

Abstract
A newly identified severe acute respiratory syndrome coronavirus (SARS-CoV), is the etiological agent responsible for the outbreak of SARS. The SARS-CoV main protease, which is a 33.8-kDa protease (also called the 3C-like protease), plays a pivotal role in mediating viral replication and transcription functions through extensive proteolytic processing of two replicase polyproteins, pp1a (486 kDa) and pp1ab (790 kDa). Here, we report the crystal structures of the SARS-CoV main protease at different pH values and in complex with a specific inhibitor. The protease structure has a fold that can be described as an augmented serine-protease, but with a Cys-His at the active site. This series of crystal structures, which is the first, to our knowledge, of any protein from the SARS virus, reveal substantial pH-dependent conformational changes, and an unexpected mode of inhibitor binding, providing a structural basis for rational drug design.
Spreading from its likely origin in Southern China, the recent epidemic of severe acute respiratory syndrome (SARS), an atypical, highly contagious pneumonia, affected 32 countries in the period from February to June, 2003. In total, ≈8,500 people were infected and >900 died from the disease.¶ Although SARS now appears to have been contained, it is possible that it may reemerge in the next cold season. The rapid transmission by means of aerosols and the high mortality rate (up to 10%) make SARS a potential global threat. At present, neither a vaccine nor an efficacious therapy is available.
Right after the SARS outbreak, a new SARS coronavirus (SARS-CoV) was discovered as the etiological agent of the disease (1-4). CoVs are positive-sense, single-stranded RNA viruses featuring the largest viral RNA genomes known to date (5-7). The SARS-CoV genome is comprised of ≈29,700 nucleotides; its replicase gene alone encompasses >21,000 nucleotides, making it larger than the whole genome of a typical picornavirus. The replicase encodes two overlapping polyproteins, pp1a (486 kDa) and pp1ab (790 kDa), which mediate all of the functions required for viral replication and transcription in CoVs (8). Functional polypeptides are released from each polyprotein through extensive proteolytic processing, largely by the 33.8-kDa main protease (Mpro) (also called the 3C-like protease, 3CLpro). The functional importance of the Mpro in the viral life cycle makes it an attractive target for the development of drugs directed against SARS and other CoV infections.
SARS-CoV Mpro has a fold that can be described as an augmented serine-protease, which is homologous to the enzymes from human CoV (HCoV) and bovine CoV, porcine transmissible gastroenteritis virus (TGEV), mouse hepatitis virus, and infectious bronchitis virus (9). Substrate studies have shown that, like the other CoV proteases, it has specificity for Gln at the P1 position (10). A theoretical model of its three-dimensional structure in monomer form, which is based on the crystal structures of the HCoV and TGEV enzymes, has been described (9).
Our crystal structures provide, to our knowledge, the first view of this viral protease, including the active site conformation. The structures determined under different pH conditions reveal a pH-dependent activity switch, essentially explaining the varying activity of Mpro at different pH values. In addition, the structure of a substrate-analogue inhibitor complex shows an unexpected mode of inhibitor binding and provides structure basis for rational drug design.
Methods
Protein Expression and Purification. The coding sequence of the SARS-CoV Mpro was cloned from the SARS-CoV BJ01 strain and inserted into the BamHI and XhoI sites of pGEX-6p-1 plasmid DNA (Amersham Biosciences). The resulting plasmid, pGEX-Mpro, was used to transform Escherichia coli BL21 (DE3) cells. The GST fusion protein, GST-SARS-CoV Mpro, was purified by GST-glutathione affinity chromatography, cleaved with GST-rhinovirus 3C protease (11), and the recombinant SARS-CoV Mpro was further purified by using anion-exchange chromatography. The purified and concentrated SARS-CoV Mpro (10 mg/ml) was stored in 10 mM Tris·HCl, pH 7.5/1 mM DTT/1 mM EDTA.
Crystallization. Apoprotein at pH 6.0. SARS-CoV Mpro was crystallized by the hanging drop vapor diffusion method at 18°C. The best plate-like crystals were obtained from 2% polyethylene glycol (PEG) 6000/3% DMSO/1 mM DTT/0.1 M [2-(N-morpholino)ethanesulfonic acid] (Mes) buffer (pH 6.0), with a protein concentration of 5 mg/ml. Crystals with the same morphology and diffraction quality could also be obtained from a similar condition, with DMSO being replaced by 2% 2-methyl-2,4-pentanediol and 5% isopropanol as additives. The cryoprotectant solution contained 30% PEG 400 and 0.1 M Mes (pH 6.0).
