

Abstract
Cell surface localization of the glucose transporter, Glut1, is a cytokine-controlled process essential to support the metabolism and survival of hematopoietic cells. Molecular mechanisms that regulate Glut1 trafficking, however, are not certain. Here we show a C-terminal PDZ-binding motif in Glut1 is critical to promote maximal cytokine-stimulated Glut1 cell surface localization and prevent Glut1 lysosomal degradation in the absence of growth factor. Disruption of this PDZ-binding sequence through deletion or point mutation sharply decreased surface Glut1 levels and led to rapid targeting of internalized Glut1 to lysosomes for proteolysis, particularly in growth factor-deprived cells. The PDZ domain protein, GIPC, bound to Glut1 in part via the Glut1 C-terminal PDZ binding motif and we found that GIPC-deficiency decreased Glut1 surface levels and glucose uptake. Unlike the Glut1 degradation observed upon mutation of the Glut1 PDZ-binding domain, however, GIPC-deficiency resulted in accumulation of intracellular Glut1 in a pool distinct from the recycling pathway of the Transferrin Receptor (TfR). Blockade of Glut1 lysosomal targeting after growth factor withdrawal also led to intracellular accumulation of Glut1, a portion of which could be rapidly restored to the cell surface after growth factor stimulation. These data indicate that the C-terminal PDZ-binding motif of Glut1 plays a key role in growth factor regulation of glucose uptake by both allowing GIPC to promote Glut1 trafficking to the cell surface and protecting intracellular Glut1 from lysosomal degradation after growth factor withdrawal, thus allowing potential for a rapid return of intracellular Glut1 to the cell surface upon re-stimulation.
Keywords: Recycling, Degradation, Lysosomes, Akt
INTRODUCTION
Cellular metabolic rates must meet biosynthetic and energetic demands to allow cell survival and function. Thus, rapidly growing and proliferating cells, such as growth factor-stimulated or cancer cells, require increased metabolism relative to their non-proliferating counterparts. Glucose in particular is preferentially utilized by proliferating cells for biosynthesis and energy production. Resting hematopoietic cells normally utilize glucose at a low rate until stimulated by a specific growth or mitogenic factor, such as the cytokines interleukin 3 (IL3) or IL7, which can promote dramatic increases in glucose uptake and consumption [1-3]. Cells deprived of these growth factors undergo a sharp decrease in glucose metabolism, initiating cellular atrophy and eventually cell death [1, 4-7]. Glucose uptake is a key regulating step in glucose metabolism and is mediated by facilitative glucose transporters (Gluts). Glut1 is the predominant glucose transporter in hematopoietic cells [4, 8, 9] and cytokines regulate glucose uptake through modulation of Glut1 protein levels and cell surface trafficking [5, 10]. The molecular mechanisms that allow growth factors to control Glut1 trafficking and degradation, however, are poorly understood.
Growth factors increase surface Glut1 in part by decreasing the rate of surface Glut1 internalization and promoting recycling of intracellular Glut1 [10]. In the absence of growth factor, Glut1 is internalized via a process that is inhibited by activation of Akt. Ultimately, internalized Glut1 is degraded in lysosomes [11]. Glut1 does not appear to normally be rapidly targeted to lysosomes, however, as this degradation can require days and growth factor deprivation of apoptosis-resistant Bak−/−Bax−/− cells has shown some Glut1 protein can persist intracellularly for several weeks after growth factor withdrawal [12]. It is not certain how targeting of Glut1 to lysosomes is controlled after growth factor withdrawal and how accumulation of intracellular Glut1 may affect regulation of glucose uptake. Prevention of general lysosomal targeting by RNAi of Rab7 or use of dominant negative Rab7, however, has been shown to protect nutrient transporters, including Glut1, from lysosomal degradation and promote growth factor-independent survival [11]. The relationship between Glut1 lysosomal degradation and cell surface levels and the potential role for intracellular stores of Glut1 remain uncertain. These issues are likely important in control of cell growth and survival as glucose metabolism can both provide energy for cell growth and influence Bcl-2 family proteins regulation of cell death [6, 13, 14]. Rapid loss of Glut1 protein after growth factor deprivation may prevent cells from responding quickly upon re-stimulation whereas inappropriate recycling of intracellular Glut1 may allow continued glucose uptake to prevent apoptosis after growth factor withdrawal [6].
It has been suggested that the C-terminal tail of Glut1 is critical to control Glut1 subcellular localization [15] and may mediate protein interactions essential for cytokine-regulated Glut1 trafficking. Yeast two-hybrid screens have identified two proteins that bind to the Glut1 C-terminal tail which may control Glut1 trafficking, namely a SUMOconjugating enzyme, Ubc9, and the PDZ domain protein, Gα interacting protein interacting protein, C-terminus (GIPC) [16, 17]. Ubc9 has been shown to interact with a specific lysine residue on the C-terminal tail of Glut1 and Glut4 but not Glut3, and to influence total Glut1 and Glut4 protein levels [17]. In addition, the C-terminal four amino acids of Glut1 (DSQV) represent a class I PDZ-binding motif [18, 19] that is required for efficient GIPC/Glut1 interaction [16]. GIPC also associates with and promotes cell surface localization of other cell surface proteins, including transforming growth factor type III receptor, human lutropin receptors, and dopamine D2 and D3 receptors [20-22]. Colocalization studies of Glut1 lacking the PDZ-binding motif have suggested that GIPC may protect Glut1 from association with late endosomes/lysosomes [23].
To better understand cytokine regulation of glucose uptake and Glut1 lysosomal targeting, we have analyzed the role of specific domains of Glut1 required for growth factor regulated Glut1 trafficking and glucose uptake. Here we show that the Glut1 C-terminal PDZ-binding motif plays critical roles in regulation of Glut1 trafficking. Importantly, internalized Glut1 was rapidly targeted for lysosomal degradation upon growth factor withdrawal when the Glut1 PDZ-binding motif was deleted or mutated. The Glut1-binding PDZ-domain protein GIPC, however, did not regulate Glut1 lysosomal targeting. Instead GIPC was required to promote return of intracellular Glut1 to the cell surface as total Glut1 levels were unaltered in GIPC-deficient cells resulting in Glut1 accumulation in an intracellular pool distinct from the endosomal recycling pathway characterized by TfR localization. Prevention of Glut1 targeting to lysosomes also maintained Glut1 protein levels upon growth factor withdrawal with accumulation of intracellular Glut1. Importantly, a portion of this intracellular Glut1 could be rapidly restored to the cell surface upon restimulation. Together these data show that the PDZ-binding motif of Glut1 plays a key role to regulate Glut1 protein levels and trafficking to both the cell surface and the lysosomes to support growth factor regulated glucose uptake and cell growth.
MATERIALS AND METHODS
Plasmids
N-terminal GFP-tagged hGlut3 was generated as previously described for GFP-rGlut1 [10]. Mutants of FLAG-rGlut1 were generated to delete the last 38 amino acids or the last 4 amino acids using the FLAG-tagged rGlut1 previously described [10]. A FLAG-tagged chimeric protein was constructed by replacing the rGlut1 C-terminal tail (38 aa) with the corresponding C-terminal tail of hGlut3 (44 aa). Point mutagenesis was used to mutate lysine 456 of FLAG-Glut1 to an alanine. A double mutant was constructed by deleting the C-terminal four amino acids of the FLAG-Glut1K456A construct. Point mutagenesis was used to mutate individual amino acids in the PDZ-binding domain of Glut1 (DSQV) to alanine residues as indicated. mRab7 shRNAi plasmid was constructed using pCR2.1 TOPO (Invitrogen, Carlsbad, CA) with hU6 promoter and published target sequence [11]. pET30a(+)-GIPC vector for expression of recombinant His6-tagged GIPC was generously provided by Dr. Brent Reed (Louisiana State University, Shreveport, LA). mGIPC shRNAi plasmids were constructed using pCR2.1 TOPO cloning (Invitrogen) with the hU6 promoter and target sequences to 486-504bp or 570-593bp of mouse GIPC. GFP shRNAi was utilized as a control plasmid for downregulation [6]. mGIPC overexpression construct was made by reverse transcription of mouse T-cell cDNA and cloned into pCR2.1 TOPO before subcloning into pEF6 vector. pEF6-myrAkt1 was previous described [10].
