

Abstract
Autoimmune diseases such as systemic lupus erythematosus are complex genetic traits with contributions from major histocompatibility complex (MHC) genes and multiple unknown non-MHC genes. Studies of animal models of lupus have provided important insight into the immunopathogenesis of disease, and genetic analyses of these models overcome certain obstacles encountered when studying human patients. Genome-wide scans of different genetic crosses have been used to map several disease-linked loci in New Zealand hybrid mice. Although some consensus exists among studies mapping the New Zealand Black (NZB) and New Zealand White (NZW) loci that contribute to lupus-like disease, considerable variability is also apparent. A variable in these studies is the genetic background of the non-autoimmune strain, which could influence genetic contributions from the affected strain. A direct examination of this question was undertaken in the present study by mapping NZB nephritis-linked loci in backcrosses involving different non-autoimmune backgrounds. In a backcross with MHC-congenic C57BL/6J mice, H2z appeared to be the strongest genetic determinant of severe lupus nephritis, whereas in a backcross with congenic BALB/cJ mice, H2z showed no influence on disease expression. NZB loci on chromosomes 1, 4, 11, and 14 appeared to segregate with disease in the BALB/cJ cross, but only the influence of the chromosome 1 locus spanned both crosses and showed linkage with disease when all mice were considered. Thus, the results indicate that contributions from disease-susceptibility loci, including MHC, may vary markedly depending on the non-autoimmune strain used in a backcross analysis. These studies provide insight into variables that affect genetic heterogeneity and add an important dimension of complexity for linkage analyses of human autoimmune disease.
Keywords: systemic lupus erythematosus, genetic linkage, murine lupus
The F1 hybrids of New Zealand Black (NZB) and New Zealand White (NZW) mice remain one of the best studied models of human systemic lupus erythematosus (1, 2). These mice spontaneously develop an autoimmune disease characterized by the production of IgG antinuclear antibodies and the development of a severe immune-complex glomerulonephritis. The disease phenotype of the F1 is distinct from that of the two parental strains, and genes from each parent are necessary for the full expression of lupus-like nephritis (1, 3). Similar to other autoimmune diseases, lupus nephritis in (NZB × NZW)F1 mice is a complex genetic trait with contributions from major histocompatibility complex (MHC) and multiple unknown non-MHC genes (4–12). Previous analyses of backcross and congenic mice have demonstrated that the most important dominant contribution from the NZW is encoded within the MHC (H2z in NZW), most likely by class II MHC genes (3, 10, 13, 14). Genome-wide linkage analyses have mapped NZB nephritis-linked loci on chromosomes 1, 4, and 7 (among others) in addition to the MHC (H2d in NZB), but considerable variability exists among these studies (4, 5, 8–12).
A genetic analysis of linkage usually involves directed breeding of the affected strain with a mouse strain that does not express the phenotype being studied and subsequent analysis of backcross or intercross mice. Limited attention has been given to the genetic background from the unaffected strain used in genetic crosses. In the present work, we directly examined whether background genes in different non-autoimmune strains could change the influence of disease-linked loci from autoimmune NZB mice in the development of severe lupus nephritis in backcross progeny. The results indicate that disease-susceptibility loci, including MHC, may vary markedly depending on the non-autoimmune strain used in the backcross analysis.
MATERIALS AND METHODS
Mice.
Parental NZB/BINJ, C57BL/6J (designated B6), and BALB/cJ (designated BALB) mice were obtained from The Jackson Laboratory and were maintained in the animal care facility at The National Jewish Center for Immunology and Respiratory Medicine. All congenic, F1, and backcross mice were bred and maintained at The National Jewish Center. Only female mice were studied for expression of disease.
