

Abstract
Bacterial lipopolysaccharide (LPS; endotoxin) is implicated in the pathogenesis of acute liver failure and several chronic inflammatory liver diseases. To evaluate the effect of hepatocyte cyclooxygenase-2 (COX-2) in LPS-induced liver injury, we generated transgenic mice with targeted expression of COX-2 in the liver by using the albumin promoter-enhancer driven vector and the produced animals were subjected to a standard experimental protocol of LPS-induced acute fulminant hepatic failure (intraperitoneal injection of low dose of LPS in combination with D-galactosamine (GalN)). The COX-2 transgenic mice exhibited earlier mortality, higher serum ALT and AST levels and more prominent liver tissue damage (parenchymal hemorrhage, neutrophilic inflammation, hepatocyte apoptosis and necrosis) than wild type mice. Western blot analysis of the liver tissues showed that LPS/D-GalN treatment for 4 hours induced much higher cleavage of PARP, caspase-3 and caspase-9 in COX-2 transgenic mice than in wild type mice. Increased hepatic expression of JNK2 in COX-2 transgenic mice suggest that upregulation of JNK2 may represent a potential mechanism for COX-2-mediated exacerbation of liver injury. Blocking the prostaglandin receptor, EP1, prevented LPS/D-GalN-induced liver injury and hepatocyte apoptosis in COX-2 transgenic mice. Accordingly, the mice with genetic ablation of EP1 showed less LPS/D-GalN-induced liver damage and less hepatocyte apoptosis with prolonged survival when compared to the wild type mice. These findings demonstrate that COX-2 and its downstream prostaglandin receptor EP1 signaling pathway accelerates LPS-induced liver injury. Therefore, blocking COX-2/EP1 pathway may represent a potential approach for amelioration of LPS-induced liver injury.
Keywords: Lipopolysaccharide, endotoxin, cyclooxygenase-2, EP1 receptor, liver, acute hepatic failure
INTRODUCTION
Despite the current progress in clinical medicine, sepsis continues to pose major clinical problems and is associated with high patient mortality(1). Bacterial lipopolysaccharide (LPS; endotoxin) is the major component of the outer membrane from the Gram-negative bacteria and is critically involved in the development of sepsis. LPS induces fever, hypotension, intravascular coagulation and finally failure of vital organs, including acute liver failure, which is often refractory to clinical treatment(2, 3). LPS has been shown to induce the expression of cyclooxygenase-2 (COX-2) in a variety of human cells including hepatic cells(4, 5); however, the biological role of COX-2 in LPS-induced tissue injury remains unknown.
COX-2 plays a key role in the pathogenesis of inflammation, for its expression is markedly upregulated by inflammatory stimuli leading to increased synthesis of prostanoids (potent lipid inflammatory mediators) in inflamed tissues. Prostaglandin E2 (PGE2) is the most abundant prostanoid detected in inflamed tissues and its effect is mediated through activation of the EP receptors located in the plasma membrane (EP1, EP2, EP3, and EP4), which belong to the G protein-coupled receptor (GPCR) superfamily of seven-transmembrane spanning proteins(6-8). In fact, the conventional view that LPS-induced fever is mediated by pyrogenic cytokines is being gradually replaced by the concept that it is initiated by the production of PGE2(9, 10).
In addition to the role of LPS in acute liver injury, LPS derived from intestinal bacteria is also implicated in the pathogenesis of chronic inflammatory liver diseases, such as chronic hepatitis, alcoholic liver disease and cirrhosis(11, 12). Given that gram-negative bacteria normally colonize the colon, the body has developed strong defensive mechanisms that tightly regulate the entry and processing of LPS(13). The liver plays a central role in this process by virtue of its dual ability not only to clear LPS, but to actively respond to LPS. Consequently, several molecular mechanisms of inflammation and cellular damage have been implicated in the pathogenesis of LPS hepatotoxicity, including those related to the overt generation of inflammatory cytokines and oxygen free radicals from Kupffer cells; however, the potential role of liver parenchymal cells in LPS-induced liver injury has not been defined.
In the present study, we hypothesized that COX-2 activation in hepatocytes might accelerate LPS-induced liver injury and, therefore, interruption of the COX-2 cascade could attenuate LPS-induced tissue damage. This hypothesis was tested by both genetic and pharmacological approaches. We generated a novel transgenic mice model with targeted expression of COX-2 in the liver and showed that hepatic expression of COX-2 sensitized the liver to LPS-induced tissue damage and liver failure. Furthermore, we showed that the LPS/DGalN-induced liver damage in the COX-2 transgenic mice is attenuated by pretreatment with NS-398, a selective COX-2 inhibitor, or ONO-8711, a specific antagonist for the prostaglandin receptor EP1. Accordingly, the mice with genetic ablation of EP1 showed less LPS/D-GalN-induced liver damage and less hepatocyte apoptosis with prolonged survival when compared to the wild type mice. These results demonstrate an important role of COX-2/EP1 signaling cascade in LPS-induced hepatic injury and suggest that blocking this pathway may represent a novel target for amelioration of LPS-induced liver injury.
EXPERIMENTAL METHODS
Animals
Transgenic mice with targeted expression of COX-2 in the liver were developed by using the well-established albumin promoter-enhancer driven vector. To construct the albumin promoter-COX-2 transgene, a 1.8 kb human COX-2 cDNA containing the entire coding region of human COX-2 was inserted into the first exon of the human growth hormone gene controlled by the mouse albumin enhancer/promoter(14, 15). This transgene was micro-injected into mouse zygotes (B6SJL/F1 eggs) at the transgenic core facility of the University of Pennsylvania according to our contract. Five transgenic lines were produced and the mice were brought back to the University of Pittsburgh animal facility for propagation. The transgenic lines were maintained by backcrossing to the C57Bl/6 wild type mice. The transgenic mice were identified by genotyping using tail DNA samples. The COX-2 transgenic mice used in this study were derived from one transgenic line that was backcrossed to C57Bl/6 wild type mice for five consecutive generations. The mice at the age of 8−12 weeks were utilized for experiments, with age- and sex-matched wild type C57Bl/6 mice as controls. The animals were kept at 22°C under a 12-h light/dark cycle and received food and water ad libitum. The handling of mice and experimental procedures were conducted in accordance with experimental animal guidelines.
The EP1 knockout mice (homozygous) were kindly provided by Dr. Beverly Koller at the University of North Carolina and Dr. John McNeish at Pfizer Inc. The animals were bred in our animal facility and were used for experiments at the age of 8−12 weeks with age- and sex-matched wild type C57Bl/6 mice as controls.
Experimental Protocol
The COX-2 transgenic mice and the age/sex-matched wild type mice were administered intraperitoneally 30 ng/g body weight of LPS (Escherichia coli O55: B6, Sigma) in combination with 800 μg/g body weight of D-galactosamine (GalN) (Sigma) to induce acute fulminant hepatic failure (the reagents were dissolved in sterile nonpyrogenic saline solution). All experimental animals used in this study were treated according to the protocol approved by the University of Pittsburgh Animal Care and Use Committee (Protocol # 0303501).
Assessment of Hepatotoxicity and Survival Rate
To determine the survival rate, the animals were monitored continuously after LPS/D-GalN injection until animal death. In other experiments, the animals were sacrificed at specific time points to obtain blood and liver tissues. The blood samples were centrifuged at 3000 rpm for 15 min, and the serum was collected and stored at −80°C. Serum alanine aminotransferase (ALT) and aspartate aminotransferase (AST) activities were measured using an automatic analyzer at the University of Pittsburgh Medical Center Chemistry Department. The liver tissues were subjected to standard formalin fixation and paraffin-embedding for histological evaluation and TUNEL stain.
Hematoxylin and Eosin Staining
For histological analysis, liver tissue was fixed in 10% neutral-buffered formalin and embedded in paraffin. Sections of 5-μm thickness were affixed to slides, deparaffinized, and stained with hematoxylin and eosin (H&E) to determine morphologic changes.
TUNEL Stain
The extent of hepatocyte apoptosis was detected by terminal deoxynucleotidy1-transferase (TdT)-mediated deoxyuridine triphosphate-digoxigenin (dUTP) nick-end labeling (TUNEL). TUNEL-positive cells were counted by randomly selecting high-power fields (× 400) distributed over 3−5 independent sections. The numbers of TUNEL-positive and -negative cells were compiled and the percentages of TUNEL-positive cells were calculated.