Cells
The early hematopoietic myeloid/lymphoid cell line, FL5.12, was cultured as previously described [10, 24] with addition of recombinant murine IL3 (500 pg/ml; (Peprotech, Rocky Hill, NJ). For inhibition of lysosomal degradation, cells were cultured in either 40 μM Chloroquine (CQ) (Sigma-Aldrich, St. Louis, MO) or 10mM ammonium chloride (NH4Cl) (Sigma-Aldrich) for duration of growth factor withdrawal. Stable expression of FLAG-Glut1, FLAG-Glut1ΔC, FLAG-Glut1Δ4, FLAG-Glut1/CtermGlut3, FLAG-Glut1K456A, and FLAG-Glut1K456AΔ4 in FL5.12 cells was achieved by transfection of linearized construct by electroporation (Kit V, Amaxa Biosystems, Gaithersburg, MD). Stable clones were identified after selection with blasticidin (Invitrogen) and flow cytometric analysis for surface FLAG positive cells. Stable expression of Bcl-xL in FLAG-Glut1 cells was achieved by retroviral transfection of pKD-GFP-hBcl-xL. Bulk populations of cells were used, gating on GFP positive cells. For growth factor withdrawal, cells were washed three times in PBS prior to resuspension in appropriate media for six hours unless otherwise noted.
2-deoxy-D-glucose Transport Assay
Glucose uptake was measured as previously described [10]. In short, cells were resuspensed in Kreb's Ringer HEPES (KRH; KRH at pH 7.4: 136 mM NaCl, 4.7 mM KCl, 1.25 mM CaCl2, 1.25 mM MgSO4, 10 mM HEPES). 2-deoxy-D-[H3]glucose (2 μCi/rxn) was added for a period of 5 min at 37°C. Reactions were quenched by addition of ice-cold 200 μM phloretin (Calbiochem, San Diego, CA) and centrifugation through an oil layer (1:1 Dow Corning 550 Silicon fluid (Motion Industries, Birmingham, AL) and Dinonyl phthalate (Sigma-Aldrich)). The oil layer was washed and the cell pellet was solubilized in 1M NaOH and radioactivity was measured with scintillation counter.
Flow Cytometry
Cells were analyzed by a FACscan (Becton Dickinson, San Jose, CA) and FlowJo software (Tree Star, INC., Ashland, OR). To determine FLAG surface expression, cells were stained and analyzed as previously described [10]. To measure internalization of FLAG-Glut1, pulse-chase assays were performed as previously described [10]. To determine TfR (CD71) surface expression, cells were incubated with anti-mouse CD71-PE (BD Biosciences PharMingen, San Diego, CA).
Fluorescence Microscopy
To image cells expressing GFP fusion proteins, cells were fixed with 1% paraformaldehyde in PBS and viewed with a Zeiss LSM410 confocal microscope (Carl Zeiss, Thornwood, NY) and MetaMorph software (Molecular Devices, Sunnyvale, CA.) To image lysosomes, cells were treated with 50nM Lysotracker RED (Invitrogen) for 30 minutes before fixation. To image TfR trafficking, cells were treated with 50 μg/ml Alexa Fluor 568-tagged Transferrin for one hour before fixation (Invitrogen).
Preparation of His6 GIPC and Pulldowns
The pET30a(+)-GIPC vector was transformed into E. coli BL21DE3 pLys cells and His6GIPC was purified as described [16]. Briefly, cells were induced and fusion protein was purified from inclusion bodies with Ni affinity chromatography (Ni-NTA Agarose, Qiagen, Valencia, CA) under denaturing conditions using guanidine HCl. The nickel-bound His6GIPC beads were washed extensively in buffer containing 10 mM Hepes pH 8.0, 1 M NaCl and 1 mM DTT. Control nickel beads were pre-blocked in 0.125 M Histidine followed by additional blocking for both control and His6GIPC beads in PBS/3% BSA. Beads were then washed with PBS/1% TritonX-100 before overnight incubation with lysates from cells transfected to transiently express FLAG-Glut1 proteins. Beads were resuspended in sample buffer and heated at 70°C for 10 minutes before loading on SDS-PAGE gel.
Western Blot
Cells were lysed for western blotting for one hour on ice in 1% Triton and 0.1% SDS in PBS with protease inhibitors (BD Biosciences PharMingen) and precleared by centrifugation. Primary antibodies used were: anti-FLAG M2 Peroxidase (Sigma-Aldrich); rabbit anti-Glut1 (Abcam, Cambridge, MA); rabbit anti-phospho Akt ser473 (Cell Signaling, Beverly, MA); mouse anti-Actin (Sigma); goat anti-GIPC (Santa Cruz, Santa Cruz, CA); rabbit anti-6X His (Abcam); mouse anti-Rab7 (Santa Cruz). Secondary antibodies used were: anti-rabbit HRP (Cell Signaling); anti-mouse HRP (BD Biosciences PharMingen); donkey anti-goat HRP (Santa Cruz). Secondary HRP conjugated antibodies were viewed by ECL-Plus (GE Healthcare, Little Chalfont, Buckinghamshire, United Kingdom).
RESULTS
The Glut1 C-terminal tail is required for maximal growth factor-stimulated surface expression
Glut1 cell surface localization in hematopoietic cells is regulated by cytokines, such as IL3, and activation of the PI3K/Akt pathway [10]. To identify mechanisms that regulate Glut1 trafficking and are specific to Glut1, we compared the ability of cytokine to regulate the trafficking of Glut1 with the glucose transporter, Glut3. Glut3 is closely homologous to Glut1 yet divergent in the C-terminus, a domain previously implicated as important in Glut1 trafficking [15]. Cells were transfected with N-terminally GFP tagged Glut1 or Glut3 fusion proteins and imaged by confocal microscopy in the presence or after a six hour withdrawal from IL3 (Figure 1A). In contrast to GFP-Glut1 which showed cell surface levels regulated by IL3 consistent with previous results [10], GFP-Glut3 was observed in an intracellular punctate pattern both in the presence and absence of IL3 with very little GFP-Glut3 present near the cell surface.
Figure 1. The Glut1 C-terminal tail is required for maximal surface Glut1 localization.

(A) FL5.12 cells were transiently transfected with GFP-Glut1 or GFP-Glut3 for 18 hours and then cultured in the presence or absence of IL3 for an additional six hours. Cells were visualized with confocal microscopy. (B) Schematic of FLAG-tagged Glut1 full length, C-terminal tail truncation (FLAG-Glut1ΔC), C-terminal four amino acid truncation (FLAG-Glut1Δ4) and Glut1 chimeric protein with C-terminus of Glut3 (Glut1/CtermGlut3). (C and D) Cells stably expressing FLAG-Glut1 constructs as shown in B, were cultured in the presence or absence of IL3 for six hours. (C) Surface levels of FLAG-Glut1 were measured by flow cytometry. (D) Total FLAG-Glut1 levels were measured by immunoblot with actin loading control. (E) Cell surface FLAG-Glut1 levels were normalized to total FLAG expression. Mean and standard deviations of triplicate samples are shown. Asterisks (*) indicate p≤ 0.001 within the experiment. Representative results are shown for three or more experiments.
The distinct C-terminal tails of Glut1 and Glut3 may have determined the different intracellular localization of these transporters and regulation by IL3. To further establish the role of the Glut1 C-terminus in protein localization, surface levels of deletion or chimeric Glut1 were analyzed. A tandem FLAG epitope tag was inserted into the exofacial loop of wild type and mutant Glut1 to allow flow cytometric measurement of surface Glut1 and trafficking without interference with any intracellular Glut1 domains [10]. FLAG-tagged deletion mutants of Glut1 were generated that lacked the last 38 amino acids of the cytoplasmic C-terminus (FLAG-Glut1ΔC) or lacked the C-terminal four amino acids (FLAG-Glut1Δ4) that constitute the PDZ-binding motif in Glut1. In addition, a FLAG-tagged chimeric protein was made by replacing the C-terminal tail of Glut1 with the corresponding amino acids of Glut3 (FLAG-Glut1/CtermGlut3) (Figure 1B). Cells stably expressing FLAG-tagged chimeric and deletion Glut1 constructs were washed and cultured in the presence or absence of IL3 for six hours and surface levels were analyzed by flow cytometry. As we have previously shown, full length FLAG-Glut1 had high surface expression in the presence of IL3 that decreased upon growth factor-withdrawal (Figure 1C and [10]). FLAG-Glut1ΔC and FLAG-Glut1Δ4 were found to have lower surface expression than full length FLAG-Glut1 and these surface levels were further decreased upon cytokine withdrawal. Replacement of the Glut1 C-terminal tail with that of Glut3 also reduced cell surface Glut1 expression to a level comparable to that of Glut1ΔC (Figure 1C).
