation of protein N-glycosylation in 2-DE: erythropoietin as a study case. Proteomics. 2007;7:4278–4291. doi: 10.1002/pmic.200700572. [DOI] [PubMed] [Google Scholar]
- 33.Yamada K, Hyodo S, Matsuno Y.-k., Kinoshita M, Maruyama S.-z., Osaka Y.-s., Casal E, Lee YC, Kakehi K. Rapid and sensitive analysis of mucin-type glycans using an in-line flow glycan-releasing apparatus. Analytical Biochemistry. 2007;371:52–61. doi: 10.1016/j.ab.2007.06.013. [DOI] [PubMed] [Google Scholar]
- 34.Zhu X, Borchers C, Bienstock RJ, Tomer KB. Mass spectrometric characterization of the glycosylation pattern of HIV-gp120 expressed in CHO cells. Biochemistry. 2000;39:11194–11204. doi: 10.1021/bi000432m. [DOI] [PubMed] [Google Scholar]
- 35.Cutalo JM, Deterding LJ, Tomer KB. Characterization of glycopeptides from HIV-ISF2 gp120 by liquid chromatography mass spectrometry. Journal of the American Society for Mass Spectrometry. 2004;15:1545–1555. doi: 10.1016/j.jasms.2004.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wada Y, Tajiri M, Yoshida S. Hydrophilic affinity isolation and MALDI multiple-stage tandem mass spectrometry of glycopeptides for glycoproteomics. Analytical Chemistry. 2004;76:6560–6565. doi: 10.1021/ac049062o. [DOI] [PubMed] [Google Scholar]
- 37.Harazono A, Kawasaki N, Kawanishi T, Hayakawa T. Site-specific glycosylation analysis of human apolipoprotein B100 using LC/ESI MS/MS. Glycobiology. 2005;15:447–462. doi: 10.1093/glycob/cwi033. [DOI] [PubMed] [Google Scholar]
- 38.Tajiri M, Yoshida S, Wada Y. Differential analysis of site-specific glycans on plasma and cellular fibronectins: application of a hydrophilic affinity method for glycopeptide enrichment. Glycobiology. 2005;15:1332–1340. doi: 10.1093/glycob/cwj019. [DOI] [PubMed] [Google Scholar]
- 39.Harazono A, Kawasaki N, Itoh S, Hashii N, Ishii-Watabe A, Kawanishi T, Hayakawa T. Site-specific N-glycosylation analysis of human plasma ceruloplasmin using liquid chromatography with electrospray ionization tandem mass spectrometry. Analytical Biochemistry. 2006;348:259–268. doi: 10.1016/j.ab.2005.10.036. [DOI] [PubMed] [Google Scholar]
- 40.Snovida SI, Chen VC, Krokhin O, Perreault H. Isolation and identification of sialylated glycopeptides from bovine alpha-1-acid glycoprotein by off-line capillary electrophoresis MALDI-TOF mass spectrometry. Analytical Chemistry. 2006;78:6556–6563. doi: 10.1021/ac060738k. [DOI] [PubMed] [Google Scholar]
- 41.Go EP, Irungu J, Zhang Y, Dalpathado DS, Liao H-X, Sutherland LL, Alam SM, Haynes BF, Desaire H. Glycosylation site-specific analysis of HIV envelope proteins (JR-FL and CON-S) reveals major differences in glycosylation site occupancy, glycoform profiles, and antigenic epitopes' accessibility. Journal of Proteome Research. 2008;7:1660–1674. doi: 10.1021/pr7006957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Iacob RE, Perdivara I, Przybylski M, Tomer KB. Mass spectrometric characterization of glycosylation of hepatitis C virus E2 envelope glycoprotein reveals extended microheterogeneity of N-glycans. Journal of the American Society for Mass Spectrometry. 2008;19:428–444. doi: 10.1016/j.jasms.2007.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kolarich D, Weber A, Pabst M, Stadlmann J, Teschner W, Ehrlich H, Schwarz HP, Altmann F. Glycoproteomic characterization of butyrylcholinesterase from human plasma. Proteomics. 2008;8:254–263. doi: 10.1002/pmic.200700720. [DOI] [PubMed] [Google Scholar]