Chloromethyl ketone (CMK) inhibitor complex. Apo Mpro crystals grown under pH 6.0 were soaked in a number of different small-molecule inhibitors. The best results were obtained by using a 5.0-mM solution of the substrate-analogue CMK inhibitor (9), Cbz-Val-Asn-Ser-Thr-Leu-Gln-CMK in 7.5% PEG 6000, 6% DMSO, and 0.1 M Mes (pH 6.0). A 3-μl aliquot of such solution was added to the drop and the crystals were soaked for 18 h.
Apoprotein at pH 7.6 and 8.0. Crystals grown at pH 6.0 were transferred, respectively, to solutions containing 30% PEG 400, 0.1 M Hepes (pH 7.6), and 30% PEG 400, 0.1 M Hepes (pH 8.0), and soaked overnight before data collection.
Data Collection. All data were collected at the Beijing Synchrotron Radiation Facility (Beijing) at a wavelength of 0.9801 Å by using a MAR345 (MAR Research, Hamburg) image plate detector at 100 K. Data integration and scaling were performed by using the programs DENZO and SCALEPACK (12). Data collection statistics are summarized in Table 1. All crystals are in space group P21. A self-rotation function search suggested the presence of two monomers in an asymmetric unit, corresponding to a Vmax of 2.6 Å3/Da and a solvent content of 52% (13).
Table 1. Data collection and refinement statistics.
| pH 6.0 | pH 7.6 | pH 8.0 | CMK complex | |
|---|---|---|---|---|
| Data collection statistics | ||||
| Wavelength, Å | 0.9801 | 0.9801 | 0.9801 | 0.9801 |
| Resolution limit, Å | 50.0—1.9 (1.97—1.90) | 50.0—2.4 (2.49—2.40) | 50.0—2.2 (2.28—2.20) | 50.0—2.5 (2.54—2.50) |
| Cell parameters | ||||
| a, Å | 52.8 | 49.3 | 49.5 | 52.6 |
| b, Å | 97.2 | 97.0 | 97.4 | 97.7 |
| c, Å | 67.7 | 67.8 | 67.5 | 67.9 |
| β, ° | 103.1 | 101.9 | 101.7 | 102.8 |
| Total reflections | 190,084 | 83,091 | 119,770 | 78,378 |
| Unique reflections | 51,775 | 24,261 | 31,275 | 22,614 |
| Completeness | 99.9 (99.5) | 99.3 (94.7) | 100.0 (100.0) | 98.4 (96.3) |
| Redundancy | 3.7 (3.6) | 3.7 (3.7) | 4.4 (4.4) | 4.2 (4.1) |
| Rmerge* | 0.107 (0.570) | 0.132 (0.652) | 0.108 (0.484) | 0.104 (0.444) |
| I/σ(I) | 10.4 (3.7) | 9.8 (3.3) | 10.5 (3.3) | 7.3 (3.1) |
| Refinement statistics | ||||
| Resolution range | 30.0-1.9 | 50.0-2.4 | 50.0-2.2 | 50.0-2.5 |
| Rwork† % | 21.1 | 22.9 | 22.6 | 21.6 |
| Rfree, % | 25.9 | 24.8 | 25.3 | 26.4 |
| rms deviation from ideal geometry | ||||
| Bonds, Å | 0.013 | 0.012 | 0.020 | 0.008 |
| Angles, ° | 1.75 | 1.57 | 2.31 | 1.40 |
| Average B factor, Å2 | ||||
| Chain A | 28.4 | 30.5 | 32.6 | 38.5 |
| Chain B | 28.1 | 36.0 | 36.3 | 37.8 |
| Solvent | 38.2 | 34.3 | 34.6 | 36.8 |
| Ramachandran plot‡ | ||||
| Favored, % | 89.3 | 81.5 | 84.4 | 82.4 |
| Allowed, % | 8.8 | 15.7 | 13.0 | 15.5 |
| Generously allowed, % | 1.0 | 1.7 | 1.5 | 1.0 |
| Disallowed, % | 1.0 | 1.1 | 1.1 | 1.1 |
Rmerge = Σh Σl|Iih — 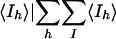 , where
, where 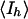 is the mean of the observations, Iih, of reflection h
is the mean of the observations, Iih, of reflection h
Rwork = Σ (∥Fobs| — Fcalc∥)/Σ|Fobs|; Rfree is the R factor for a subset (10%) of reflections that was selected prior to refinement calculations and was not included in the refinement
Ramachandran plots were generated by using the program PROCHECK (14)
Structure Determination, Refinement, and Model Building. The native pH 6.0 structure was determined by molecular replacement by using a single monomer of HCoV 229E Mpro (PDB ID code 1P9S) (9) as a search model. Cross-rotation function and translation function searches were performed with the program CNS (15), and a clear solution for a symmetric dimer was found. The residues that differ between HCoV 229E Mpro and SARS-CoV Mpro were then replaced,and geometric adjustments were made with the program O (16) under the guidance of 2 Fo - Fc and Fo - Fc difference maps, followed by cycles of iterative rebuilding