The Glut1 C-terminus may have affected Glut1 surface levels by altering Glut1 trafficking and/or protein levels. To address this, total cellular expression of wild type Glut1, deletions and chimeric Glut1 proteins was determined by immunoblot (Figure 1D). Since slight differences of total FLAG-Glut1 expression levels in the presence of IL3 were observed, surface FLAG-Glut1 levels were normalized to total FLAG-Glut1 expression levels. The decreased surface levels of the truncation mutants or chimera in the presence of IL3 reflected true changes in Glut1 distribution or trafficking when normalized to total levels (Figure 1E). Interestingly, while withdrawal from cytokine for six hours did not substantially affect total FLAG-Glut1 levels, FLAG-Glut1ΔC and FLAG-Glut1Δ4 protein levels were sharply reduced upon cytokine withdrawal (Figure 1D), suggesting possible rapid targeting of internalized Glut1 deletion mutants for degradation.
Differences in protein levels of Glut1 deletion mutants were most pronounced when cells were deprived of growth factors. We have previously shown that Akt is a critical mediator of growth factor receptor signals to regulate Glut1 trafficking [10]. We tested, therefore, if Akt was sufficient to rescue expression and prevent degradation of the FLAG-Glut1Δ4 mutant. FLAG-Glut1 and FLAG-Glut1Δ4 cells were transiently transfected with a constitutively active form of Akt (myristoylated, myrAkt) and cultured in the presence or absence of IL3 for 6 hours (Figure 2A and 2B). Activated Akt increased Glut1 protein levels and prevented the degradation of both full length Glut1 and Glut1Δ4 in the presence or absence of IL3 (Figure 2A). In addition, activated Akt was sufficient to prevent the decrease in surface levels of FLAG-Glut1Δ4 during growth factor withdrawal (Figure 2B). Expression of activated Akt could also protect Glut1 lacking the entire C-terminus from internalization and degradation (data not shown). Akt activation was not sufficient, however, to rescue the total and surface levels of the FLAG-Glut1Δ4 mutant relative to the level of wildtype FLAG-Glut1. Therefore, active Akt can maintain surface Glut1 in the absence of growth factor independent of the Glut1 C-terminus, possibly by inhibiting Glut1 internalization [10], but the PDZ-binding motif of Glut1 is required for both maximal total and surface levels.
Figure 2. Akt can prevent degradation of Glut1Δ4.

(A) Total levels of FLAG-Glut1 and phospho-Akt (ser473) were measured by immunoblot with actin loading control. (B) Surface FLAG-Glut1 levels were measured by flow cytometry. Mean and standard deviations of triplicate samples are shown. Asterisks (*) indicate p≤ 0.001 within the experiment. Representative results are shown for three or more experiments.
Glut1 lysine 456 regulates Glut1 levels
In addition to the C-terminal four amino acids of Glut1, Ubc9 modification of lysine 456 of the Glut1 C-terminal tail has been proposed to regulate Glut1 protein levels [17]. To determine if lysine residue 456 of Glut1 C-terminal tail regulated Glut1 degradation and if it modulated the effect of the four amino acid truncation mutant, lysine 456 was mutated to alanine (FLAG-Glut1K456A) and a double mutant containing the K456A point mutation and truncation of the last four amino acids was generated (FLAG-Glut1K456A/Δ4). Cells stably expressing FLAG-Glut1K456A and FLAG-Glut1K456A/Δ4 were cultured in the presence or absence of IL3 for 6 hours and total and surface FLAG-Glut1 levels were compared to both full length and four amino acid truncation mutant of FLAG-Glut1 (Figure 3A). The FLAG-Glut1K456A mutant exhibited a large increase in total FLAG-Glut1 expression levels when compared to full length FLAG-Glut1 and FLAG-Glut1Δ4, whereas the double mutant, FLAG-Glut1K456A/Δ4, had a phenotype similar to FLAG-Glut1Δ4 with decreased total protein levels. FLAG-Glut1K456A exhibited higher surface levels both in the presence and absence of IL3 when compared to full length FLAG-Glut1 whereas FLAG-Glut1K456A/Δ4 had diminished surface levels similar to the four amino acid truncation mutant (Figure 3B). Although surface and total levels of Glut1 were increased upon mutation of lysine 456, the glucose uptake of FLAG-Glut1K456A was similar to full length Glut1, whereas the double mutant had comparable glucose uptake to the four amino acid truncation mutant (Figure 3C). Taken together these data suggest that possible modification of lysine 456 may favor Glut1 degradation but that the C-terminal four amino acids ultimately determine Glut1 fate.
Figure 3. Glut1 Lysine 456 and the C-terminal PDZ-binding motif regulate Glut1 degradation.

Cells stably expressing FLAG-Glut1, FLAG-Glut1Δ4, FLAG-Glut1K456A, and FLAG-Glut1K456AΔ4 were cultured in the presence or absence of IL3 for six hours. (A) Total FLAG expression levels were measured by immunoblot with actin as a loading control. (B) Surface FLAG levels were analyzed by flow cytometry. (C) Glucose uptake was measured. Mean and standard deviations of triplicate samples are shown. Asterisks (*) indicate p≤ 0.005 and (**) p≤ 0.01 within the experiment. Representative results are shown for three or more experiments.
Targeting of Glut1 to lysosomes is regulated by the Glut1 C-terminal four amino acids
Degradation of internalized Glut1 upon growth factor withdrawal likely occurred in lysosomes [11]. To test if lysosomal proteolysis of Glut1 was regulated by the C-terminal four amino acids of Glut1, N-terminal GFP-tagged Glut1 and Glut1Δ4 were analyzed by fluorescence microscopy for colocalization with Lysotracker RED, a dye that stains lysosomes on the basis of low lysosomal pH level. Cells were transiently transfected with either GFP-Glut1 or GFP-Glut1Δ4 and cultured in the presence or absence of IL3 for 8 hours. To ensure activation of cell death pathways did not alter protein trafficking during the growth factor withdrawal cells were also transiently transfected with the anti-apoptotic protein, hBcl-xL. Glut1 remained on the cell surface in the presence of IL3 with very little intracellular staining [10], however, intracellular Glut1 protein was observed to colocalize with lysosomes upon growth factor withdrawal (Figure 4A). In contrast, the four amino acid truncation mutant of Glut1 resided mainly intracellular and largely colocalized with lysosomes both in the presence and absence of the cytokine IL3. These data suggest that the C-terminal four amino acids may be critical to protect Glut1 from lysosomal targeting following growth factor deprivation.
Figure 4. Targeting of Glut1 to lysosomes is regulated by the Glut1 C-terminal four amino acids.
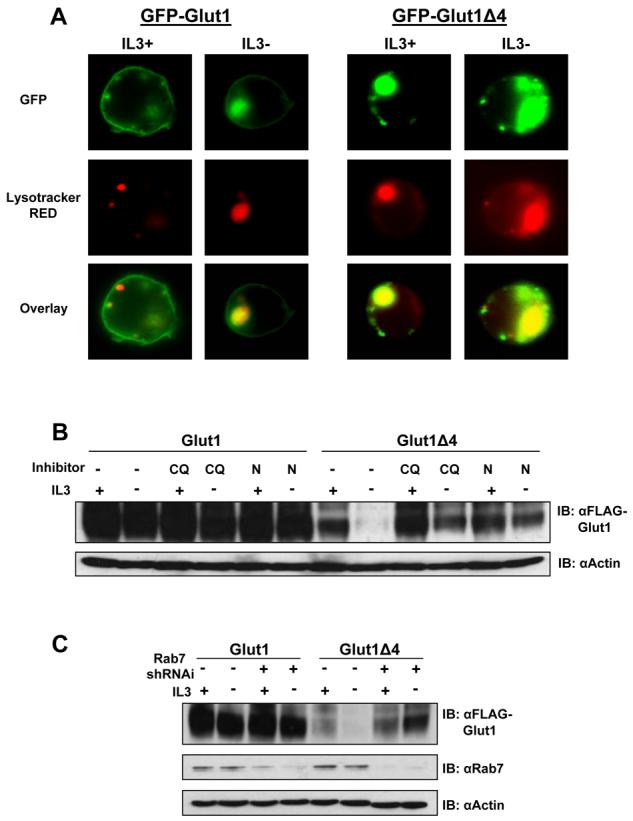
(A) FL5.12 cells were co-transfected with either GFP-Glut1 or GFP-Glut1Δ4 and hBcl-xL to maintain cell viability for 18 hours then cultured in the presence or absence of IL3 for an additional eight hours. Lysotracker RED was added to culture in the final 30 minutes followed by fixation and visualization with fluorescence microscopy. (B) FLAG-Glut1 and FLAG-Glut1Δ4 cells were withdrawn from IL3 for six hours and vehicle control, Chloroquine (CQ) or NH4Cl (N) was added to cells for the duration of the IL3-withdrawal. Total FLAG expression levels were measured by immunoblot with actin as a loading control. (C) FLAG-Glut1 and FLAG-Glut1Δ4 cells were transiently transfected with control shRNAi or Rab7 shRNAi for 24 hours followed by a six hour growth factor withdrawal. Total FLAG and Rab7 protein levels were measured via immunoblot with actin as a loading control. Representative results are shown for three or more experiments.