C57BL/6J or BALB/cJ were made congenic for H2z by mating these mice with NZW mice, and backcrossing the progeny to C57BL/6J or BALB/cJ mice, respectively. Inheritance of H2z was monitored both by immunofluorescence analysis of I-Az expression and by screening for a simple sequence length polymorphism in the tumor necrosis factor α gene (Tnfa) (15). Each congenic strain was made homozygous for H2z after 12 generations, and these strains are designated B6.H2z and BALB.H2z, respectively. All F1 (and subsequent backcross) mice were bred using mice homozygous for H2z. B6.H2z and BALB.H2z congenic strains were analyzed for the length of the congenic NZW interval bred onto the respective parental strains. In relation to the MHC, analysis of markers ≈4 centimorgans (cM) proximal to MHC on chromosome 17 (D17Mit51; ≈14.5 cM from the centromere), within the MHC [Tnfa (15) or H2z Alu repeat (http://www-mickey.utmem.edu); ≈19 cM from the centromere], and 3 cM distal to MHC (D17Mit49 or D17Mit50; ≈22.3 cM from the centromere) showed alleles inherited from NZW in the B6.H2z and BALB.H2z congenic strains. These data, therefore, show that the entire NZW MHC was inherited in both congenic strains.
Evaluation of Renal Disease and Collection of Tissue.
Mice were studied from 4 to 12 months of age, and were evaluated for proteinuria at bimonthly intervals using tetrachlorophenol-tetrabromosulfophthalein paper (Chemstrip; Boehringer Mannheim) as described (3). A scoring system of 0 to 3+ was used, as follows: 0 or trace, <30 mg/dl; 1+, ≈30 mg/dl; 2+, ≈100 mg/dl; 3+, >300 mg/dl. A score of 2+ or greater was considered indicative of severe proteinuria, and mice exhibiting severe proteinuria on two or more successive occasions, or at the final evaluation before death or sacrifice, were considered positive for renal disease. As in previous studies (3, 8, 11, 12), the majority of these mice died before 1 year of age. A negative phenotype was ascribed to mice which did not exhibit proteinuria during the 12 months of follow-up, and these mice appeared healthy at the time of sacrifice. Mice that produced proteinuria >100 mg/dl at least once, but whose proteinuria values thereafter fluctuated above and below 100 mg/dl, were excluded from further analysis. All of the mice with this intermediate phenotype were alive at 12 months of age. In the (B6.H2z × NZB)F1 × NZB backcross, 12% of 152 study mice were excluded on this basis. In the (BALB.H2z × NZB)F1 × NZB backcross, 12% of 184 mice were excluded. The tip of the tail from all backcross mice was excised at 4 months of age. The liver and kidneys were collected at the time of death or sacrifice at 12 months of age. All tissues were stored at −70°C, and DNA was extracted as described previously (8).
Genomic Mapping Using Simple Sequence Length Polymorphisms (SSLPs).
Oligonucleotide primers for SSLP mapping analysis were either purchased from a commercial vendor (Research Genetics, Huntsville, AL) or synthesized by The Molecular Resource Center at the National Jewish Center using an Applied Biosystems model 392 DNA synthesizer. Primer nucleotide sequences (http://www-genome.wi.mit.edu/), and the methods for SSLP screening and mapping have been previously described (8, 11, 12, 16, 17). Positions of SSLP markers (and genetic loci) are given in accordance with the Mouse Chromosome Committee Reports (distributed by R. Williams, University of Tennessee, http://www.mickey.utmem.edu).
Amplification of the simple sequence repeats was achieved using the polymerase chain reaction (PCR) in a PTC-100 thermal cycler (MJ Research, Cambridge, MA). 20 μl reactions were conducted using 35 cycles consisting of 30 sec at 94°C, 1 min at 55°C, and 30 sec at 72°C. PCR product (10–15 μl) was loaded onto a 15% polyacrylamide gel and electrophoresed at 12 V/cm for 2–4 h. Gels were then soaked in a 500 ng/ml solution of ethidium bromide in water and examined under ultraviolet transillumination at 254 nm. Polymorphisms were scored in comparison to results from parental PCR products.
Statistical Analysis.