Immunohistochemical stain for caspase-3
Formalin-fixed, paraffin-embedded sections of the liver tissues were subjected to immunohistochemical analysis for caspase-3. 5-μm-thick tissue sections were deparaffinized and rehydrated, followed by microwave retrieval of antigen according to standard procedures. The slides were sequentially blocked with avidin and biotin and then incubated at 4°C overnight with 1:100 diluted rabbit anti-caspase-3 (Cell Signaling Catalog #9661). Following repeated washings, the slides were incubated at room temperature for 30 minutes with biotinylated goat anti-rabbit (Vector Laboratories Catalog #BA-1000, 1:200 dilution with 3× goat serum). The slides were then washed and incubated with ABC/HRP at room temperature for 30 minutes (Vector Laboratories PK-6100) according to the manufacture's instruction. AEC substrate/chromogen was used for color development followed by counterstaining with hematoxylin.
Analysis of Cytokine Production
To analyze cytokine levels in the liver, mice were sacrificed 4 hours after LPS/D-GalN injection and the liver was removed immediately. The liver tissue was homogenized in PBS containing protease inhibitors. The obtained supernatants were stored at −80°C and analyzed for cytokine levels at the University of Pittsburgh Luminex Core Facility by the Luminex mouse cytokine bead immunoassay kit (Birsource, Camarillo, CA) according to the manufacture's instructions.
Measurement of PGE2 level
The liver tissues from wild type and COX-2 transgenic mice were homogenized and then extracted in 100 mg Amprep C18 minicolumn. The eluted samples were dried and the amounts of PGE2 were determined by specific enzymeimmune assay as we previously described(16).
Immunoblotting
Mouse liver tissues were homogenized with cold phosphate-buffered saline (PBS) containing 0.5 mM PMSF and 10μg/ml leupeptin and resuspended in 5-fold volume of hypotonic buffer consisting of 50 mM HEPES pH 7.55, 1 mM EDTA, 1 mM DTT and protease inhibitor cocktail tablets (Roche Diagnostics GmbH). The cell lysate was collected by centrifugation at the speed of 15,000g at 4°C for 10 minutes to remove cell debris and stored in aliquots at −20°C until use. The protein concentrations in the cell extracts were determined by the Bio-Rad protein assay (Bio-Rad, CA). 30 μg of cellular protein was subjected to SDS-PAGE and the separated proteins were electrophoretically transferred onto the nitrocellulose membranes (BioRad, CA). Nonspecific binding was blocked with PBS-T (0.5% Tween 20 in PBS) containing 5% non-fat milk for 1 hr at room temperature. The membranes were then incubated overnight at 4°C with antibodies against JNK2, phospho-JNK, caspase-3, caspase-9 and PARP in PBS-T containing 1% non-fat milk at the dilutions specified by the manufacturers. Following three washes with PBS-T, the membranes were then incubated with the horseradish peroxidase-conjugated secondary antibodies at 1:10,000 dilution in PBS-T containing 1% non-fat milk for 1 hour at room temperature. The membranes were then washed 3 times with PBS-T and the protein bands were visualized with the ECL Western blotting detection system according to the manufacturer's instructions. β-actin was used as the loading control.
RESULTS
To determine the effect of hepatocyte COX-2 in LPS-induced liver injury, we developed transgenic mice with targeted expression of COX-2 in the liver by using the well-established albumin promoter-enhancer driven vector (Figure 1). These mice were generated by microinjection of the COX-2 transgene (complete COX-2 cDNA cloned into the albumin promoter-driven vector(14, 15)) into mouse zygotes. The function of expressed COX-2 is supported by elevated production of PGE2 in the liver tissue homogenates of COX-2 transgenic mice (57.17 ± 7.72 pg/mg liver tissue in COX-2 transgenic mice versus 24.26 ± 5.24 pg/mg liver tissue in wild type mice; p<0.01, n=6) (Figure 2). These COX-2 transgenic mice develop normally with no significant liver inflammation or histologic abnormality under normal housing conditions. However, the COX-2 transgenic mice exhibited early mortality than wild type mice when the animals were subjected to a standard experimental protocol of LPS-induced acute fulminant hepatic failure (intraperitoneal injection of low dose of LPS in combination with D-galactosamine (GalN))(17-23). In COX-2 transgenic group (n=16), mortality became apparent at 5 to 6 h and all mice died by 9 h (Table 1). In wild type mice (n=16), no death was observed at 6 h; first animal death was observed at 7 h and all animals died by 11 h.
Figure 1. Generation of COX-2 transgenic mice.
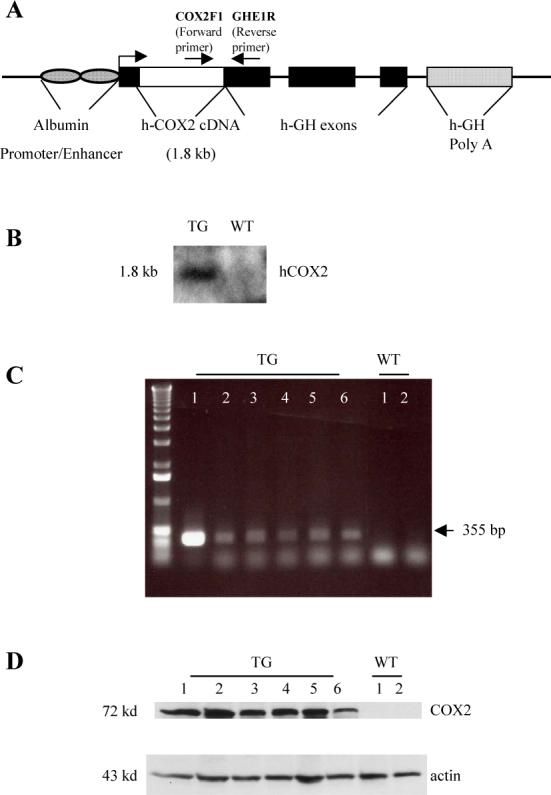
The COX-2 transgene (complete human COX-2 cDNA cloned into the albumin promoter-driven vector) was microinjected into mouse zygotes to generate transgenic mice by established method. (A) Schematic representation of the human COX-2 transgene with albumin promoter. (B) Southern blot analysis of tail genomic DNA showing the presence of COX-2 transgene in transgenic (TG) but not in wild-type (WT) littermates. The DNA samples were digested with Bam H1; the blot was hybridized with [32P]-labeled human COX-2 cDNA probe. (C) PCR analysis of tail genomic DNA showing the presence of COX-2 transgene in transgenic (TG) mice but not in wild-type (WT) littermates. (D) Western blot showing successful expression of COX-2 protein in the liver tissues from COX-2 transgenic mice. Equal amounts of the mouse liver tissue proteins from transgenic mice (TG) and wild type (WT) littermates were subjected to SDS-PAGE and Western blotting using anti-human COX-2 antibody.
Figure 2. PGE2 levels in the liver tissues from wild type and COX-2 transgenic mice.
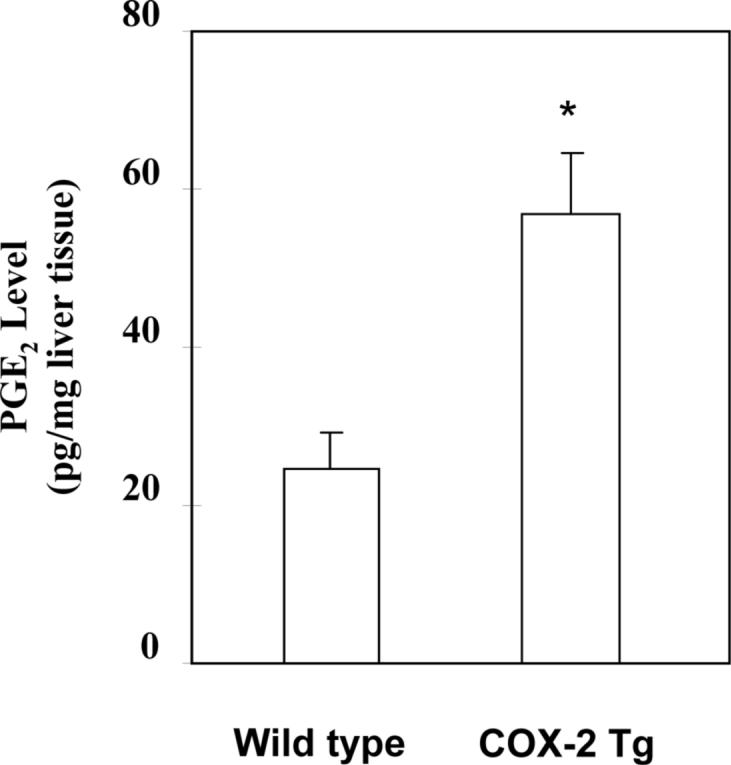
Liver tissues were homogenized and extracted in 100 mg Amprep C18 minicolumn. The eluted samples were dried and the amounts of PGE2 were determined by specific enzymeimmune assay. The data are presented as mean±SD of pg/mg liver tissue (*ρ<0.01 compared to wild type mice, n=6).