The role of lysosomes in degradation of Glut1Δ4 was further analyzed using pharmacologic and genetic techniques. Chloroquine (CQ) and ammonium chloride (NH4Cl) can neutralize lysosomal pH to inhibit lysosomal proteolytic capacity [25-27]. FLAG-Glut1 and FLAG-Glut1Δ4 expressing cells were cultured in the presence or absence of IL3 for 6 hours with vehicle control, CQ or NH4Cl for the duration of the growth factor withdrawal (Figure 4B). Chemical inhibition of lysosomes did not alter full length FLAG-Glut1 total protein levels in the presence or absence of cytokine IL3 for the time points analyzed here. An increase in protein levels, however, was observed in the Glut1 four amino acid truncation mutant when lysosomal degradation was inhibited in the presence or absence of IL3. A genetic approach was also employed to confirm a role for lysosomal targeting for degradation of Glut1Δ4. The small GTPase Rab7 has been implicated in targeting proteins for lysosomal degradation [11, 28-30], therefore we utilized a shRNAi construct containing a previously published and validated target sequence to downregulate Rab7 expression [11]. Control shRNAi or Rab7 shRNAi constructs were transiently transfected into FLAG-Glut1 and FLAG-Glut1Δ4 cells to reduce Rab7 expression and cells were subjected to a 6 hour growth factor withdrawal (Figure 4C). Similar to chemical inhibition of lysosomes, Rab7-deficiency largely prevented the degradation of FLAG-Glut1Δ4. Neither pharmacologic or genetic approaches to block lysosomal degradation fully rescued FLAG-Glut1Δ4 protein levels, thereby indicating that each approach was not fully efficient or that alternate proteolytic pathways may also regulate Glut1. Taken together, these data show that Glut1 degradation by lysosomes is regulated by the last four amino acids, DSQV, which are critical to protect intracellular Glut1 from lysosomal degradation.
Amino acids dictating PDZ-binding specificity regulate Glut1 total and surface levels
It has been demonstrated that individual amino acids within a class I PDZ-binding motif are critical in determining what protein-protein interactions occur and a given PDZ-binding motif may interact with a number of PDZ domain containing proteins. In particular, binding specificity of class I PDZ binding motifs is dependent on C-terminal amino acids in the 0 and −2 position [31]. To further characterize the role of the Glut1 PDZ-binding motif, each amino acid was mutated and total and surface levels of FLAG-Glut1 were observed (Figure 5A and 5B). Mutation of all four amino acids to alanine residues resulted in a phenotype similar to the four amino acid truncation mutant with low total and surface levels. Point mutation of the −2 (DAQV) and −3 (ASQV) positions did not impact total FLAG-Glut1 levels but caused a slight decrease in surface FLAG-Glut1 levels. In contrast, point mutation of the 0 (DSQA), −1 (DSAV) or 0 and −2 (DAQA) positions each significantly decreased total protein levels and surface levels. Even though total levels of the −1 point mutation (DSAV) were almost undetectable by immunoblot, surface levels of FLAG-Glut1-DSAV were detected at levels higher than background staining control by flow cytometry. While Q to A (−1) mutations represent a charge change that may in principle affect protein folding, the V to A (0) mutation is structurally well-conserved but also led to a loss of Glut1 protein. These data support a necessary role for the Glut1 PDZ-binding domain and possible interaction with class I PDZ domain proteins as essential for regulation of Glut1 trafficking.
Figure 5. The Glut1 PDZ-binding motif is critical to regulate Glut1 total and surface levels.

(A, B, and C) Cells were transiently transfected with FLAG-Glut1 constructs containing point mutations in the PDZ-binding motif, as noted in bold print, for 18 hours and then cells were cultured in the presence or absence of IL3 for an additional six hours. (A) Total FLAG-Glut1 was measured by immunoblot with actin as a loading control. (B) Flow cytometric analysis was utilized to measure surface FLAG-Glut1 levels. FL5.12 cells without FLAG-Glut1 were used for a negative stain control. (C) Cells were stained with anti-FLAG antibody, washed and cultured at 37 °C for indicated times prior to staining with fluorescent secondary antibody to determine the fraction of surface label remaining after various times incubation. (D) Cells were transiently transected with FLAG-Glut1, FLAG-Glut1Δ4 and FLAG-Glut1point mutants (DSQA, DAQV, DSAV ASQV) constructs for 24 hours and lysates were prepared. Lysates were then incubated with purified uncoated or nickel-bound His6GIPC beads. Anti- 6XHis immunoblot compares uncoated beads to nickel-bound His6GIPC beads used for pull-down. After washing, beads were boiled with sample buffer and FLAG levels were measured via immunoblot. U = uncoated beads, G = nickel-bound His6GIPC beads, I = input cell lysate, P = pull-down with HIS6GIPC beads. Mean and standard deviations of triplicate samples are shown. Asterisks (*) indicate p< 0.0005 and (**) p≤ 0.005 within the experiment. Representative results are shown for three or more experiments.
Glut1 levels may be regulated through control of either Glut1 internalization or targeting to lysosomes and mutations in the Glut1 PDZ-binding domain may have affected either or both processes. To test if this PDZ-binding domain affected Glut1 internalization, a pulse-chase assay was performed to measure the internalization rate of full length FLAG-Glut1, FLAG-Glut1Δ4 and FLAG-Glut1-DSQA in the presence or absence of growth factor (Figure 5C). Cells were stained with anti-FLAG primary antibody and placed at 37°C for indicated times prior to addition of fluorescently labeled secondary antibody. Flow cytometry was used to measure cell fluorescence and determine the fraction of FLAG-tagged Glut1 and primary antibody remaining on the cell surface at each time point. As we have previously shown, internalization of full-length Glut1 was more rapid in growth factor-deprived cells than in the presence of growth factor [10]. Glut1 lacking the C-terminal four amino acids and Glut1-DSQA both internalized more rapidly than full length Glut1 in the presence of growth factor and had an even more rapid loss of cell surface levels in the absence of growth factor. While the Glut1 PDZ-binding motif may play multiple roles in Glut1 trafficking, these data suggest that this domain contributes to regulation of Glut1 internalization.
The PDZ-domain protein GIPC has been described to associate with the C-terminus of Glut1 and these interactions may mediate the effects of the Glut1 PDZ-binding domain in Glut1 trafficking. To determine if GIPC associated with full length Glut1 but not PDZ-domain mutants of Glut1, His6-tagged GIPC was purified and used to coat nickel beads (Figure 5D). Cells were transiently transfected to express FLAG-tagged Glut1, Glut1Δ4, or Glut1 point mutations DSQA, DAQV, DSAV, or ASQV and cell lysates were mixed with GIPC labeled or unlabeled beads for pull-down analyses (Figure 5D). As expected, full length Glut1 bound GIPC labeled beads but not unlabeled beads. Glut1Δ4 or Glut1 DSQA also showed binding to GIPC, although in each case binding was reduced compared to that of full length Glut1 (note quantitations of the ratio of input Glut1 to GIPC-bound Glut1 indicated below each band). As shown above, Glut1 DSQV was not readily detectable and could not be quantitated, but the Glut1 DAQV and ASQV mutants each bound GIPC with similar or greater efficiency than wild type Glut1. Thus, while the Glut1 PDZ-binding domain does not appear to be the sole determinant of Glut1 association with GIPC, it does provide a significant contribution to this interaction.