Linkage of different loci with nephritis was examined by χ2 analysis, using a standard [2 × 2] contingency matrix (8, 17) and χ2 values were used to determine p values. As recommended by Lander and Kruglyak (18), the threshold used for suggestive linkage was P < 3.4 × 10−3 (χ2 > 8.58; 1 df), and for probable linkage was P < 1.0 × 10−4 (χ2 > 15.13; 1 df). A value of P < 0.01 was used for confirmation of linkage if a colocalizing locus had previously been reported as associated at this level in a separate data set (18). Odds ratios (OR) were calculated as:
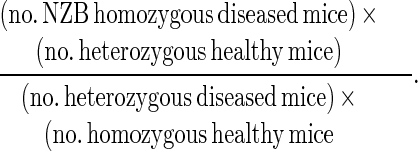 |
Comparisons of ORs were performed using the Breslow–Day test for homogeneity (19), calculated using a statistics software package (stat exact; CYTEL Software, Cambridge, MA).
RESULTS
To study the possible influence of non-autoimmune background genes on contributions from disease-linked loci, we designed two backcrosses—(B6.H2z × NZB)F1 × NZB and (BALB.H2z × NZB)F1 × NZB—that are designated B6 and BALB backcrosses, respectively. BALB/cJ and C56BL/6J mice were chosen as two strains unrelated to each other or to NZB, and these strains do not show any evidence of autoimmune disease. A region encompassing the NZW MHC was bred onto these non-autoimmune backgrounds, and 12th generation mice congenic and homozygous for H2z were used in the present studies. Thus, in each backcross, about half of the mice will express an MHC type (H2d/z) shown to be associated with increased expression of lupus nephritis compared with mice expressing H2d/d (the other half of backcross mice) (3, 5, 10, 13).
Female (B6.H2z × NZB)F1 and (BALB.H2z × NZB)F1 mice and respective backcross mice were followed for 1 year for the development of severe nephritis as described previously (3, 8, 11, 12). None of the F1 mice demonstrated proteinuria or other evidence of disease (n > 22 for each F1 strain), indicating that MHC type is not sufficient for disease expression and that the C57BL/6 and BALB/c backgrounds, compared with NZB or NZW backgrounds, contain disease resistance alleles.
Overall, 20% of the B6 backcross (n = 152) and 17% of the BALB backcross (n = 184) mice developed severe proteinuria, and, as in previous studies, most of these mice died from their renal disease before 1 year of age (data not shown). Surprisingly, a strong influence of MHC type on disease expression was seen only in the B6 cross (Fig. 1). At 12 months of age, 34% of the H2d/z mice had developed disease compared with 8% of the H2d/d mice (P < 0.001, Fig. 1). In contrast, the BALB backcross mice showed no difference in disease expression related to MHC type, and the influence of MHC in the B6 versus BALB backcross was significantly different (P = 0.02) as determined by a comparison of ORs. It is emphasized that in both crosses, H2d/d mice are being compared with H2d/z mice, and analysis of the B6 and BALB congenic strains showed that similar levels of surface I-Az were expressed (data not shown). Furthermore, in both congenic strains, the entire NZW MHC region was encompassed within the congenic intervals bred on chromosome 17 (NZW markers extended from 4 cM proximal through at least 3 cM distal of MHC).
Figure 1.
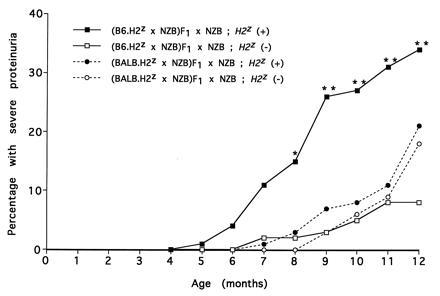
Cumulative incidence of severe nephritis in backcross mice in relation to MHC type. The numbers of mice in each group are given in Table 1. *P < 0.01 and **P < 0.001 when comparing (B6.H2z × NZB)F1 × NZB backcross mice typed as H2z/d versus H2d/d. There was no difference in disease expression when (BALB.H2z × NZB)F1 × NZB backcross mice, typed as either H2z/d versus H2d/d, were compared.