and refinement with the program CNS. Noncrystallographic symmetry restrain was applied to the model in the early stage of refinement. In the final stages of refinement, a composite omit map was calculated to eliminate model bias and waters were allocated to a maxima of >4 σ in the Fo - Fc map. The final pH 6.0 structure consists of residues 3-303 (of a total of 306 residues) in monomer A, 1-302 in monomer B, and 300 water molecules. The pH 7.6, pH 8.0, and CMK complex structures were determined based on the apo pH 6.0 structure by using a similar protocol to the above. In the CMK complex, an inhibitor molecule could be identified, which was bound to each of the two protease protomers from the difference electron density maps. In the final refined models, the residue Tyr-A154, Asp-B33, Asn-B84, Tyr-B154, Thr-B169, and Val-B171 were of unfavorable backbone conformations, albeit the corresponding electron density was well defined. Final refinement statistics are shown in Table 1.
Enzyme Activity Assay. The effects of pH on enzyme activity were measured by using a peptide cleavage assay (17). The sequence of peptide substrate used was NH2-Thr-Ser-Ala-Val-Leu-Gln-Ser-Gly-Phe-Arg-COOH. Cleavage reactions were incubated at 298 K and contained 1.5 μM Mpro, 20 mM Na2HPO4, 200 mM NaCl, 1 mM EDTA, 1 mM DTT, and 0.5 mM peptide substrate in a total volume of 60 μl. The pH value ranged from 5.5 to 8.5. Aliquots of reaction mixture were removed at 90 min, added to an equal volume of 2% trifluoroacetic acid, and immersed in liquid N2, before being stored at 193 K. The samples were centrifuged for 10 min at 15,000 × g before analysis by reverse-phase HPLC on a C18 column (3.9 × 150 mm). Cleavage products were resolved by using a 15 min, 5-60% linear gradient of acetonitrile in 0.1% trifluoroacetic acid. The absorbance was determined at 215 nm, and peak areas were calculated by integration.
Results and Discussion
Overall Structure of the SARS-CoV Mpro. SARS-CoV Mpro forms a dimer in the crystal (and in solutions at concentrations of >1 mg/ml) with the two protomers (denoted as ”A” and ”B”) oriented almost at right angles to each other (Fig. 1 A and B), in an arrangement that is similar to the HCoV and TGEV Mpro structures (9, 18).
Fig. 1.

The SARS-CoV Mpro dimer structure complexed with a substrate-analogue hexapeptidyl CMK inhibitor. (A) The SARS-CoV Mpro dimer structure is presented as ribbons, and inhibitor molecules are shown as ball-and-stick models. Protomer A (the catalytically competent enzyme) is red, protomer B (the inactive enzyme) is blue, and the inhibitor molecules are yellow. The N-finger residues of protomer B are green. The molecular surface of the dimer is superimposed. (B) A cartoon diagram illustrating the important role of the N-finger in both dimerization and maintenance of the active form of the enzyme.
A protomer of the crystal structure is composed of three domains. Domains I (residues 8-101) and II (residues 102-184) have an antiparallel β-barrel structure, which is similar to other CoV proteases and reminiscent of the trypsin-like serine proteases. Domain III (residues 201-303) contains five α-helices arranged into a largely antiparallel globular cluster, and is connected with domain II by means of a long loop region (residues 185-200). SARS-CoV Mpro has a Cys-His catalytic dyad, and the substrate-binding site is located in a cleft between domains I and II.
The crystallographic data here reveal the limitations of the theoretical model (9). The rms deviation of the Cα atoms is 3.8 Å between the crystal structure and the theoretical model (PDB ID code 1P9T) (9), which provided a monomer only. This difference is much larger than that between the two protomers (residues 3-302) in our pH 6.0 crystal structure (0.95 Å), one of which appears to be active and the other inactive.