GIPC does not regulate Glut1 degradation or internalization but is required for efficient surface Glut1 localization
Based on the critical role of the Glut1 PDZ-binding domain in regulation of Glut1 lysosomal degradation and the association of GIPC with this domain (Figure 5D) [16, 23], we sought to determine if GIPC regulated Glut1 and was necessary to protect Glut1 from lysosomal degradation. Two individual shRNAi constructs were generated to target GIPC and decrease GIPC expression. Glucose uptake was significantly reduced in GIPC-deficient cells in the presence of IL3 relative to cells expressing a control shRNAi construct (Figure 6A). A more modest yet significant difference was observed in IL3 withdrawn cells. To determine if GIPC was necessary to maintain Glut1 protein levels and prevent Glut1 lysosomal targeting, total levels of endogenous Glut1 were measured by immunoblot (Figure 6B). In contrast to the loss of Glut1 protein observed by deletion of the C-terminal binding motif of Glut1, no loss of total Glut1 protein was detected when GIPC was downregulated. These data demonstrate that GIPC association with the Glut1 C-terminal PDZ-binding motif is required to promote maximal glucose uptake, but is not required to protect Glut1 from lysosomal degradation.
Figure 6. GIPC-deficiency decreases glucose uptake and surface Glut1 levels but does not cause Glut1 degradation.

(A and B) FL5.12 cells were transiently transfected with either control shRNAi, GIPC shRNAi1or GIPC shRNAi2 constructs for 48 hours and then were cultured in the presence or absence of IL3 for six hours. (A) Glucose uptake was measured. (B) Total endogenous Glut1 and GIPC levels were measured by immunoblot with actin as a loading control. (C, D, E) FLAG-Glut1 and FLAG-Glut1Δ4 expressing cells were transiently transfected with control shRNAi, GIPC shRNAi1 or GIPC shRNAi2 constructs for 48 hours followed by culture in the presence or absence of IL3 for six hours. (C) Total FLAG-Glut1 and GIPC levels were measured by immunoblot with actin as a loading control. (D) Total surface FLAG-Glut1 levels were measured by flow cytometry and surface levels were normalized to total FLAG expression levels. (E) Surface FLAG-Glut1Δ4 levels were measured with flow cytometry. (F) FLAG-Glut1 cells were transfected with control shRNAi or GIPC shRNAi1 constructs for 48 hours and cultured in the presence of IL3. Cells were stained with anti-FLAG antibody, washed and cultured at 37 °C for indicated times prior to staining with fluorescent secondary antibody. Mean and standard deviations of triplicate samples are shown. Asterisks (*) indicate p≤ 0.005 and (**) p<0.05 within the experiment. Representative results are shown for three or more experiments.
The decreased glucose uptake but maintenance of Glut1 protein levels in GIPC-deficient cells suggested that the Glut1 C-terminus may interact with GIPC to regulate non-lysosomal trafficking of Glut1. To determine if Glut1 trafficking was affected by GIPC-deficiency, surface expression of FLAG-Glut1 was determined in control and GIPC-deficient cells in the presence of IL3 or after a six hour IL3 withdrawal and normalized to total FLAG-Glut1 levels (Figures 6C and 6D). GIPC-deficiency led to markedly decreased surface FLAG-Glut1 in the presence of IL3 that decreased further after IL3 withdrawal (Figure 6D). Importantly, loss of surface FLAG-Glut1 in GIPC-deficient cells did not reflect a loss of Glut1 protein as FLAG-Glut1 levels were the same or modestly increased in the absence of GIPC (Figure 6C). This decrease in surface FLAG-Glut1 relative to control RNAi was observed with two independent RNAi constructs targeting GIPC (Figure 6D). In addition, decreased surface FLAG-Glut1 levels caused by GIPC-deficiency were specific to full length FLAG-Glut1 as surface levels of Glut1 lacking the C-terminal four amino acids critical for interaction with GIPC were unaffected by loss of GIPC (Figure 6E).
Reduced surface levels of Glut1 may have been due to more rapid internalization or failure of intracellular Glut1 to recycle back to the cell surface. Having shown that Glut1 lacking four amino acids or with mutation in the C-terminus has more rapid internalization (Figure 5C), we tested the role for GIPC in Glut1 internalization. A pulse-chase assay for surface Glut1 internalization was performed on cells transfected with control or GIPC shRNAi (Figure 6F). GIPC-deficiency did not alter the internalization rate of Glut1 compared to cells transfected with control RNAi. We have previously shown that recycling of Glut1 is essential for maximal surface levels [10]. The decreased surface Glut1 levels, normal Glut1 internalization and normal total cellular levels of Glut1, suggest that GIPC may be essential for trafficking of intracellular Glut1 back to the cell surface.
Disruption of GIPC alters Glut1 subcellular localization and suggests a Glut1 recycling pool
GIPC-deficiency decreased surface FLAG-Glut1 leading to accumulation of intracellular Glut1. To characterize how GIPC affected Glut1 intracellular trafficking and this pool of intracellular Glut1, we transiently transfected cells with GFP-Glut1 and control shRNAi or GIPC shRNAi1 for 48hrs in the presence of IL3. Lysotracker RED was added 30 minutes prior to fixation and cells were visualized with fluorescent microscopy to compare GFP-Glut1 localization to lysosomes (Figure 7A). Partial colocalization of GFP-Glut1 with lysosomes was observed in both control shRNAi and GIPC shRNAi1 cells, however, majority of GFP-Glut1 resided on the cell surface in control cells whereas in GIPC-deficient cells, GFP-Glut1 resided in intracellular compartments that were distinct from lysosomes. These data suggested that GIPC-deficiency may lead to Glut1 accumulation in the endosomal recycling pool. Given that this recycling pathway is commonly associated with trafficking of the TfR, we investigated if GFP-Glut1 colocalized with TfR in control and GIPC-deficient cells (Figure 7B). Cells were transfected with GFP-Glut1 and treated with fluorescently labeled transferrin to label TfR and its trafficking patterns. Importantly, GFP-Glut1 did not colocalize with transferrin in either control or GIPC-deficient cells suggesting that Glut1 exists in an independent or specialized pool. This effect of GIPC on Glut1 trafficking was not due to a general disruption of endosomal trafficking because GIPC-deficiency did not decrease, but rather slightly increased the cell surface levels of TfR as measured by flow cytometry (Figure 7C). Overall, these data suggest that GIPC is important for Glut1 cell surface localization and that Glut1 can exist in intracellular pools independent from general recycling pools containing transferrin and the TfR.
Figure 7. Disruption of GIPC suggests that Glut1 can exist in a recycling pool distinct from TfR.
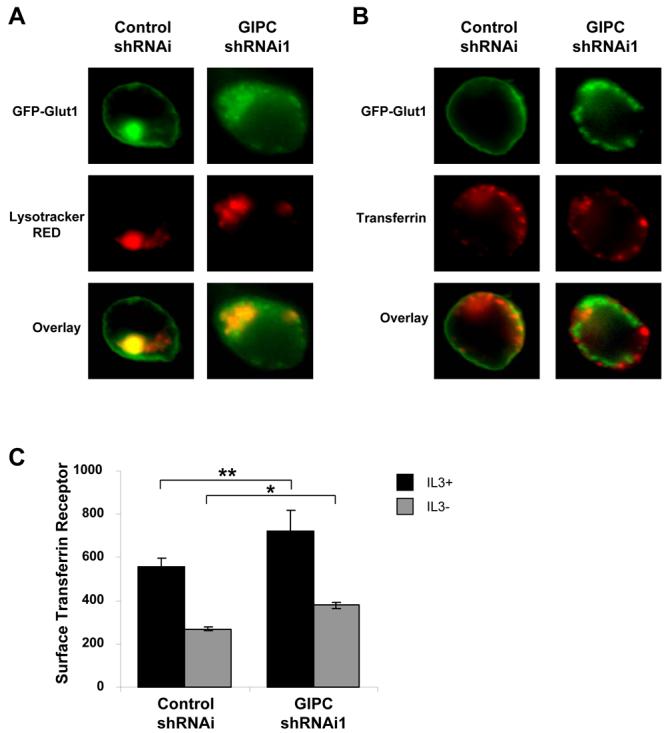
(A and B) FL5.12 cells were transfected with GFP-Glut1 and control shRNAi or GIPC shRNAi1 constructs for 48 hours and were incubated with (A) 50 nM Lysotracker RED for 30 minutes or (B) 50μg/ml Alexa Fluor 568-tagged Transferrin for one hour followed by fixation and visualization with fluorescence microscopy. (C) Cells were transfected with control shRNAi or GIPC shRNAi constructs for 48 hours and then were cultured in the presence or absence of IL3 for an additional six hours. Cells were stained with anti-TfR antibody and surface levels of TfR were measured via flow cytometry. Mean and standard deviations of triplicate samples are shown. Asterisks (*) indicate p< 0.0005 and (**) p≤ 0.05 within the experiment. Representative results are shown for three or more experiments.