Mapping of non-MHC loci was directed to NZB chromosomal regions that have previously been suggested to be linked with nephritis and/or autoantibody production (8–12, 20). Table 1 shows loci that were found to have at least suggestive linkage in one or both backcrosses. Data are presented as P values, determined from χ2 analysis; the ORs are also shown. The strongest linkage with nephritis was seen on distal chromosome 1 at Crp/Sap, (≈95 cM from the centromere), and is designated Nba2 (New Zealand black autoimmunity 2) because it represents confirmation of a previous mapping (11). This was the only locus showing at least a trend for linkage with disease in both backcrosses. Furthermore, the contribution from this locus was greatest in H2z-positive mice in each backcross, which is reflected in the P values as well as the different ORs in H2z-positive versus H2z-negative mice (7.29 versus 3.0 in the B6 cross; 4.48 versus 0.74 in the BALB cross). The latter was significantly different at P < 0.05 by the Breslow–Day test for homogeneity. Overall, the chromosome 1 locus showed suggestive linkage with disease when all mice from both crosses were grouped together, and met criteria for probable linkage in all H2z-positive mice.
Table 1.
NZB loci linked with nephritis
| Marker* | Position* | (B6.H2z ×
NZB)F1 × NZB
|
(BALB.H2z ×
NZB)F1 × NZB
|
Both crosses
|
|||
|---|---|---|---|---|---|---|---|
| P value | OR | P value | OR | P value | OR | ||
| All mice (n = 134) | All mice (n = 158) | All mice (n = 292) | |||||
| D1Mit111 | 1, 91 | 0.16 | 1.79 | 0.48 | 1.40 | 0.13 | 1.61 |
| Crp/Sap | 1, 94 | 1.0 × 10−3† | 4.58 | 0.071 | 2.17 | 3.3 × 10−4† | 3.09 |
| D1Mit206 | 1, 96 | 3.0 × 10−3† | 4.50 | NT | NT | NA | NA |
| D4Mit9 | 4, 43 | 0.17 | 1.07 | 2.2 × 10−3† | 3.65 | 0.020 | 2.01 |
| D4Mit58/D4Mit28 | 4, 50 | 0.23 | 1.67 | 2.9 × 10−3† | 3.42 | 2.6 × 10−3† | 2.44 |
| D4Mit70 | 4, 65 | 0.71 | 1.17 | 0.020 | 2.51 | 0.052 | 1.74 |
| D11Mit84/D11Mit2 | 11, 17/11, 9 | 0.63 | 1.24 | 0.011 | 2.90 | 0.026 | 1.96 |
| D14Mit41/D14Mit34 | 14, 43/14, 40 | 0.097 | 2.05 | 0.044 | 2.26 | 8.8 × 10−3 | 2.16 |
| H2§ | 17, 19 | 4.4 × 10−4† | 5.56 | 0.55 | 1.28 | 4.0 × 10−3 | 2.44 |
| H2z-positive (n = 74) | H2z-positive (n = 90) | H2z-positive (n = 164) | |||||
| D1Mit111 | 1, 91 | 0.012 | 3.56 | 0.15 | 2.39 | 2.9 × 10−3† | 3.11 |
| Crp/Sap | 1, 94 | 3.0 × 10−4† | 7.29 | 5.5 × 10−3 | 4.48 | 2.6 × 10−6‡ | 5.97 |
| D1Mit206 | 1, 96 | 8.4 × 10−4† | 7.13 | NT | NT | NA | NA |
| D4Mit9 | 4, 43 | 0.99 | 1.00 | 0.045 | 2.88 | 0.14 | 1.71 |
| D4Mit58/D4Mit28 | 4, 50 | 0.17 | 2.02 | 0.051 | 2.74 | 0.015 | 2.40 |
| D4Mit70 | 4, 65 | 0.68 | 1.13 | 0.096 | 2.34 | 0.12 | 1.74 |
| D11Mit84/D11Mit2 | 11, 17/11, 9 | 0.87 | 0.92 | 2.4 × 10−3† | 5.02 | 0.022 | 2.31 |
| D14Mit41/D14Mit34 | 14, 43/14, 40 | 0.45 | 1.47 | 2.9 × 10−3† | 4.69 | 4.8 × 10−3 | 2.76 |
Mice were classified as positive for nephritis if proteinuria was detected at ≥100 mg/dl on two or more successive occasions, or ≥100 mg/dl proteinuria at the testing immediately prior to death. Mice which never exhibited proteinuria at ≥100 mg/dl were classified as negative for nephritis. The percentages of mice developing nephritis in each backcross are shown in Fig. 1. NT, not tested; NA, not applicable.