- 44.Zhang Y, Go EP, Desaire H. Maximizing coverage of glycosylation heterogeneity in MALDI-MS analysis of glycoproteins with up to 27 glycosylation sites. Analytical Chemistry. 2008;80:3144–3158. doi: 10.1021/ac702081a. [DOI] [PubMed] [Google Scholar]
- 45.An HJ, Peavy TR, Hedrick JL, Lebrilla CB. Determination of N-glycosylation sites and site heterogeneity in glycoproteins. Analytical Chemistry. 2003;75:5628–5637. doi: 10.1021/ac034414x. [DOI] [PubMed] [Google Scholar]
- 46.Clowers BH, Dodds ED, Seipert RR, Lebrilla CB. Site determination of protein glycosylation based on digestion with immobilized nonspecific proteases and Fourier transform ion cyclotron resonance mass spectrometry. Journal of Proteome Research. 2007;6:4032–4040. doi: 10.1021/pr070317z. [DOI] [PubMed] [Google Scholar]
- 47.Seipert RR, Dodds ED, Clowers BH, Beecroft SM, German JB, Lebrilla CB. Factors that influence fragmentation behavior of N-linked glycopeptide ions. Analytical Chemistry. 2008;80:3684–3692. doi: 10.1021/ac800067y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Juhasz P, Martin SA. The utility of nonspecific proteases in the characterization of glycoproteins by high-resolution time-of-flight mass spectrometry. International Journal of Mass Spectrometry. 1997;169:217–230. [Google Scholar]
- 49.Wuhrer M, Koeleman CAM, Hokke CH, Deelder AM. Protein glycosylation analyzed by normal-phase nano-liquid chromatography-mass spectrometry of glycopeptides. Analytical Chemistry. 2005;77:886–894. doi: 10.1021/ac048619x. [DOI] [PubMed] [Google Scholar]
- 50.Wuhrer M, Balog CI, Koeleman CA, Deelder AM, Hokke CH. New features of site-specific horseradish peroxidase (HRP) glycosylation uncovered by nano-LC-MS with repeated ion-isolation/fragmentation cycles. Biochimica et Biophysica Acta. 2005;1723:229–239. doi: 10.1016/j.bbagen.2005.02.013. [DOI] [PubMed] [Google Scholar]
- 51.Temporini C, Perani E, Calleri E, Dolcini L, Lubda D, Caccialanza G, Massolini G. Pronase-immobilized enzyme reactor: an approach for automation in glycoprotein analysis by LC/LC-ESI/MSn. Analytical Chemistry. 2007;79:355–363. doi: 10.1021/ac0611519. [DOI] [PubMed] [Google Scholar]
- 52.Yu YQ, Fournier J, Gilar M, Gebler JC. Identification of N-linked glycosylation sites using glycoprotein digestion with pronase prior to MALDI tandem time-of-flight mass spectrometry. Analytical Chemistry. 2007;79:1731–1738. doi: 10.1021/ac0616052. [DOI] [PubMed] [Google Scholar]
- 53.Axen R, Porath J, Ernback S. Chemical coupling of peptides and proteins to polysaccharides by means of cyanogen halides. Nature. 1967;214:1302–1304. doi: 10.1038/2141302a0. [DOI] [PubMed] [Google Scholar]
- 54.Seipert RR, Dodds ED, Lebrilla CB. Exploiting differential dissociation chemistries of O-linked glycopeptide ions for the localization of mucin-type protein glycosylation. Journal of Proteome Research. doi: 10.1021/pr8007072. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Clowers BH, Dodds ED, Seipert RR, Lebrilla CB. Dual polarity accurate mass calibration for electrospray ionization and matrix-assisted laser desorption/ionization mass spectrometry using maltooligosaccharides. Analytical Biochemistry. 2008;381:205–213. doi: 10.1016/j.ab.2008.06.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fu D, Chen L, O'Neill RA. A detailed structural characterization of ribonuclease B oligosaccharides by 1H NMR spectroscopy and mass spectrometry. Carbohydrate Research. 1994;261:173–186. doi: 10.1016/0008-6215(94)84015-6. [DOI] [PubMed] [Google Scholar]