Substrate-Binding Sites. In contrast to common serine proteases, which have a Ser-His-Asp catalytic triad, SARS Mpro has a Cys-His catalytic dyad (Cys-145 and His-41), which is similar to TGEV Mpro (Cys-144 and His-41) and HCoV Mpro (Cys-144 and His-41) (9, 18). In the active sites and substrate-binding pockets, all four protease protomers in the pH 7.6 and 8.0 structures are similar to protomer A at pH 6.0, but show considerable differences to protomer B. Therefore, we will consider only the two protomers at pH 6.0 for discussion. At this pH, the rms deviation of Cα atoms is 0.95 Å between the two protomers (residues 3-302), with slight differences in some surface loops, especially in domains I and II. However, distinct differences are observed in the substrate-binding sites. Whereas protomer A is similar to other homologous active protease structures, and, hence, resembles a catalytically competent conformation, in protomer B, both the active site and the substrate-binding pocket collapse (Fig. 2 A-C). In protomer A, the Sγ atom of the catalytic residue, Cys-145, is 3.9 Å from the Nε2 atom of the general base, His-41. The Nδ1 of His-41 also donates a 2.7-Å hydrogen bond to a tightly bound water molecule (Fig. 2 A and C). In contrast, in protomer B, the Cys-145 side chain adopts two well defined conformations (i.e., gauche+ and trans), which appear to be equally populated (Fig. 2 B and C). The distances between His-41 Nε2 and the two different positions of Cys-145 Sγ are 3.2 and 3.9 Å, respectively, and the first distance is significantly shorter than that seen in any other viral protease of known structures. As in protomer A, there is a water molecule interacting with His-41 Nδ1 (3.05 Å). Neither of the active-site cysteines show indications of sulfur oxidation, in contrast to what has been reported in other CoV Mpro structures (9, 18).
Fig. 2.

Conformational variations in the S1 substrate-binding pocket. (A) A stereoview of the active site of protomer A built into the 1.9-Å electron density (2 Fo - Fc, contoured at 1.0 σ). The oval-shaped piece of electron density, which is red, is assigned to a water molecule. In S1 subsite of protomer A, Glu-A166 is red, His-A163 and His-A172 are yellow, and the other residues are green. Protomer B is cyan. The amino acid residues of the protein are labeled in single letters; for example, H163A stands for His-163 of monomer A (i.e., His-A163). (B) A stereo image showing the collapsed active site of protomer B built into electron density (2Fo - Fc, contoured at 1.0 σ). The oxyanion hole collapses, the N-finger of chain A is not anchored to its binding site on protomer B, Phe-B140 is directed out into bulk solvent, and Glu-B166 switches conformation to block the substrate-binding site. (C) A schematic presentation of the conformational variations and altered hydrogen-bonding networks in active sites. (Upper) The oxyanion hole (for protomer A) and N-finger of protomer B docked to its binding site. (Lower) The corresponding view of the collapsed active site in protomer B. The N-finger is not docked to its binding site, with the following consequences: (i) the oxyanion hole collapses; (ii) Phe-B140 protrudes into bulk solvent; and (iii) Glu-B166 switches conformation to block the S1 substrate-binding subsite. (D) Comparison of four SARS-CoV Mpro structures. A stereo figure is shown of the substrate-binding pocket of protomer B, with their Cα superimposed. The coloring is as follows: pH 6.0, yellow; pH 7.6, cyan; pH 8.0, green; and CMK inhibitor complex, pink. Side chains are shown as ball-and-stick models for the residues Tyr-B118, Phe-B140, Cys-B145, His-B163, Glu-B166, and His-B172. Note the dramatic conformational changes for Tyr-B118, Phe-B140, Cys-B145, and Glu-B166 when the pH changes from 6.0 to higher pH values.
The substrate-binding subsite, S1, of a CoV protease confers absolute specificity for the Gln-P1 substrate residue on the enzyme (10). The S1 specificity site in protomer A, consisting of the side chains of His-A163 and Phe-A140 and the main-chain atoms of Met-A165, Glu-A166, and His-A172 (Fig. 2 A), is open to substrate binding. The Glu-A166 side chain forms a salt bridge with His-A172, and also interacts with the amide group of Ser-B1, i.e., the N terminus of protomer B in the dimer. Squeezing in between domain III of the parent protomer and domain II of the neighboring protomer in the dimer, the N-terminal residues 1-7 (N-finger) play an important role in the dimerization and formation of the active site of Mpro (Figs. 1B and 2 A and B). The NH group of Ser-B1 donates hydrogen bonds to the carboxylate of Glu-A166, as well as to the main-chain carbonyl of Phe-A140 (Fig. 2 A and C). The latter residue, in turn, stacks against His-A163, presumably ensuring that this histidine will remain uncharged at physiological pH, to facilitate optimal interaction with the Gln-P1 of the substrate. The importance of the N-finger and dimerization is supported by the fact that a deletion mutant of the related TGEV Mpro that lacks residues 1-5 is almost completely inactive (18).