Markers and their respective positions are presented. For some chromosomal positions, it was necessary to use different markers for the two backcrosses because of the absence of polymorphism with NZB. This is indicated by the presence of two marker names separated by a slash. In these cases the marker used for the (B6.H2z × NZB)F1 × NZB backcross is presented to the left of the slash, and the marker used for the (BALB.H2z × NZB)F1 × NZB backcross is presented to the right of the slash.
Loci showing suggestive linkage with nephritis (χ2 > 8.58; P < 0.0034).
Loci showing probable linkage with nephritis (χ2 > 15.13; P < 0.0001).
The disease susceptibility allele for this locus, H2z, is contributed by the B6.H2z or BALB.H2z strain. The inverse of the odds ratio is presented for this locus for ease of comparison with all other loci, for which the disease susceptibility allele is contributed by the NZB mouse.
Table 1 also shows that three other loci, on chromosomes 4, 11, and 14, met criteria for suggestive linkage, but only in the BALB backcross. The locus on chromosome 4 is particularly interesting since a colocalizing locus (Nba1) has been mapped previously in several studies (Table 2) (8–12). Thus, the present mapping results in the BALB backcross also represent a confirmation of linkage, but this locus showed no trend for linkage in the B6 backcross and its contribution in the BALB versus B6 backcross was significantly different (P = 0.04). Linkage at the chromosome 4 locus appeared to be independent of MHC type, with no increase in OR when H2z-positive and -negative mice are compared. In contrast, NZB loci on chromosomes 11 and 14 showed greater evidence for linkage in the H2z-positive mice, and the interaction with MHC was statistically significant at P = 0.03 and P = 0.06, respectively, by comparison of ORs.
Table 2.
NZB loci linked with nephritis in different studies of New Zealand hybrid mice
| Cross | Reference | Linkage
with loci on chromosome
|
MHC influence* | |||||
|---|---|---|---|---|---|---|---|---|
| 1 | 4 | 7 | 11 | 14 | 17 | |||
| (B6.H2z × NZB)F1 × NZB | This work | ++ | —† | ++ | d/z > d/d | |||
| (C57BL/6 × NZM)F1 × NZM‡ | 9 | +++ | +++ | +++ | +++ | b/z > z/z | ||
| (BALB.H2z × NZB)F1 × NZB | This work | + | ++ | —† | ++ | ++ | d/z ≈ d/d | |
| (NZB × SM/J)F1 × NZW | 11 | ++ | v/z ≈ d/z | |||||
| (NZB × NZW)F1 × NZW | 8, 12 | ++ | ++§ | d/z > z/z | ||||
| (NZB × NZW)F1 × NZW | 22 | ND | —¶ | ND | ND | ND | ++ | d/z > z/z |
| (NZB × NZW)F2 | 10 | ++ | ++‖ | +++ | d/z > d/d = z/z | |||
+, P ≤ 0.01; ++, P ≤ 0.0034; +++, P ≤ 0.0001. ND, not determined.
Influence of MHC haplotypes on expression of lupus nephritis in each study.
Loci on mid and proximal chromosome 7 were screened and no trend for linkage was found.
NZM (NZM/Aeg2410) is a recombinant inbred strain of NZB and NZW backgrounds. The loci shown to be linked with disease are not known to be of NZB or NZW origin, although the disease-linked locus on chromosome 1 appears to be contained within an NZW interval (9, 21).
Recent results with additional mice showed linkage (χ2 ≥ 14.7; P ≤ 0.0001) of the NZB H2d with nephritis (12).
A trend for linkage (P < 0.02) with nephritis was found for inheritance of a colocalizing NZB locus on distal chromosome 4 (22). We have not indicated linkage in this table since it did not meet criteria at P < 0.01 (18).