Prevention of Glut1 degradation allows more rapid recovery of cell surface Glut1 levels
It was unclear how intracellular accumulation of Glut1 and inhibition of Glut1 degradation following growth factor withdrawal would affect surface Glut1 levels. In principle, accumulation of excess intracellular Glut1 may lead to growth factor-independent return of Glut1 to the cell surface. Alternatively, excess Glut1 may remain intracellular, but allow enhanced recovery of surface Glut1 levels upon re-stimulation. To test this, we transfected cells with control or Rab7 shRNAi to prevent Glut1 lysosomal targeting and measured surface levels of FLAG-Glut1 in the presence or absence of IL3 for 6 hours (Figure 8A). At this early time point, inhibiting lysosomal targeting of Glut1 by Rab7-deficiency modestly increased surface FLAG-Glut1 levels compared to control cells. To analyze how prevention of Glut1 lysosomal degradation affected Glut1 trafficking at later time points, Bcl-xL was expressed to prevent cell death and cells were growth factor withdrawn for one day prior to analysis. After one day without growth factor surface Glut1 levels were greatly diminished regardless of control shRNAi or Rab7 shRNAi or chloroquine treatment, although inhibition of Glut1 degradation did provide a small increase in surface Glut1 levels (Figure 8B and 8C, note the scales). Importantly, a portion of this intracellular Glut1 was capable of rapid return to the cell surface after growth factor re-stimulation of both Rab7 shRNAi transfected and chloroquine treated cells. Increased intracellular Glut1 levels, therefore, both enhances growth factor-independent surface Glut1 levels and allows more rapid recovery of surface Glut1 after growth factor stimulation.
Figure 8. Inhibition of Glut1 degradation does not lead to high surface Glut1 levels but allows a portion of Glut1 to rapidly recycle following growth factor stimulation.

(A) FLAG-Glut1 cells were transiently transfected with control shRNAi or Rab7 shRNAi plasmids for 24 hours and cultured for an additional six hours in the presence or absence of IL3. Surface FLAG-Glut1 levels were analyzed by flow cytometry. (B) FLAG-Glut1 cells stably expressing hBcl-xL were transfected with control shRNAi or Rab7 shRNAi for 24 hours followed by a 24 hour growth factor withdrawal. Surface FLAG-Glut1 levels were measured by flow cytometry after 0, 15, 30 and 60 minutes of IL3 re-addition. (C) FLAG-Glut1 cells stably expressing hBcl-xL were withdrawn from IL3 for 24 hours with either vehicle or 40μM Chloroquine added during the last 6 hours of starve before re-addition of IL3. Surface FLAG-Glut1 levels were measured by flow cytometry 15 minutes after re-addition of IL3. Mean and standard deviations of triplicate samples are shown. Asterisks (*) indicate p< 0.005 within the experiment. Representative results are shown for three or more experiments.
DISCUSSION
In hematopoietic cells, growth factors and cytokines regulate not only Glut1 synthesis [13, 32, 33], but also control Glut1 trafficking to and from the cell surface [10] to regulate glucose uptake. It has been suggested that internalized Glut1 is ultimately recycled back to the cell surface or degraded in lysosomes [11, 23]. Little is known, however, about this process and direct evidence for Glut1 degradation and regulation of this degradation has been lacking. We show here that the Glut1 C-terminal tail is critical for determining the fate of internalized Glut1 to either accumulate intracellularly or be degraded by lysosomes. Utilizing truncation and point mutants of Glut1, we demonstrated that the C-terminal four amino acid PDZ-binding domain of Glut1 is necessary for maximal surface localization and to avoid lysosomal degradation. A PDZ-domain protein, GIPC, could bind this C-terminal motif in Glut1 and was necessary for efficient return of internalized Glut1 back to the cell surface as GIPC-deficiency resulted in accumulation of an intracellular pool of Glut1.We suggest, therefore, that the C-terminal four amino acids of Glut1 serve as a critical platform for protein-protein interactions that regulate multiple aspects of Glut1 trafficking and may control levels of intracellular Glut1 that may return to the cell surface upon restimulation.
Growth factor regulation of Glut1 localization relies on proper control of Glut1 internalization, recycling, and lysosomal targeting. The balance and how growth factors, such as IL3, influence each of these processes, however, has been unclear. In addition to previous work showing that IL3 and Akt can inhibit Glut1 internalization, we show here inhibition of endosomal recycling by shRNAi of GIPC or expression of dominant negative Rab11a [10] strongly decreased surface Glut1 levels in growth factor-stimulated cells. These data suggest an important role for recycling of Glut1 to support growth factor stimulated glucose uptake. Growth factors were also found to suppress Glut1 targeting to lysosomes, as lysosomal degradation of Glut1 appeared to increase in the absence of growth factor, particularly if the C-terminal PDZ-binding domain of Glut1 was mutated. Thus, growth factor signaling appears to attenuate Glut1 internalization, promote Glut1 recycling, and inhibit Glut1 degradation. The signaling mechanisms mediating this growth factor regulation of Glut1 trafficking are not clear. Akt activation can prevent Glut1 internalization but did not fully rescue protein levels of Glut1 lacking the PDZ-binding domain. PDZ domain interactions with Glut1, therefore, appear likely to regulate Glut1 levels and glucose uptake independent of Akt.
Despite coordinated growth factor control, Glut1 recycling and lysosomal targeting were not directly linked and inhibition of either process led to accumulation of intracellular Glut1 rather than to the opposite fate. Inhibition of Glut1 lysosomal targeting led largely to accumulation of intracellular Glut1 rather than recycling of Glut1 back to the cell surface. Similarly, loss of GIPC led to accumulation of intracellular Glut1 rather than targeting of Glut1 to lysosomes. This intracellular pool of Glut1 was independent of the common TfR recycling pool and may represent a novel Glut1 trafficking mechanism. Importantly, a portion of this intracellular Glut1 could rapidly return to the cell surface following growth factor stimulation. That only a portion of Glut1 could rapidly recycle back to the cell surface after growth factor stimulation may indicate that much of the Glut1 was trafficked to a compartment incapable of rapid recycling. Alternatively, the protein machinery necessary for rapid recycling may be diminished after a one day growth factor withdrawal. These findings contrast somewhat with previous studies that have shown Rab7-deficiency to prevent targeting of nutrient transporters to lysosomes which allowed maintenance of nutrient uptake via recycling of transporters back to the cell surface even in the absence of growth factors [11]. It is possible, however, that Glut1 is regulated differently than other nutrient transporters through specific interaction with multiple PDZ domain proteins such as GIPC. In support of this notion are findings that Glut1 trafficking is rapamycin insensitive [10], while other nutrient transporters are rapamycin sensitive [34]. Alternatively, deficiency in lysosomal targeting via Rab7 may affect other cellular processes that can sustain cell metabolism and viability independent of Glut1.
Mutation of the Glut1 C-terminal four amino acid PDZ-binding motif had a profound effect on Glut1 and resulted in decreased total and surface Glut1 levels. Interactions with PDZ domain containing proteins, therefore, are likely critical in regulation of Glut1. The PDZ domain-mediated interaction between Glut1 and GIPC appeared to play a key role in regulation of Glut1 recycling. The intracellular punctate localization close to the plasma membrane and association of GIPC with cytoskeletal elements, such as Myo6 and actin [23, 35, 36], have previously suggested that GIPC may play a role in vesicle sorting to the cell surface. Consistent with this proposed role for GIPC, we found increased intracellular levels of Glut1 in GIPC-deficient cells, particularly in the presence of growth factor. It is not clear, however, if or how GIPC may be regulated by growth factor to promote Glut1 recycling. Given the role of GIPC in trafficking of other cell surface receptors to the plasma membrane [20-22], it will be important in future studies to determine if growth factor signaling pathways may also affect their recycling via regulation of GIPC.