- 57.Holland JW, Deeth HC, Alewood PF. Proteomic analysis of kappa-casein micro-heterogeneity. Proteomics. 2004;4:743–752. doi: 10.1002/pmic.200300613. [DOI] [PubMed] [Google Scholar]
- 58.Holland JW, Deeth HC, Alewood PF. Analysis of O-glycosylation site occupancy in bovine kappa-casein glycoforms separated by two-dimensional gel electrophoresis. Proteomics. 2005;5:990–1002. doi: 10.1002/pmic.200401098. [DOI] [PubMed] [Google Scholar]
- 59.Sheares BT. Site-specific glycosylation in animal cells - substitution of glutamine for asparagine 293 in chicken ovalbumin does not allow glycosylation of asparagine 312. Journal of Biological Chemistry. 1988;263:12778–12782. [PubMed] [Google Scholar]
- 60.Küster B, Wheeler SF, Hunter AP, Dwek RA, Harvey DJ. Sequencing of N-linked oligosaccharides directly from protein gels: in-gel deglycosylation followed by matrix-assisted laser desorption/ionization mass spectrometry and normal-phase high-performance liquid chromatography. Analytical Biochemistry. 1997;250:82–101. doi: 10.1006/abio.1997.2199. [DOI] [PubMed] [Google Scholar]
- 61.Suzuki T, Kitajima K, Emori Y, Inoue Y, Inoue S. Site-specific de-N-glycosylation of diglycosylated ovalbumin in hen oviduct by endogenous peptide: N-glycanase as a quality control system for newly synthesized proteins. Proceedings of the National Academy of Sciences. 1997;94:6244–6249. doi: 10.1073/pnas.94.12.6244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Landberg E, Pahlsson P, Lundblad A, Arnetorp A, Jeppsson JO. Carbohydrate composition of serum transferrin isoforms from patients with high alcohol consumption. Biochemical and Biophysical Research Communications. 1995;210:267–274. doi: 10.1006/bbrc.1995.1656. [DOI] [PubMed] [Google Scholar]
- 63.Flahaut C, Michalski JC, Danel T, Humbert MH, Klein A. The effects of ethanol on the glycosylation of human transferrin. Glycobiology. 2003;13:191–198. doi: 10.1093/glycob/cwg016. [DOI] [PubMed] [Google Scholar]
- 64.Valmu L, Kalkkinen N, Husa A, Rye PD. Differential susceptibility of transferrin glycoforms to chymotrypsin: a proteomics approach to the detection of carbohydrate-deficient transferrin. Biochemistry. 2005;44:16007–16013. doi: 10.1021/bi051749v. [DOI] [PubMed] [Google Scholar]
- 65.Sanz-Nebot V, Balaguer E, Benavente F, Neususs C, Barbosa J. Characterization of transferrin glycoforms in human serum by CE-UV and CE-ESI-MS. Electrophoresis. 2007;28:1949–1957. doi: 10.1002/elps.200600648. [DOI] [PubMed] [Google Scholar]
- 66.Spiro RG, Bhoyroo VD. Structure of the O-glycosidically linked carbohydrate units of fetuin. Journal of Biological Chemistry. 1974;249:5704–5717. [PubMed] [Google Scholar]
- 67.Takasaki S, Kobata A. Asparagine-linked sugar chains of fetuin: occurrence of tetrasialyl triantennary sugar chains containing the Gal beta 1-3GlcNAc sequence. Biochemistry. 1986;25:5709–5715. doi: 10.1021/bi00367a054. [DOI] [PubMed] [Google Scholar]
- 68.Green ED, Adelt G, Baenziger JU, Wilson S, Van Halbeek H. The asparagine-linked oligosaccharides on bovine fetuin - structural analysis of N-glycanase-released oligosaccharides by 500-megahertz 1H NMR spectroscopy. Journal of Biological Chemistry. 1988;263:18253–18268. [PubMed] [Google Scholar]
- 69.Huang Y, Mechref Y, Novotny MV. Microscale nonreductive release of O-linked glycans for subsequent analysis through MALDI mass spectrometry and capillary electrophoresis. Analytical Chemistry. 2001;73:6063–6069. doi: 10.1021/ac015534c. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.