Although this study did not find evidence for linkage with nephritis, results showed suggestive linkage with IgG autoantibodies to chromatin.
Table 2 presents a summary of different studies that have screened for NZB genes having linkage with nephritis. Several previous studies have noted the strong influence of MHC, especially when mice heterozygous (H2d/z or H2b/z) were compared with mice homozygous for the H2d or H2z haplotypes (5, 9, 10, 12). Not shown in Table 2, an analysis of (NZB × NZW)F1 × NZB backcross mice also showed that H2d/z versus H2d/d genotype was the major genetic influence in the development of lupus nephritis (3), and congenic studies involving NZB and NZW backgrounds also demonstrated the importance of H2d/z versus H2d/d for expression of disease (13). Thus, the absence of influence of MHC type (i.e., H2d/z versus H2d/d) in our BALB backcross is striking.
As noted above, a distal chromosome 1 locus (here designated Nba2) linked with nephritis was also apparent in a previous study that examined NZB genes after crossing to the non-autoimmune SM/J strain (11) (Table 2). It is interesting that this NZB contribution has not been found in studies that included only NZB and NZW backgrounds. This may imply that this locus is codominant or shared between NZB and NZW autoimmune strains. Consistent with this hypothesis, a study of recombinant NZM mice showed a contribution from an NZW interval on distal chromosome 1 (9, 21). Table 2 also shows that several studies have identified loci linked with nephritis on chromosome 4 with maximal significance in a region about 50–70 cM from the centromere. None of the studies listed in Table 2 found nephritis-linked loci on chromosomes 11 or 14. However, mapping was directed to these positions in the present work because NZB and/or NZW loci in similar chromosomal positions were noted to be linked with IgG autoantibody production (10, 12, 20).
DISCUSSION
In the analysis of genes contributing to disease in animal models of autoimmunity, the affected strain is usually outcrossed to another strain that does not express the phenotype being studied. Whether the genes from the unaffected or non-autoimmune strain can change the results of linkage analyses has not been directly studied. McAleer et al. (23) analyzed genes contributing to insulin-dependent diabetes in NOD mice in outcrosses to MHC-congenic NON mice and compared results to previous outcrosses with C57BL/10 mice. Background genetic influences from NON versus C57BL/10 strains were apparent, but the fact that NOD and NON are related backgrounds likely influenced some of the findings. Furthermore, the comparison was retrospective, which could have introduced another variable. In our study, we used two well-studied and unrelated normal strains, C57BL/6J and BALB/cJ, and all backcross progeny were bred and followed for disease concomitantly. Our results indicate that the genetic background of the non-autoimmune strain can markedly alter the genes showing linkage with disease in a backcross analysis.
We believe that it is extremely unlikely that linkage in the current study reflects false positive results. Despite screening only a subset of loci, we used statistical thresholds suggested for genome-wide scans for linkage [as recommended by Lander and Kruglyak (18)]. Most of the loci mapped met criteria for suggestive linkage (P < 0.0034), but sometimes only when H2z-positive mice were separated from total mice. Overall, only the locus on chromosome 1 (Nba2) met criteria for probable linkage (P < 0.0001) in H2z-positive mice. However, because of the numerous studies that have implicated MHC (particularly H2d/z) in the development of nephritis in this animal model and because of previous studies showing that colocalizing loci on chromosomes 1 and 4 are linked with lupus nephritis, we believe that these loci have been proven to be involved in the disease process. Loci on chromosomes 11 and 14, although colocalizing with loci linked with IgG autoantibody production in previous studies (10, 12, 20), will require confirmation of their linkage with nephritis in separate data sets. It is of interest that the locus on chromosome 14 colocalizes with the T cell receptor alpha-chain locus (Tcra), for which an interaction with MHC could be envisioned mechanistically.