Table 1.
Survival Rate of wild type, COX-2 transgenic and EP1 knockout mice after LPS/GlaN injection
| Hours after injection |
Wild Type Number of viable mice |
COX-2 Transgenic Number of viable mice |
EP1 Knockout Number of viable mice |
Saline Injection Number of viable mice |
|---|---|---|---|---|
| 5 | 16 | 16 | 15 | 8 |
| 6 | 16 | 10 | 15 | 8 |
| 7 | 14 | 6 | 13 | 8 |
| 8 | 8 | 4 | 9 | 8 |
| 9 | 2 | 0 | 7 | 8 |
| 10 | 2 | 0 | 3 | 8 |
| 11 | 0 | 0 | 3 | 8 |
| 12 | 0 | 0 | 1 | 8 |
| 24 | 0 | 0 | 1 | 8 |
The COX-2 transgenic, EP1 knockout and wild type mice at 6−8 weeks of age received a single intraperitoneal injection of LPS/GalN. The animals were closely monitored for activity and mortality was documented at hourly intervals (up to 24 hours).
Based on the survival rate, additional COX-2 transgenic and wild type mice were sacrificed 4 hours after LPS/D-GalN administration to obtain blood samples and liver tissues for liver enzyme and tissue analyses. The COX-2 transgenic mice showed significantly higher serum alanine transaminase (ALT) and aspartate transaminase (AST) levels than wild type mice (Figure 3). Histological examination of the liver tissues revealed more prominent liver damage in the COX-2 transgenic than in wild type mice (Figure 4). In the COX-2 transgenic group, massive hemorrhagic necrosis and hepatocyte apoptosis were observed, with prominent vascular congestion and neutrophil infiltration; only residual areas of surviving hepatocytes were present, showing vacuolar degeneration and cytoplasmic swelling. In contrast, only mild scattered necrosis and apoptosis were observed in the wild type mice. The number of TUNEL-positive hepatocytes in the COX-2 transgenic mice is significantly higher than in the wild type mice (47.42±2.34% vs 11.77±0.04%, p<0.01) (Figure 4). Since a clear distinction between apoptosis and necrosis during liver injury cannot always be made with certainty based on the TUNEL assay(24), especially considering that some of the TUNEL-positive hepatocytes show significant cellular swelling, we further performed immunohistochemical stain for caspase-3 in the liver tissue sections. As shown in Figure 4, the number of caspase-3 positive cells in the COX-2 transgenic mice is also significantly higher than in wild type mice (33.18±0.39% vs 8.80±0.02%, p<0.01). The caspase-3 positive counts are slightly lower than TUNEL positive counts, suggesting that some of the TUNEL-positive cells might reflect necrotic hepatocytes. The liver tissues from the COX-2 transgenic mice and wild type mice treated with LPS/D-GalN for four hours were then analyzed by western blot analysis to determine the levels of PARP, caspase-3 and caspase-9. As shown in Figure 5, LPS/D-GalN treatment for 4 hours induced much more prominent cleavage of PARP, caspase-3 and caspase-9 in COX-2 transgenic mice than in wild type mice. The presence of weak caspase-3 and PARP cleavage in wild type mice at this time point indicates the occurring of hepatocyte apoptosis in these control animals, which is consistent with the documented caspase activation in LPS/D-GalN-induced liver injury(25-30). The relatively weak intensity of the cleaved caspase-3 and PARP in our study may be due to several factors, including the early time point in our study, the different concentrations of LPS/D-GalN, the different antibody sources and/or titers, or the different methods for apoptotic detection. Collectively, our findings from the histopathological evaluation, TUNEL assay, caspase-3 immunocytochemical stain and immunoblotting all indicate the presence of increased hepatocyte apoptosis in COX-2 transgenic mice, despite that some of the injured hepatocyte may represent hepatocyte necrosis or aponecrosis. Thus, although hepatic overexpression of COX-2 did not cause liver inflammation or hepatocyte injury under baseline conditions, it enhanced LPS-induced hepatocyte apoptosis/necrosis, tissue damage and liver failure. These observations suggest a potential role of hepatocyte COX-2 in the pathogenesis of LPS-induced liver injury.
Figure 3. Hepatic expression of COX-2 enhances serum transaminase levels induced by LPS.

The COX-2 transgenic mice and the age/sex-matched wild type mice were administered intraperitoneally 30 ng/g body weight of LPS in combination with 800 μg/g body weight of D-galactosamine (D-GalN). The animals were sacrificed 4 hours after injection. Blood samples were collected and sera were separated for transaminase analysis. The COX-2 transgenic mice show significantly higher serum ALT and AST levels than the wild type mice after LPS/D-GalN treatment (p<0.01 compared to wild type mice treated with LPS/D-GalN) (similar transaminase levels were observed between COX-2 transgenic and wild type mice when the animals were not subjected to LPS/D-GalN treatment).
Figure 4. Hepatic expression of COX-2 enhances LPS-induced liver injury.
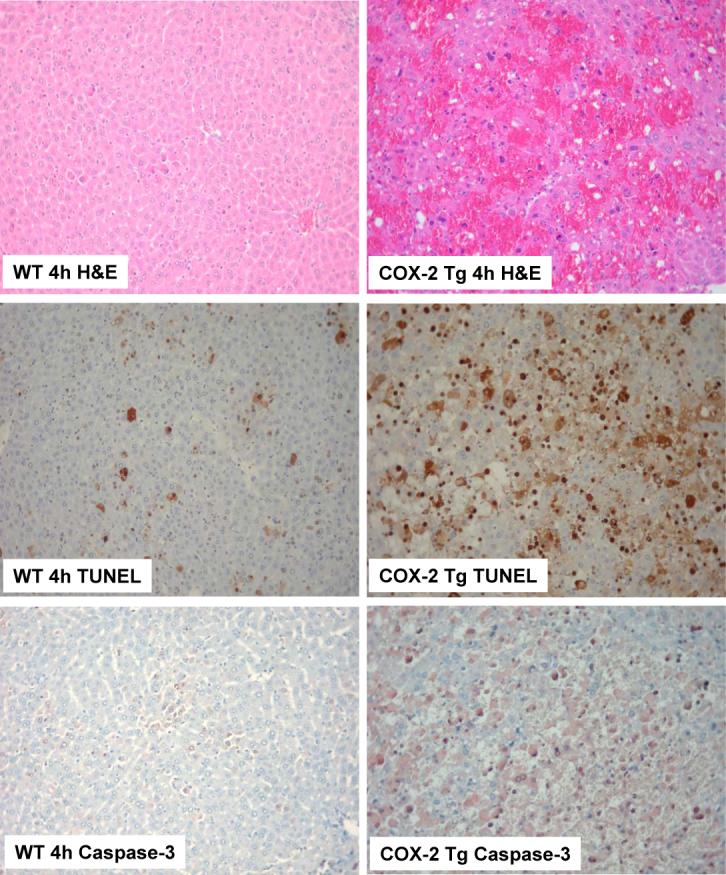
The COX-2 transgenic mice and the age/sex-matched wild type mice were administered intraperitoneally 30 ng/g body weight of LPS in combination with 800 μg/g body weight of D-GalN. The animals were sacrificed 4 hours after injection and the liver tissues were harvested for histological evaluation. Formalin-fixed and paraffin-embedded sections (5 μm thick) were stained with hematoxylin and eosin (H&E), terminal deoxynucleotidy1-transferase-mediated deoxyuridine triphosphate-digoxigenin nick-end labeling (TUNEL), and caspase-3. (Upper panels) Histopathological characteristics of the liver tissues (H&E stain, 200X). The livers of COX-2 transgenic mice (right panel) exhibit more prominent hemorrhage necrosis, hepatocyte apoptosis and degeneration when compared to the livers of wild type mice (left panel). (Mid panels). TUNEL stain (200X) in liver tissues of LPS-treated mice. The number of TUNEL-positive hepatocytes in COX-2 transgenic mice is significantly higher than in wild type mice. (Lower panels) Caspase-3 immunostain (200X) in liver tissues. COX-2 transgenic mice show significantly higher numbers of caspase-3-positive apoptotic hepatocytes than wild type mice.