In addition to recycling, Glut1 also required the PDZ-binding domain to be protected from rapid targeting to lysosomes for degradation after growth factor withdrawal. Deficiency of GIPC, however, did not lead to Glut1 degradation similar to that observed in Glut1 lacking the PDZ-binding motif or with mutation of the C-terminal valine, a residue known to be important in specificity of PDZ binding [31]. These data suggest that an additional protein may interact with the Glut1 PDZ-binding motif and may associate with Glut1 to prevent Glut1 lysosomal degradation. Reminiscent of this model is the β2-adrenergic receptor, which can interact with multiple PDZ domain proteins and requires association with the PDZ domain protein, EBP50, to prevent β2-adrenergic receptor targeting to lysosomes for destruction [37]. It remains unclear, however, how the Glut1 PDZ-binding motif may protect intracellular Glut1 from lysosomal targeting and degradation.
Sumoylation of lysine 456 of the Glut1 C-terminal tail by Ubc9 has also been shown to regulate total Glut1 protein levels [17]. Mutation of lysine 456 to alanine altered Glut1 levels, with accumulation of both surface and total Glut1 that contrasted starkly with the loss of Glut1 protein observed with mutation of the PDZ-binding domain. Importantly, mutation of both critical sites in the Glut1 C-terminus (K456A/Δ4) presented with the phenotype of the PDZ-binding domain mutant. The Glut1 PDZ-binding motif and possible interactions with PDZ domain proteins, therefore, represent the dominant determinant in Glut1 fate. It may be that Glut1 interaction with a PDZ protein is affected by lysine modification. This type of regulation has been described for the transcriptional co-repressor, CtBP, which is regulated by antagonistic PDZ protein binding and sumoylation [38], and may also occur for Glut1.
Here we demonstrate that the Glut1 C-terminal four amino acids are a key component in growth factor regulation of Glut1. Interestingly, this PDZ-binding motif was essential to allow existence of intracellular Glut1 after growth factor withdrawal, a portion of which could be rapidly returned to the cell surface upon re-stimulation. Similar to Glut4, which is stored intracellularly in specialized storage vesicles [39, 40], Glut1 may also exist in intracellular pools that are independent of standard endosomal and recycling pathways that encompass TfR recycling and dependent on GIPC to return to the cell surface. This trafficking pattern would be less dynamic than that observed in insulin regulated Glut4 trafficking, but nevertheless provides a mechanism to rapidly retrieve some Glut1 to the cell surface should a growth signal be received. Together, these data show that Glut1 is highly regulated through multiple modifications or interactions. As Glut1 is a central glucose transporter in many tissues, these points of Glut1 regulation are important for further study as they may impact cellular glucose homeostasis in both normal and pathological states.
ACKNOWLEDGEMENTS
We thank Jonathan Coloff and Yuxing Zhao for helpful comments in preparation of this manuscript. We would also like to thank Dr. Brent Reed (Louisiana State University, Shreveport, LA) for generously providing the pET30a(+)-GIPC plasmid. We would also like to thank the Department of Pharmacology and Cancer Biology shared microscopy facility and the Duke University Flow Cytometry Shared Resource. This work was funded by NIAID R01 (R01AI063345).
Abbreviations
- NH4Cl
Ammonium Chloride
- CQ
Chloroquine
- GFP
Green Fluorescent Protein
- GIPC
Gα interacting protein interacting protein, C-terminus
- HRP
Horseradish Peroxidase
- IL3
Interleukin-3
- IL7
Interleukin-7
- myrAkt
myristoylated Akt
- PBS
Phosphate Buffered Saline
- PI3K
Phosphatidylinositol 3-kinase
- shRNAi
small hairpin RNA interference
- TfR
Transferrin Receptor
- 2-DOG
2-deoxy-D-glucose
REFERENCES
- 1.Kan O, Baldwin SA, Whetton AD. Apoptosis is regulated by the rate of glucose transport in an interleukin 3 dependent cell line. J Exp Med. 1994;180:917–923. doi: 10.1084/jem.180.3.917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rathmell JC, Farkash EA, Gao W, Thompson CB. IL-7 enhances the survival and maintains the size of naive T cells. J Immunol. 2001;167:6869–6876. doi: 10.4049/jimmunol.167.12.6869. [DOI] [PubMed] [Google Scholar]
- 3.Bentley J, Itchayanan D, Barnes K, McIntosh E, Tang X, Downes CP, Holman GD, Whetton AD, Owen-Lynch PJ, Baldwin SA. Interleukin-3-mediated cell survival signals include phosphatidylinositol 3-kinase-dependent translocation of the glucose transporter GLUT1 to the cell surface. J Biol Chem. 2003;278:39337–39348. doi: 10.1074/jbc.M305689200. [DOI] [PubMed] [Google Scholar]
- 4.Rathmell JC, Vander Heiden MG, Harris MH, Frauwirth KA, Thompson CB. In the absence of extrinsic signals, nutrient utilization by lymphocytes is insufficient to maintain either cell size or viability. Mol Cell. 2000;6:683–692. doi: 10.1016/s1097-2765(00)00066-6. [DOI] [PubMed] [Google Scholar]
- 5.Rathmell JC, Fox CJ, Plas DR, Hammerman PS, Cinalli RM, Thompson CB. Akt-directed glucose metabolism can prevent Bax conformation change and promote growth factor-independent survival. Mol Cell Biol. 2003;23:7315–7328. doi: 10.1128/MCB.23.20.7315-7328.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhao Y, Altman BJ, Coloff JL, Herman CE, Jacobs SR, Wieman HL, Wofford JA, Dimascio LN, Ilkayeva O, Kelekar A, Reya T, Rathmell JC. Glycogen synthase kinase 3alpha and 3beta mediate a glucose-sensitive antiapoptotic signaling pathway to stabilize Mcl-1. Mol Cell Biol. 2007;27:4328–4339. doi: 10.1128/MCB.00153-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Edinger AL, Thompson CB. Akt maintains cell size and survival by increasing mTOR-dependent nutrient uptake. Mol Biol Cell. 2002;13:2276–2288. doi: 10.1091/mbc.01-12-0584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Joost HG, Thorens B. The extended GLUT-family of sugar/polyol transport facilitators: nomenclature, sequence characteristics, and potential function of its novel members (review) Mol Membr Biol. 2001;18:247–256. doi: 10.1080/09687680110090456. [DOI] [PubMed] [Google Scholar]
- 9.Macheda ML, Rogers S, Best JD. Molecular and cellular regulation of glucose transporter (GLUT) proteins in cancer. J Cell Physiol. 2005;202:654–662. doi: 10.1002/jcp.20166. [DOI] [PubMed] [Google Scholar]
- 10.Wieman HL, Wofford JA, Rathmell JC. Cytokine stimulation promotes glucose uptake via phosphatidylinositol-3 kinase/Akt regulation of Glut1 activity and trafficking. Mol Biol Cell. 2007;18:1437–1446. doi: 10.1091/mbc.E06-07-0593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Edinger AL, Cinalli RM, Thompson CB. Rab7 prevents growth factor-independent survival by inhibiting cell-autonomous nutrient transporter expression. Dev Cell. 2003;5:571–582. doi: 10.1016/s1534-5807(03)00291-0. [DOI] [PubMed] [Google Scholar]
- 12.Lum JJ, Bauer DE, Kong M, Harris MH, Li C, Lindsten T, Thompson CB. Growth factor regulation of autophagy and cell survival in the absence of apoptosis. Cell. 2005;120:237–248. doi: 10.1016/j.cell.2004.11.046. [DOI] [PubMed] [Google Scholar]