We did not complete a genome-wide scan of the two backcrosses. Screening was directed to chromosomal regions that have shown linkage with autoimmune phenotypes, and 37 markers (≈26% of the genome) and 35 markers (≈32% of the genome) were covered in the B6 and BALB backcrosses, respectively. Still, if the incidence of nephritis in NZB mice congenic for H2d/z averages 50–70% (3, 14, 24), calculations of genotypic risk ratios and total risk ratios (25) estimate that greater than 90% of the risk in the B6 cross is accounted for by Nba2 and H2z (25). In support of this estimate, recent preliminary studies with additional markers that cover about 90% of the B6 backcross have not mapped any additional loci linked (at P < 0.02) to nephritis (S.J.R., T.J.V., J. Akolt, and B.L.K., unpublished results). Especially related to the directed genetic screening and extent of genetic risk identified in this study, our results support the conclusion that the pool of lupus etiologic alleles in NZB mice is limited in size. Importantly, our conclusions regarding the effects of the non-autoimmune background would not be altered by mapping more loci linked with disease.
The mechanism for the influence of normal background on disease susceptibility loci is unclear and may be different, depending on the disease-contributing gene. One straightforward possibility is that C57BL/6 and BALB/c strains express polymorphic alleles of the genes conferring increased risk for disease. For example, NZB and C57BL/6 may share a similar allele at the disease gene on chromosome 4, which is different compared with BALB/c. However, this explanation cannot apply to the different effects of MHC haplotype in the two backcrosses, since the MHC haplotypes are identical in both crosses, inherited from NZB and NZW (i.e., NZW congenic interval). In the case of MHC, the influence of non-autoimmune background therefore must be related to gene(s) at a distance from the MHC.
Genetic heterogeneity is apparent in a genetic analysis when separate combinations of disease-linked loci can independently cause disease. Thus, the different combinations of NZB loci that contribute to nephritis in the two backcrosses is indicative of genetic heterogeneity, which is dependent on the different non-autoimmune backgrounds used in the breeding. The results are consistent with prior studies of lupus-prone New Zealand mice, which found that different combinations of disease-linked loci can independently determine disease susceptibility and that nephritis is inherited as a threshold genetic liability (9, 11). Although these previous studies each involved one strain combination, the variable genetic background among backcross progeny likely determined which susceptibility alleles operate in an individual mouse.
Genome-wide mapping studies in human systemic lupus erythematosus are being planned and initiated. Problems anticipated to be encountered include the variability of phenotypes expressed by different patients, the likelihood that different disease-susceptibility genes may operate in different populations (another type of genetic heterogeneity), and the limited number of multiplex families expressing a similar and well-defined phenotype. Clearly, the present results add another issue of complexity when considering the likelihood of success in linkage analyses of human systemic lupus erythematosus. However, our findings may also provide a guide to those genetic contributions in the human disease that will be least affected by genetic background influences.
Acknowledgments
This work was supported by National Institutes of Health Grant AR37070.
Footnotes
Abbreviations: MHC, major histocompatibility complex; cM, centimorgan(s); NZB, New Zealand Black; NZW, New Zealand White; OR, odds ratio; SSLP, simple sequence length polymorphism.