Figure 5. Liver tissue analysis for PARP, caspase-3 and caspase-9.

The COX-2 transgenic mice, EP1 knockout mice and matched wild type mice were subjected to LPS and D-GalN injection (i.p.). The animals were sacrificed 4 hours after injection. The liver tissues were obtained and the cellular proteins were subjected to SDS-PAGE and Western blot analysis to determine the levels of PARP, caspase-3 and caspase-9 as described in the Methods. LPS/DGalN treatment for 4 hours induced much higher cleavage of PARP, caspase-3 and caspase-9 in COX-2 transgenic mice than in wild type or EP1 knockout mice.
Given the documented role of inflammatory cytokines in LPS/D-GalN-induced liver injury(31, 32), we examined the expression of key inflammatory cytokines in liver tissues by using the Luminex cytokine bead immunoassays. As shown in Table 2, although LPS/D-GalN treatment significantly increased the production of a repertoire of inflammatory cytokines including IL-1α, IL-1β, TNF-α, IP-10, MIP-1α, IL-10, IL-12, MCP-1, KC and MIG in the wild type mice, the cytokine profiles were not significantly different between the COX-2 transgenic and wild type groups. These findings suggest that COX-2-mediated enhancement of liver injury most likely involves mechanism independent of cytokine production.
Table 2.
Cytokine production in liver tissues of wild type and COX-2 transgenic mice with or without LPS/D-GalN injection
| Treatment | Vehicle |
LPS/D-GalN |
||
|---|---|---|---|---|
| wild type | COX-2 Tg | wild type | COX-2 Tg | |
| FGF | 21.81±5.62 | 18.13±3.22 | 20.91±1.98 | 25.68±5.08 |
| IFN-γ | ND | ND | ND | ND |
| IL-12(p40/p70) | 0.29±0.02 | 0.61±0.10 | 1.59±0.51* | 0.68±0.05 |
| TNF-α | ND | ND | 0.20±0.05** | 0.25±0.15 |
| VEGF | 2.43±0.22 | 2.60±0.70 | 1.12±0.14 | 1.00±0.06 |
| IL-2 | 0.88±0.11 | 1.00±0.15 | 1.21±0.20 | 0.84±0.09 |
| IL-4 | ND | ND | ND | ND |
| IL-5 | ND | ND | 0.99±0.08 | 1.05±0.46 |
| IL-10 | 3.78±1.25 | 3.50±0.43 | 9.48±2.04* | 3.81±0.35 |
| IP-10 | 2.68±0.40 | 3.89±1.09 | 386.00±129.13** | 139.27±6.96 |
| KC | 1.12±0.12 | 1.38±0.14 | 3.57±0.42** | 1.41±0.02 |
| MCP-1 | 0.19±0.02 | 0.21±0.02 | 2.77±0.50** | 1.22±0.13 |
| MIG | 0.32±0.04 | 0.39±0.07 | 4.44±0.12** | 5.45±1.24 |
| MIP-1α | 0.35±0.05 | 0.52±0.10 | 8.96±0.93** | 4.04±0.06 |
| IL-1β | 0.50±0.07 | 0.60±0.10 | 6.71±0.99** | 4.07±1.28 |
| IL-6 | 3.13±0.43 | 3.50±0.54 | 3.10±0.57 | 2.35±0.49 |
| GM-CSF | ND | ND | ND | ND |
| IL-1α | 0.47±0.13 | 0.52±0.10 | 51.27±14.81** | 12.22±0.08 |
| IL-13 | 1.32±0.19 | 1.70±0.31 | 1.97±0.49 | 1.54±0.34 |
| IL-17 | 0.30±0.05 | 0.41±0.11 | 0.48±0.16 | 0.29±0.08 |
Values are in pg/mg wet weight of liver tissue and as expressed as mean ± SD from 3−5 experiments (*p <0.05, **p<0.01; compared to wild type animals treated with vehicle alone). ND, undetectable (below detectable limit of the assay). FGF, fibroblast growth factor; GM-CSF, granulocyte macrophage colony-stimulating factor; IL, interleukin; IFN-γ, interferon-γ; IP-10; interferon-inducible protein-10; KC, keratinocyte-derived chemokine (KC); MCP-1, monocyte chemoattractant protein-1; MIG, monokine induced by interferon-γ; MIP-1α, macrophage inflammatory protein-1α; TNF-1α, tumor necrosis factor-α; VEGF, vascular endothelial growth factor.
The c-Jun NH2-terminal kinase (JNK) is a member of the MAPK family which is known to trigger apoptosis in response to environmental stresses as well as inflammatory cytokines(33). The JNK signaling pathway is activated in various forms of liver injury(34-38). Recently, several studies, based on the gene-knockout approach, have convincingly demonstrated the critical role of JNK in hepatocyte apoptosis, which was induced by concanavalin A, a methionine- and choline-deficient diet, or LPS/D-GalN(36-39). Since JNK2 plays an essential role in LPS/D-GalN-induced liver injury through direct activation of caspase(37), we examined whether overexpression of COX-2 in hepatocytes might activate JNK2 in our system. Indeed, the COX-2 transgenic mice express significantly higher level of JNK2 in the liver when compared to the wild type mice (Figure 6). Higher phosphorylation of p54-JNK is also observed in the COX-2 Tg livers when compared to the wild type controls after LPS/D-GalN injection (Figure 7). Therefore, upregulation of JNK2 may represent an important mechanism for COX-2-mediated exacerbation of liver injury. Nonetheless, in light of the complexity of LPS/D-GalN-induced liver injury, the possibility of other mechanisms cannot be excluded.
Figure 6. Increased expression of JNK2 in COX-2 transgenic mice.
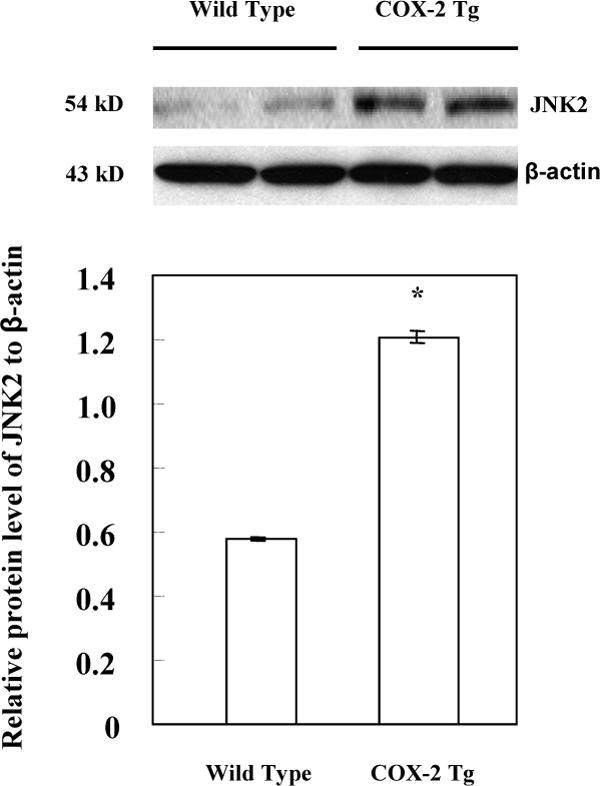
The liver tissues from the COX-2 transgenic mice and their matched wild type mice were homogenized. The cellular proteins were subjected to SDS-PAGE and Western blot analysis to determine the protein level of JNK2. Western blot for β-actin was used as the loading control. Higher level of JNK2 was observed in the liver tissues from the COX-2 transgenic mice when compared to the wild type mice. The lower panel represents the ratio between JNK2 and β-actin by densitometry analysis (* p < 0.01).
Figure 7. Increased phosphorylation of JNK in COX-2 transgenic mice treated with LPS/D-GalN.