- 13.Plas DR, Talapatra S, Edinger AL, Rathmell JC, Thompson CB. Akt and Bcl-xL promote growth factor-independent survival through distinct effects on mitochondrial physiology. J Biol Chem. 2001;276:12041–12048. doi: 10.1074/jbc.M010551200. [DOI] [PubMed] [Google Scholar]
- 14.Gottlob K, Majewski N, Kennedy S, Kandel E, Robey RB, Hay N. Inhibition of early apoptotic events by Akt/PKB is dependent on the first committed step of glycolysis and mitochondrial hexokinase. Genes Dev. 2001;15:1406–1418. doi: 10.1101/gad.889901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Verhey KJ, Hausdorff SF, Birnbaum MJ. Identification of the carboxy terminus as important for the isoform-specific subcellular targeting of glucose transporter proteins. J Cell Biol. 1993;123:137–147. doi: 10.1083/jcb.123.1.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bunn RC, Jensen MA, Reed BC. Protein interactions with the glucose transporter binding protein GLUT1CBP that provide a link between GLUT1 and the cytoskeleton. Mol Biol Cell. 1999;10:819–832. doi: 10.1091/mbc.10.4.819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Giorgino F, de Robertis O, Laviola L, Montrone C, Perrini S, McCowen KC, Smith RJ. The sentrin-conjugating enzyme mUbc9 interacts with GLUT4 and GLUT1 glucose transporters and regulates transporter levels in skeletal muscle cells. Proc Natl Acad Sci U S A. 2000;97:1125–1130. doi: 10.1073/pnas.97.3.1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Songyang Z, Fanning AS, Fu C, Xu J, Marfatia SM, Chishti AH, Crompton A, Chan AC, Anderson JM, Cantley LC. Recognition of unique carboxyl-terminal motifs by distinct PDZ domains. Science. 1997;275:73–77. doi: 10.1126/science.275.5296.73. [DOI] [PubMed] [Google Scholar]
- 19.Vaccaro P, Brannetti B, Montecchi-Palazzi L, Philipp S, Helmer Citterich M, Cesareni G, Dente L. Distinct binding specificity of the multiple PDZ domains of INADL, a human protein with homology to INAD from Drosophila melanogaster. J Biol Chem. 2001;276:42122–42130. doi: 10.1074/jbc.M104208200. [DOI] [PubMed] [Google Scholar]
- 20.Blobe GC, Liu X, Fang SJ, How T, Lodish HF. A novel mechanism for regulating transforming growth factor beta (TGF-beta) signaling. Functional modulation of type III TGF-beta receptor expression through interaction with the PDZ domain protein, GIPC. J Biol Chem. 2001;276:39608–39617. doi: 10.1074/jbc.M106831200. [DOI] [PubMed] [Google Scholar]
- 21.Hirakawa T, Galet C, Kishi M, Ascoli M. GIPC binds to the human lutropin receptor (hLHR) through an unusual PDZ domain binding motif, and it regulates the sorting of the internalized human choriogonadotropin and the density of cell surface hLHR. J Biol Chem. 2003;278:49348–49357. doi: 10.1074/jbc.M306557200. [DOI] [PubMed] [Google Scholar]
- 22.Jeanneteau F, Diaz J, Sokoloff P, Griffon N. Interactions of GIPC with dopamine D2, D3 but not D4 receptors define a novel mode of regulation of G protein-coupled receptors. Mol Biol Cell. 2004;15:696–705. doi: 10.1091/mbc.E03-05-0293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Reed BC, Cefalu C, Bellaire B, Cardelli JA, Louis T, Salamon J, Bloecher MA, Bunn RC. GLUT1CBP(TIP2/GIPC1) Interactions with GLUT1 and Myosin VI: Evidence Supporting an Adapter Function for GLUT1CBP. Mol Biol Cell. 2005 doi: 10.1091/mbc.E04-11-0978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vander Heiden MG, Chandel NS, Williamson EK, Schumacker PT, Thompson CB. Bcl-xL regulates the membrane potential and volume homeostasis of mitochondria. Cell. 1997;91:627–637. doi: 10.1016/s0092-8674(00)80450-x. [DOI] [PubMed] [Google Scholar]
- 25.Suzuki N, Shibata Y, Urano T, Murohara T, Muramatsu T, Kadomatsu K. Proteasomal degradation of the nuclear targeting growth factor midkine. J Biol Chem. 2004;279:17785–17791. doi: 10.1074/jbc.M310772200. [DOI] [PubMed] [Google Scholar]
- 26.Pfister MF, Ruf I, Stange G, Ziegler U, Lederer E, Biber J, Murer H. Parathyroid hormone leads to the lysosomal degradation of the renal type II Na/Pi cotransporter. Proc Natl Acad Sci U S A. 1998;95:1909–1914. doi: 10.1073/pnas.95.4.1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Furuchi T, Aikawa K, Arai H, Inoue K. Bafilomycin A1, a specific inhibitor of vacuolar-type H(+)-ATPase, blocks lysosomal cholesterol trafficking in macrophages. J Biol Chem. 1993;268:27345–27348. [PubMed] [Google Scholar]
- 28.Meresse S, Gorvel JP, Chavrier P. The rab7 GTPase resides on a vesicular compartment connected to lysosomes. J Cell Sci. 1995;108(Pt 11):3349–3358. doi: 10.1242/jcs.108.11.3349. [DOI] [PubMed] [Google Scholar]
- 29.Bucci C, Thomsen P, Nicoziani P, McCarthy J, van Deurs B. Rab7: a key to lysosome biogenesis. Mol Biol Cell. 2000;11:467–480. doi: 10.1091/mbc.11.2.467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chavrier P, Parton RG, Hauri HP, Simons K, Zerial M. Localization of low molecular weight GTP binding proteins to exocytic and endocytic compartments. Cell. 1990;62:317–329. doi: 10.1016/0092-8674(90)90369-p. [DOI] [PubMed] [Google Scholar]
- 31.van Ham M, Hendriks W. PDZ domains-glue and guide. Mol Biol Rep. 2003;30:69–82. doi: 10.1023/a:1023941703493. [DOI] [PubMed] [Google Scholar]
- 32.Vander Heiden MG, Plas DR, Rathmell JC, Fox CJ, Harris MH, Thompson CB. Growth factors can influence cell growth and survival through effects on glucose metabolism. Mol Cell Biol. 2001;21:5899–5912. doi: 10.1128/MCB.21.17.5899-5912.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Frauwirth KA, Thompson CB. Regulation of T lymphocyte metabolism. J Immunol. 2004;172:4661–4665. doi: 10.4049/jimmunol.172.8.4661. [DOI] [PubMed] [Google Scholar]
- 34.Edinger AL, Linardic CM, Chiang GG, Thompson CB, Abraham RT. Differential effects of rapamycin on mammalian target of rapamycin signaling functions in mammalian cells. Cancer Res. 2003;63:8451–8460. [PubMed] [Google Scholar]
- 35.Aschenbrenner L, Lee T, Hasson T. Myo6 facilitates the translocation of endocytic vesicles from cell peripheries. Mol Biol Cell. 2003;14:2728–2743. doi: 10.1091/mbc.E02-11-0767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Naccache SN, Hasson T, Horowitz A. Binding of internalized receptors to the PDZ domain of GIPC/synectin recruits myosin VI to endocytic vesicles. Proc Natl Acad Sci U S A. 2006;103:12735–12740. doi: 10.1073/pnas.0605317103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cao TT, Deacon HW, Reczek D, Bretscher A, von Zastrow M. A kinase-regulated PDZ-domain interaction controls endocytic sorting of the beta2-adrenergic receptor. Nature. 1999;401:286–290. doi: 10.1038/45816. [DOI] [PubMed] [Google Scholar]
- 38.Lin X, Sun B, Liang M, Liang YY, Gast A, Hildebrand J, Brunicardi FC, Melchior F, Feng XH. Opposed regulation of corepressor CtBP by SUMOylation and PDZ binding. Mol Cell. 2003;11:1389–1396. doi: 10.1016/s1097-2765(03)00175-8. [DOI] [PubMed] [Google Scholar]
- 39.Hou JC, Pessin JE. Ins (endocytosis) and outs (exocytosis) of GLUT4 trafficking. Curr Opin Cell Biol. 2007;19:466–473. doi: 10.1016/j.ceb.2007.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rea S, James DE. Moving GLUT4: the biogenesis and trafficking of GLUT4 storage vesicles. Diabetes. 1997;46:1667–1677. doi: 10.2337/diab.46.11.1667. [DOI] [PubMed] [Google Scholar]