References
- 1.Theofilopoulos A N, Dixon F J. Adv Immunol. 1985;37:269–390. doi: 10.1016/s0065-2776(08)60342-9. [DOI] [PubMed] [Google Scholar]
- 2.Kotzin B L. Cell. 1996;85:303–306. doi: 10.1016/s0092-8674(00)81108-3. [DOI] [PubMed] [Google Scholar]
- 3.Kotzin B L, Palmer E. J Exp Med. 1987;165:1237–1251. doi: 10.1084/jem.165.5.1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vyse T J, Todd J A. Cell. 1996;85:311–318. doi: 10.1016/s0092-8674(00)81110-1. [DOI] [PubMed] [Google Scholar]
- 5.Drake C G, Rozzo S J, Vyse T J, Palmer E, Kotzin B L. Immunol Rev. 1995;144:51–74. doi: 10.1111/j.1600-065x.1995.tb00065.x. [DOI] [PubMed] [Google Scholar]
- 6.Shirai T. Immunol Today. 1982;3:187–194. [Google Scholar]
- 7.Theofilopoulos A N. Immunol Today. 1995;16:150–159. doi: 10.1016/0167-5699(95)80133-2. [DOI] [PubMed] [Google Scholar]
- 8.Drake C G, Babcock S K, Palmer E, Kotzin B L. Proc Natl Acad Sci USA. 1994;91:4062–4066. doi: 10.1073/pnas.91.9.4062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Morel L, Rudofsky U H, Longmate J A, Schiffenbauer J, Wakeland E K. Immunity. 1994;1:219–229. [PubMed] [Google Scholar]
- 10.Kono D H, Burlingame R W, Owens D G, Kuramochi A, Balderas R S, Balomenos D, Theofilopoulos A N. Proc Natl Acad Sci USA. 1994;91:10168–10172. doi: 10.1073/pnas.91.21.10168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Drake C G, Rozzo S J, Hirschfeld H F, Smarnworawong N P, Palmer E, Kotzin B L. J Immunol. 1995;154:2441–2447. [PubMed] [Google Scholar]
- 12.Vyse T J, Drake C G, Rozzo S J, Roper E, Izui S, Kotzin B L. J Clin Invest. 1996;98:1762–1772. doi: 10.1172/JCI118975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hirose S, Ueda G, Noguchi K, Okada T, Sekigawa I, Sato H, Shirai T. Eur J Immunol. 1986;16:1631–1633. doi: 10.1002/eji.1830161226. [DOI] [PubMed] [Google Scholar]
- 14.Chiang B L, Bearer E, Ansari A, Dorshkind K, Gershwin M E. J Immunol. 1990;145:94–101. [PubMed] [Google Scholar]
- 15.Jongeneel C V, Acha-Orbea H, Blankenstein T. J Exp Med. 1990;171:2141–2146. doi: 10.1084/jem.171.6.2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Love J M, Knight A M, McAleer M A, Todd J A. Nucleic Acids Res. 1990;18:4123–4130. doi: 10.1093/nar/18.14.4123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dietrich W F, Miller J, Steen R, Merchant M A, Damron-Boles D, et al. Nature (London) 1996;380:149–151. doi: 10.1038/380149a0. [DOI] [PubMed] [Google Scholar]
- 18.Lander E, Kruglyak L. Nat Genet. 1995;11:241–247. doi: 10.1038/ng1195-241. [DOI] [PubMed] [Google Scholar]
- 19.Breslow N E, Day N E. In: Statistical Methods in Cancer Research. Breslow N E, Day N E, editors. Vol. 1. Lyons, France: Int. Agency Res. Cancer Sci. Publ.; 1980. pp. 5–338. [PubMed] [Google Scholar]
- 20.Vyse T J, Morel L, Tanner F J, Wakeland E K, Kotzin B L. J Immunol. 1996;157:2719–2727. [PubMed] [Google Scholar]
- 21.Morel L, Yu Y, Blenman K R, Caldwell R A, Wakeland E K. Mamm Genome. 1996;7:335–339. doi: 10.1007/s003359900098. [DOI] [PubMed] [Google Scholar]
- 22.Hirose S, Tsurui H, Nishimura H, Jiang Y, Shirai T. Int Immunol. 1994;6:1857–1864. doi: 10.1093/intimm/6.12.1857. [DOI] [PubMed] [Google Scholar]
- 23.McAleer M A, Reifsnyder P, Palmer S M, Prochazka M, Love J M, Copeman J B, Powell E E, Rodrigues N R, Prins J-B, Serreze D V, DeLarato N H, Wicker L S, Peterson L B, Schork N J, Todd J A, Leiter E H. Diabetes. 1995;44:1180–1195. doi: 10.2337/diab.44.10.1186. [DOI] [PubMed] [Google Scholar]
- 24.Hirose S, Kinoshita K, Nozawa S, Nishimura H, Shirai T. Int Immunol. 1990;2:1091–1095. doi: 10.1093/intimm/2.11.1091. [DOI] [PubMed] [Google Scholar]
- 25.Risch N, Ghosh S, Todd J A. Am J Hum Genet. 1993;53:702–714. [PMC free article] [PubMed] [Google Scholar]