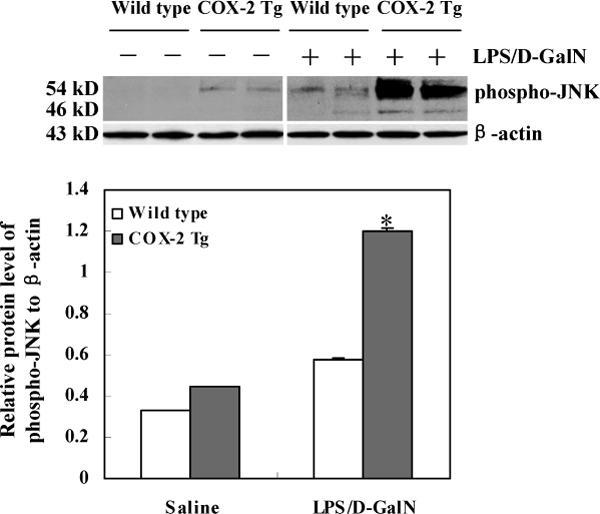
The COX-2 transgenic mice and matched wild type mice were sacrificed 4 hours after LPS/D-GalN injection. The liver tissues were homogenized and the extracted proteins were subjected to SDS-PAGE and Western blot analysis using the antibody against phospho-JNK (Cell Signaling Technology, Danvers, MA). Western blot for β-actin was used as the loading control. The lower panel represents the ratio between phosphorylated p54-JNK2 and β-actin by densitometry analysis. *ρ<0.01 compared to wild type mice treated LPS/D-GalN or COX-2 Tg mice treated with saline.
The effect of COX-2 is mediated by prostanoids that bind their G protein coupled receptors. The most abundant prostanoid in the liver is PGE2, which exerts actions through binding its membrane EP receptors(6-8). Although all four different EP receptor subtypes (EP1−4) are expressed in hepatic cells, studies have suggested a potential role of EP1 receptor in primary and transformed hepatocytes(40, 41). To determine whether EP1 receptor mediates COX-2 effect in LPS-induced liver injury, we utilized both pharmacological and genetic approaches to inhibit EP1 function and expression. Pretreatment of COX-2 transgenic mice with the specific EP1 receptor antagonist ONO-8711 for 45 minutes prevented LPS/D-GalN-induced transaminase increase as well as hepatocyte damage (Figure 8). The protective effect by ONO-8711 appears similar to that by NS-398, a selective COX-2 inhibitor (Figure 8). Accordingly, mice with genetic ablation of EP1 showed prolonged survival (n=15) compared to wild type mice after LPS/D-GalN injection (Table 1). The LPS-induced hepatocyte apoptosis and liver damage in EP1 knockout mice was notably less than in the wild type mice. Four hours after LPS/D-GalN injection, the TUNEL-positive hepatocytes in EP1 knockout mice (3.42±0.02%) was significantly lower than in wild type mice (11.77±0.04%, p < 0.01) (Figure 9). Similarly, the number of caspase-3 positive cells in the EP1 knockout mice (2.12±0.01%) was also significantly lower than in wild type mice (8.80±0.02%, p<0.01). These findings indicate that EP1 receptor may play a role in LPS-induced liver injury. It appears that JNK2 may be involved in EP1 effect, given the decreased level of JNK2 in the liver tissues from LPS/D-GalN treated EP1 mice (Figure 10).
Figure 8. The LPS/D-GalN-induced liver injury in COX-2 transgenic is attenuated by the EP1 receptor antagonist, ONO-8711 and by the COX-2 inhibitor, NS-398.
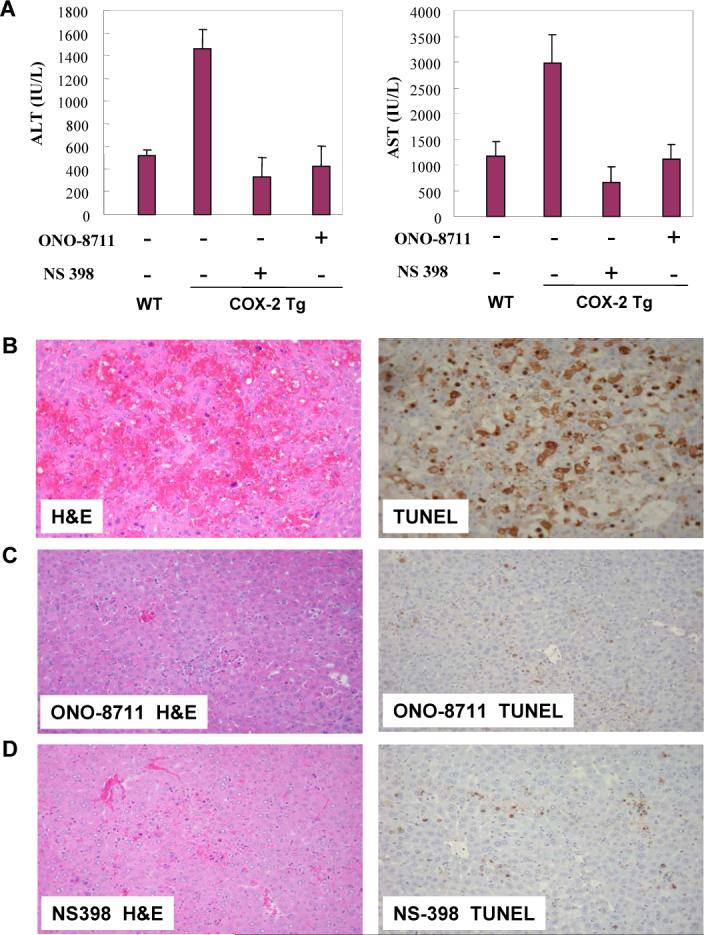
The COX-2 transgenic mice received intraperitoneal injection of the EP1 receptor antagonist, ONO-8711 (2.5 μg/g body weight) or the COX-2 inhibitor, NS-398 (5 μg/g body weight), 45 minutes before administration of LPS/D-GalN. The animals were sacrificed 4 hours after LPS/D-GalN injection. Upon sacrifice the blood samples were collected for transaminase analysis, whereas the liver tissues were harvested for histopathological examination. (A) Serum ALT and AST levels in COX-2 transgenic mice with or without ONO-8711 or NS-398 pretreatment (wild type mice were included as control; all the mice received LPS/D-GalN injection). (B-D) Representative H&E and TUNEL stains (200X) of the liver tissues from COX-2 transgenic mice pretreated with ONO-8711 (C), NS-398 (D) or without pretreatment (B) (all the mice received LPS/D-GalN injection). The TUNEL-positive hepatocytes in the mice pretreated with ONO-8711 (5.59±0.01%) or NS-398 (13.75±0.07%) is significantly lower than in mice without pretreatment (47.78±0.73%, p < 0.01).
Figure 9. Genetic ablation of EP1 receptor prevents LPS-induced liver injury.
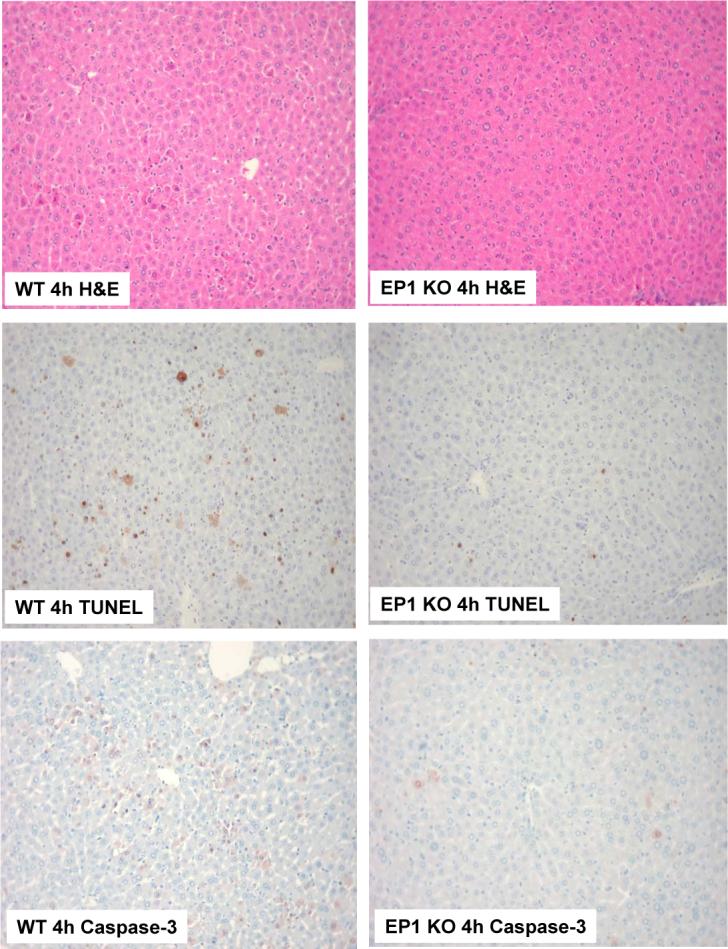
EP1 knockout mice and matched wild type mice were subjected to LPS and D-GalN injection (i.p.). The animals were sacrificed 4 hours after injection and the liver tissues were harvested for histological evaluation. Formalin-fixed and paraffin-embedded sections (5 μm thick) were stained with H&E, TUNEL, and caspase-3. (Upper panels) H&E stain of the liver tissues from LPS-treated mice (200X). (Mid panels) TUNEL stain (200X) of the liver tissues. The number of TUNEL-positive hepatocytes in EP1 knockout mice is less than in wild type mice. (Lower panels) Caspase-3 immunostain (200X) of the liver tissues. The EP1 knockout mice show less caspase-3-positive hepatocytes than wild type mice.
Figure 10. Decreased expression of JNK2 in EP1 knockout mice treated with LPS/D-GalN.
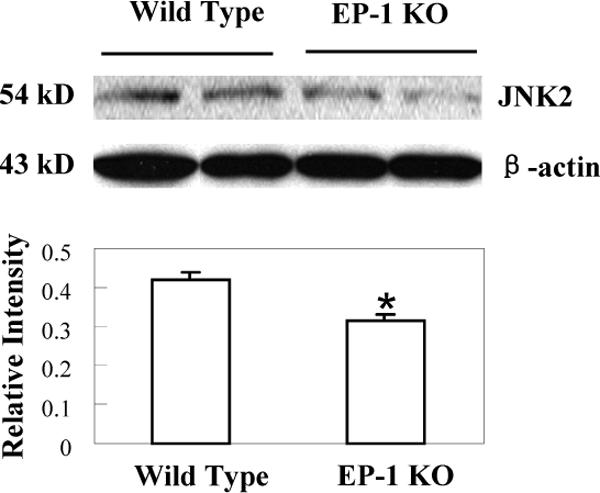
EP1 knockout mice and matched wild type mice were sacrificed 4 hours after LPS and D-GalN injection. The liver tissues were homogenized and the extracted proteins were subjected to SDS-PAGE and Western blot analysis to determine the protein level of JNK2. Western blot for β-actin was used as the loading control. Reduced JNK2 protein was observed in the EP1 knockout livers when compared to the wild type controls. The lower panel represents the ratio between JNK2 and β-actin by densitometry analysis (* p < 0.01 compared to wild type mice).
DISCUSSION
LPS/D-GalN-induced liver injury is a well-established model of acute liver failure in mice. In this model D-GalN blocks gene transcription in the liver and LPS in turn induces an acute cytokine-dependent liver inflammation accompanied by massive liver apoptosis and death of the animals(11, 17, 18, 42, 43). LPS activates Kupffer cells, resulting in overproduction of large amounts of cytokines, which subsequently trigger liver inflammation and tissue damage(31, 32). In addition to cytokines, prostaglandins have also been suggested to participate in LPS-induced liver injury(44). The synthesis of prostaglandins is tightly controlled by cyclooxygenases (including COX-1 and COX-2), which catalyze the conversion of arachidonic acid to PGs. Whereas COX-1 is constitutively expressed in most tissues, COX-2 is inducible by a variety of factors including cytokines and endotoxin. Indeed, the expression of COX-2 protein is induced in LPS-treated liver from rats(44). These results indicate that COX-2 may contribute to liver injury. However, much remains unknown about the involvement of hepatic COX-2 in liver injury during endotoxemia. In this study, we provide novel evidence for an important role of COX-2 in hepatocytes for LPS-induced liver injury. Our data indicate that COX-2 transgenic mice develop accelerated liver injury in response to LPS/D-GalN treatment. Additionally, we have shown that the LPS/D-GalN-induced liver damage in the COX-2 transgenic mice is alleviated by pharmacologic inhibition of COX-2 and EP1. Furthermore, the EP1 knockout mice exhibit less LPS/D-GalN-induced liver damage and less hepatocyte apoptosis with prolonged survival when compared to the wild type mice. These results demonstrate an important role of COX-2/EP1 signaling cascade in LPS-induced hepatic injury.
It is worth mentioning that in our transgenic mice model the expression of COX-2 is driven by the albumin promoter and is exclusively present in hepatocytes. Therefore, the accelerated liver injury observed in the COX-2 transgenic mice indicates the contribution of COX-2 in hepatocytes to LPS-induced liver injury. Additionally, it is of note that COX-2 induction occurs predominantly in Kupffer cells in response to LPS and the effects of PGE2 on hepatocytes are paracrine, whereas in our COX-2 transgenic model the effects are autacrine. Although adult hepatocytes do not express COX-2 under normal conditions, the level of COX-2 in hepatocytes is increased during chronic inflammatory liver diseases(45-48). Therefore, our findings in this study suggest that elevated hepatic COX-2 during chronic inflammatory liver diseases may represent a predisposing risk factor for LPS-induced liver failure.
The liver tissues from the COX-2 transgenic and wild type mice showed a similar cytokine expression profile under baseline conditions (without LPS/D-GalN treatment). These findings, along with histological evaluation of the liver tissues, suggest that overexpression of COX-2 alone in hepatocytes does not significantly alter hepatic cytokine production or inflammatory response. We observed that LPS/D-GalN treatment of wild type mice significantly increased the production of several key inflammatory cytokines (including IL-1α, IL-1β, TNF-α, IP-10, MIP-1α, IL-10, IL-12, MCP-1, KC and MIG); these data are consistent with the documented role of inflammatory cytokines in LPS-induced liver inflammation and tissue damage. It is of note that transgenic overexpression of COX-2 in hepatocytes did not further enhance cytokine production after LPS/D-GalN treatment, suggesting that COX-2-mediated enhancement of liver injury is likely mediated by mechanism independent of cytokine production. In this context, it is noteworthy that our data point toward the involvement of JNK2 in this process.
Prostanoids exert their biological actions primarily via their respective G protein-coupled receptors (GPCR) superfamily of seven-transmembrane spanning proteins on the cell surface membrane(7, 8). PGE2 can potentially interact with four types of receptors (EP1, EP2, EP3, and EP4). The EP1 receptor is coupled with Gq protein and thus signals through phospholipase C and intracellular Ca2+; the EP2 and EP4 receptors are coupled with Gs protein, signaling through elevation of intracellular cAMP level and activation of protein kinase A (PKA); the EP3 receptor is coupled with Gi protein and signals through reduction of intracellular cAMP. In this study, we show that the LPS/D-GalN-induced liver injury is blocked by the EP1 receptor antagonist, ONO-8711. Furthermore, the EP1 knockout mice show reduced liver injury after LPS/D-GalN challenge. These findings suggest the involvement of EP1 receptor in LPS/D-GalN-induced liver injury.
Our results in this study suggest that inhibition of COX-2/EP1 signaling pathway may reduce LPS-induced liver injury. Since COX-2 inhibitors are associated with cardiovascular side effect, which is believed to be largely due to inhibition of the antithrombotic prostacyclin(49-51), blocking EP1 receptor is expected to effectively prevent LPS-related liver injury without inhibiting prostacyclin and thus incurring no significant side effects. Therefore, inhibiting EP1 may represent a potentially effective and safe approach for amelioration of LPS-induced liver damage. The availability of transgenic mice with hepatic expression of COX-2 will allow future studies to determine the role of COX-2-controlled prostaglandin signaling in the pathogenesis of various inflammatory and neoplastic liver diseases.
ACKNOWLEDGEMENTS
We would like to thank Dr. Beverly Koller at the University of North Carolina and Dr. John McNeish at Pfizer Inc. for providing the EP1 knockout mice. The EP1 receptor antagonist ONO-8711 was kindly provided by the Ono Pharmaceutical Co., Ltd, Japan.
This work was partially supported by the National Institutes of Health R01 grants CA106280, CA102325 and DK077776 (to T.W.) and DK054411 (to C.R.G.). C.H. is the recipient of the Cancer Research and Prevention Foundation Scholar Award. G.L. is supported in part by the China Scholarship Council.
REFERENCES
- 1.Hotchkiss RS, Karl IE. The pathophysiology and treatment of sepsis. N Engl J Med. 2003;348:138–150. doi: 10.1056/NEJMra021333. [DOI] [PubMed] [Google Scholar]
- 2.Williams R. Classification, etiology, and considerations of outcome in acute liver failure. Semin Liver Dis. 1996;16:343–348. doi: 10.1055/s-2007-1007247. [DOI] [PubMed] [Google Scholar]
- 3.Ostapowicz G, Lee WM. Acute hepatic failure: a Western perspective. J Gastroenterol Hepatol. 2000;15:480–488. doi: 10.1046/j.1440-1746.2000.02074.x. [DOI] [PubMed] [Google Scholar]
- 4.Kang YJ, Wingerd BA, Arakawa T, Smith WL. Cyclooxygenase-2 gene transcription in a macrophage model of inflammation. J Immunol. 2006;177:8111–8122. doi: 10.4049/jimmunol.177.11.8111. [DOI] [PubMed] [Google Scholar]
- 5.Callejas NA, Bosca L, Williams CS, Du BR, Martin-Sanz P. Regulation of cyclooxygenase 2 expression in hepatocytes by CCAAT/enhancer-binding proteins. Gastroenterology. 2000;119:493–501. doi: 10.1053/gast.2000.9374. [DOI] [PubMed] [Google Scholar]
- 6.Funk CD. Prostaglandins and leukotrienes: advances in eicosanoid biology. Science. 2001;294:1871–1875. doi: 10.1126/science.294.5548.1871. [DOI] [PubMed] [Google Scholar]
- 7.Narumiya S, FitzGerald GA. Genetic and pharmacological analysis of prostanoid receptor function. J Clin Invest. 2001;108:25–30. doi: 10.1172/JCI13455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Breyer RM, Bagdassarian CK, Myers SA, Breyer MD. Prostanoid receptors: subtypes and signaling. Annu Rev Pharmacol Toxicol. 2001;41:661–690. doi: 10.1146/annurev.pharmtox.41.1.661. [DOI] [PubMed] [Google Scholar]
- 9.Ivanov AI, Romanovsky AA. Prostaglandin E2 as a mediator of fever: synthesis and catabolism. Front Biosci. 2004;9:1977–1993. doi: 10.2741/1383. [DOI] [PubMed] [Google Scholar]
- 10.Oka T. Prostaglandin E2 as a mediator of fever: the role of prostaglandin E (EP) receptors. Front Biosci. 2004;9:3046–3057. doi: 10.2741/1458. [DOI] [PubMed] [Google Scholar]
- 11.Jirillo E, Caccavo D, Magrone T, Piccigallo E, Amati L, Lembo A, Kalis C, Gumenscheimer M. The role of the liver in the response to LPS: experimental and clinical findings. J Endotoxin Res. 2002;8:319–327. doi: 10.1179/096805102125000641. [DOI] [PubMed] [Google Scholar]
- 12.Han DW. Intestinal endotoxemia as a pathogenetic mechanism in liver failure. World J Gastroenterol. 2002;8:961–965. doi: 10.3748/wjg.v8.i6.961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Su GL. Lipopolysaccharides in liver injury: molecular mechanisms of Kupffer cell activation. Am J Physiol Gastrointest Liver Physiol. 2002;283:G256–265. doi: 10.1152/ajpgi.00550.2001. [DOI] [PubMed] [Google Scholar]
- 14.Bell A, Chen Q, DeFrances MC, Michalopoulos GK, Zarnegar R. The five amino acid-deleted isoform of hepatocyte growth factor promotes carcinogenesis in transgenic mice. Oncogene. 1999;18:887–895. doi: 10.1038/sj.onc.1202379. [DOI] [PubMed] [Google Scholar]
- 15.Wang X, DeFrances MC, Dai Y, Pediaditakis P, Johnson C, Bell A, Michalopoulos GK, Zarnegar R. A Mechanism of Cell Survival. Sequestration of Fas by the HGF Receptor Met. Mol Cell. 2002;9:411–421. doi: 10.1016/s1097-2765(02)00439-2. [DOI] [PubMed] [Google Scholar]
- 16.Wu T, Han C, Lunz JG, 3rd, Michalopoulos G, Shelhamer JH, Demetris AJ. Involvement of 85-kd cytosolic phospholipase A(2) and cyclooxygenase-2 in the proliferation of human cholangiocarcinoma cells. Hepatology. 2002;36:363–373. doi: 10.1053/jhep.2002.34743. [DOI] [PubMed] [Google Scholar]
- 17.Galanos C, Freudenberg MA, Reutter W. Galactosamine-induced sensitization to the lethal effects of endotoxin. Proc Natl Acad Sci U S A. 1979;76:5939–5943. doi: 10.1073/pnas.76.11.5939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Leist M, Gantner F, Bohlinger I, Tiegs G, Germann PG, Wendel A. Tumor necrosis factor-induced hepatocyte apoptosis precedes liver failure in experimental murine shock models. Am J Pathol. 1995;146:1220–1234. [PMC free article] [PubMed] [Google Scholar]
- 19.Bohlinger I, Leist M, Gantner F, Angermuller S, Tiegs G, Wendel A. DNA fragmentation in mouse organs during endotoxic shock. Am J Pathol. 1996;149:1381–1393. [PMC free article] [PubMed] [Google Scholar]
- 20.Deutschman CS, Haber BA, Andrejko K, Cressman DE, Harrison R, Elenko E, Taub R. Increased expression of cytokine-induced neutrophil chemoattractant in septic rat liver. Am J Physiol. 1996;271:R593–600. doi: 10.1152/ajpregu.1996.271.3.R593. [DOI] [PubMed] [Google Scholar]
- 21.Kosai K, Matsumoto K, Funakoshi H, Nakamura T. Hepatocyte growth factor prevents endotoxin-induced lethal hepatic failure in mice. Hepatology. 1999;30:151–159. doi: 10.1002/hep.510300102. [DOI] [PubMed] [Google Scholar]
- 22.Jiang W, Sun R, Wei H, Tian Z. Toll-like receptor 3 ligand attenuates LPS-induced liver injury by down-regulation of toll-like receptor 4 expression on macrophages. Proc Natl Acad Sci U S A. 2005;102:17077–17082. doi: 10.1073/pnas.0504570102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Masaki T, Chiba S, Tatsukawa H, Noguchi H, Kakuma T, Endo M, Seike M, Watanabe T, Yoshimatsu H. The role of histamine H1 receptor and H2 receptor in LPS-induced liver injury. Faseb J. 2005;19:1245–1252. doi: 10.1096/fj.04-3195com. [DOI] [PubMed] [Google Scholar]
- 24.Jaeschke H, Lemasters JJ. Apoptosis versus oncotic necrosis in hepatic ischemia/reperfusion injury. Gastroenterology. 2003;125:1246–1257. doi: 10.1016/s0016-5085(03)01209-5. [DOI] [PubMed] [Google Scholar]
- 25.Wang H, Xu DX, Lv JW, Ning H, Wei W. Melatonin attenuates lipopolysaccharide (LPS)-induced apoptotic liver damage in D-galactosamine-sensitized mice. Toxicology. 2007;237:49–57. doi: 10.1016/j.tox.2007.04.021. [DOI] [PubMed] [Google Scholar]
- 26.Harstad EB, Klaassen CD. Tumor necrosis factor-alpha-null mice are not resistant to cadmium chloride-induced hepatotoxicity. Toxicol Appl Pharmacol. 2002;179:155–162. doi: 10.1006/taap.2001.9362. [DOI] [PubMed] [Google Scholar]
- 27.Wang YM, Feng GH, Huang F, Li Y, Zhao GZ. [Tumor necrosis factor-alpha, caspase-3 expression and hepatocyte apoptosis in fulminanting hepatic failure]. Zhonghua Nei Ke Za Zhi. 2003;42:566–570. [PubMed] [Google Scholar]
- 28.Sass G, Soares MC, Yamashita K, Seyfried S, Zimmermann WH, Eschenhagen T, Kaczmarek E, Ritter T, Volk HD, Tiegs G. Heme oxygenase-1 and its reaction product, carbon monoxide, prevent inflammation-related apoptotic liver damage in mice. Hepatology. 2003;38:909–918. doi: 10.1053/jhep.2003.50386. [DOI] [PubMed] [Google Scholar]
- 29.Hoglen NC, Chen LS, Fisher CD, Hirakawa BP, Groessl T, Contreras PC. Characterization of IDN-6556 (3-[2-(2-tert-butyl-phenylaminooxalyl)-amino]-propionylamino]-4-oxo-5-(2,3 ,5,6-tetrafluoro-phenoxy)-pentanoic acid): a liver-targeted caspase inhibitor. J Pharmacol Exp Ther. 2004;309:634–640. doi: 10.1124/jpet.103.062034. [DOI] [PubMed] [Google Scholar]
- 30.Qiu Z, Kwon AH, Tsuji K, Kamiyama Y, Okumura T, Hirao Y. Fibronectin prevents D-galactosamine/lipopolysaccharide-induced lethal hepatic failure in mice. Shock. 2006;25:80–87. doi: 10.1097/01.shk.0000185797.04589.5c. [DOI] [PubMed] [Google Scholar]
- 31.Luster MI, Germolec DR, Yoshida T, Kayama F, Thompson M. Endotoxin-induced cytokine gene expression and excretion in the liver. Hepatology. 1994;19:480–488. [PubMed] [Google Scholar]
- 32.Chensue SW, Terebuh PD, Remick DG, Scales WE, Kunkel SL. In vivo biologic and immunohistochemical analysis of interleukin-1 alpha, beta and tumor necrosis factor during experimental endotoxemia. Kinetics, Kupffer cell expression, and glucocorticoid effects. Am J Pathol. 1991;138:395–402. [PMC free article] [PubMed] [Google Scholar]
- 33.Ip YT, Davis RJ. Signal transduction by the c-Jun N-terminal kinase (JNK)--from inflammation to development. Curr Opin Cell Biol. 1998;10:205–219. doi: 10.1016/s0955-0674(98)80143-9. [DOI] [PubMed] [Google Scholar]
- 34.Trautwein C, Rakemann T, Brenner DA, Streetz K, Licato L, Manns MP, Tiegs G. Concanavalin A-induced liver cell damage: activation of intracellular pathways triggered by tumor necrosis factor in mice. Gastroenterology. 1998;114:1035–1045. doi: 10.1016/s0016-5085(98)70324-5. [DOI] [PubMed] [Google Scholar]
- 35.Bendinelli P, Piccoletti R, Maroni P, Bernelli-Zazzera A. The MAP kinase cascades are activated during post-ischemic liver reperfusion. FEBS Lett. 1996;398:193–197. doi: 10.1016/s0014-5793(96)01228-8. [DOI] [PubMed] [Google Scholar]
- 36.Schattenberg JM, Singh R, Wang Y, Lefkowitch JH, Rigoli RM, Scherer PE, Czaja MJ. JNK1 but not JNK2 promotes the development of steatohepatitis in mice. Hepatology. 2006;43:163–172. doi: 10.1002/hep.20999. [DOI] [PubMed] [Google Scholar]
- 37.Wang Y, Singh R, Lefkowitch JH, Rigoli RM, Czaja MJ. Tumor necrosis factor-induced toxic liver injury results from JNK2-dependent activation of caspase-8 and the mitochondrial death pathway. J Biol Chem. 2006;281:15258–15267. doi: 10.1074/jbc.M512953200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chang L, Kamata H, Solinas G, Luo JL, Maeda S, Venuprasad K, Liu YC, Karin M. The E3 ubiquitin ligase itch couples JNK activation to TNFalpha-induced cell death by inducing c-FLIP(L) turnover. Cell. 2006;124:601–613. doi: 10.1016/j.cell.2006.01.021. [DOI] [PubMed] [Google Scholar]
- 39.Maeda S, Chang L, Li ZW, Luo JL, Leffert H, Karin M. IKKbeta is required for prevention of apoptosis mediated by cell-bound but not by circulating TNFalpha. Immunity. 2003;19:725–737. doi: 10.1016/s1074-7613(03)00301-7. [DOI] [PubMed] [Google Scholar]
- 40.Kimura M, Osumi S, Ogihara M. Prostaglandin E(2) (EP(1)) receptor agonist-induced DNA synthesis and proliferation in primary cultures of adult rat hepatocytes: the involvement of TGF-alpha. Endocrinology. 2001;142:4428–4440. doi: 10.1210/endo.142.10.8450. [DOI] [PubMed] [Google Scholar]
- 41.Han C, Michalopoulos GK, Wu T. Prostaglandin E(2) receptor EP(1) transactivates EGFR/MET receptor tyrosine kinases and enhances invasiveness in human hepatocellular carcinoma cells. J Cell Physiol. 2006;207:261–270. doi: 10.1002/jcp.20560. [DOI] [PubMed] [Google Scholar]
- 42.Lehmann V, Freudenberg MA, Galanos C. Lethal toxicity of lipopolysaccharide and tumor necrosis factor in normal and D-galactosamine-treated mice. J Exp Med. 1987;165:657–663. doi: 10.1084/jem.165.3.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sass G, Heinlein S, Agli A, Bang R, Schumann J, Tiegs G. Cytokine expression in three mouse models of experimental hepatitis. Cytokine. 2002;19:115–120. doi: 10.1006/cyto.2002.1948. [DOI] [PubMed] [Google Scholar]
- 44.Ganey PE, Barton YW, Kinser S, Sneed RA, Barton CC, Roth RA. Involvement of cyclooxygenase-2 in the potentiation of allyl alcohol-induced liver injury by bacterial lipopolysaccharide. Toxicol Appl Pharmacol. 2001;174:113–121. doi: 10.1006/taap.2001.9183. [DOI] [PubMed] [Google Scholar]
- 45.Kondo M, Yamamoto H, Nagano H, Okami J, Ito Y, Shimizu J, Eguchi H, Miyamoto A, Dono K, Umeshita K, Matsuura N, Wakasa K, Nakamori S, Sakon M, Monden M. Increased expression of COX-2 in nontumor liver tissue is associated with shorter disease-free survival in patients with hepatocellular carcinoma. Clin Cancer Res. 1999;5:4005–4012. [PubMed] [Google Scholar]
- 46.Morinaga S, Yamamoto Y, Noguchi Y, Imada T, Rino Y, Akaike M, Sugimasa Y, Takemiya S, Kameda Y, Takanashi Y. Cyclooxygenase-2 mRNA is up-regulated in cirrhotic or chronic hepatitis liver adjacent to hepatocellular carcinoma. J Gastroenterol Hepatol. 2002;17:1110–1116. doi: 10.1046/j.1440-1746.2002.02836.x. [DOI] [PubMed] [Google Scholar]
- 47.Cheng J, Imanishi H, Iijima H, Shimomura S, Yamamoto T, Amuro Y, Kubota A, Hada T. Expression of cyclooxygenase 2 and cytosolic phospholipase A(2) in the liver tissue of patients with chronic hepatitis and liver cirrhosis. Hepatol Res. 2002;23:185–195. doi: 10.1016/s1386-6346(01)00177-2. [DOI] [PubMed] [Google Scholar]
- 48.Mohammed NA, Abd El-Aleem SA, El-Hafiz HA, McMahon RF. Distribution of constitutive (COX-1) and inducible (COX-2) cyclooxygenase in postviral human liver cirrhosis: a possible role for COX-2 in the pathogenesis of liver cirrhosis. J Clin Pathol. 2004;57:350–354. doi: 10.1136/jcp.2003.012120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fosslien E. Cardiovascular complications of non-steroidal anti-inflammatory drugs. Ann Clin Lab Sci. 2005;35:347–385. [PubMed] [Google Scholar]
- 50.Grosser T, Fries S, FitzGerald GA. Biological basis for the cardiovascular consequences of COX-2 inhibition: therapeutic challenges and opportunities. J Clin Invest. 2006;116:4–15. doi: 10.1172/JCI27291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Warner TD, Mitchell JA. Cyclooxygenases: new forms, new inhibitors, and lessons from the clinic. Faseb J. 2004;18:790–804. doi: 10.1096/fj.03-0645rev. [DOI] [PubMed] [Google Scholar]