In protomer B, the S1 pocket partly collapses, resulting from the following variations: On one hand, in contrast to protomer A, Phe-140 no longer stacks against His-163, but undergoes a dramatic conformation variation, with the phenyl ring moving by as much as ≈14 Å (Fig. 2D). On the other hand, Glu-166 no longer forms a salt bridge with His-172 but reorientates to interact instead with His-B163 (Fig. 2 B and C). It is plausible that protonation of His-B163 at pH 6.0 modulates the conformation variation seen between the two protomers. The resulting reorientation of Glu-B166 has further consequences: the interaction with the N-finger of protomer A is broken, and the N-finger residues become disordered (no electron density is visible for residues A1 and A2). Furthermore, the whole 138-143 loop undergoes a dramatic rearrangement and becomes partly disordered. These structural variations have consequences for catalysis at residues in this region, particularly Gly-143 and Cys-145, which directly participate in the formation of the oxyanion hole. Gly-B143 moves by ≈3 Å toward the active site, which is relative to its counterpart in protomer A, leaving no space to accommodate a tetrahedral reaction intermediate, whereas the main-chain amide group of Cys-B145 moves away from the oxyanion hole (Fig. 2B). Thus, we conclude that protomer B within the dimer is inactivated through collapse of both the S1 substrate pocket and oxyanion hole. It is important to note that none of the described conformational differences are due to direct influence of crystal packing. Symmetry-related contacts between dimers mainly involve domain III of both protomers, whereas the only residues in domains I and II involved in significant crystal contacts include His-A134, Asn-B53, Asp-B56, Ile-B59, Arg-B60, and His-B134.
pH-Triggered Activity Switch. The inactive conformation of one protomer observed at low pH correlates well with the enzymatic activity shown in Fig. 3. At pH 7.3-8.5, the SARS-CoV Mpro exhibits the highest proteolytic activity on a decapeptide corresponding to the N-terminal autocleavage site, whereas it displays only 50% activity at pH 6.0. In the structure at both pH 7.6 and 8.0, the two protomers in the dimer become more symmetrical. Both active sites are in the catalytically competent conformation, with an intact oxyanion hole and substrate pocket, which is similar to that observed in protomer A of the pH 6.0 structure. Thus, SARS-CoV Mpro appears to possess a pH-triggered activation switch, which may regulate proteolytic activity in different cellular compartments. Such a pH switch would provide the virus with a mechanism to synchronize the polyprotein processing with the microenvironment. Further mutagenesis and biochemical analysis on the details of the switch apparatus are clearly desirable.
Fig. 3.

The effect of pH on Mpro enzyme activity. (A) A plot of production against pH. Production is defined as the ratio of (P1 + P2):(P1 + P2 + S), where P1 and P2 represent the product of the substrate that was cleaved and S represents the substrate leftover. See Methods for further details of the pH activity assay. (B) A profile of the proteolytic reaction for determination of the enzymatic activity of SARS-CoV Mpro. Data are shown for pH 6.0. The extent of peptide cleavage was analyzed by reverse HPLC. The P1 and P2 peaks represent the product peptides that were cleaved, and the S peak represents the substrate leftover.
Unexpected Binding Mode of the Substrate-Analogue Inhibitor. Enzyme-substrate interactions were visualized in the structure of a Mpro crystal soaked in the substrate-analogue hexapeptidyl CMK inhibitor, Cbz-Val-Asn-Ser-Thr-Leu-Gln-CMK (IC50 ca. 2 mM) at pH 6.0. In each protomer, a covalent bond between the Sγ atom of Cys-145 and the methylene group of the CMK, and hydrogen bonds between the residues in the substrate-binding site and the inhibitor stabilize the conformation of substrate-analogue CMK inhibitor in the substrate-binding site (Fig. 4). The structure of the complex at 2.5-Å resolution reveals unexpected modes of inhibitor binding. In protomer A, which possesses active conformation, the side-chain carbonyl of Gln-P1 accepts a 2.8-Å hydrogen bond from the Nε2 atom of His-A163 conserved among CoV Mpro. The side-chain Nε2 of Gln-P1 donates a 2.8-Å hydrogen bond to the side-chain carbonyl of conserved Glu-A166 (Fig. 4A), resulting in the specificity for Gln-P1 in the S1 subsite. However, Leu-P2 fails to bind to the S2 subsite in the vicinity of Asp-187 and becomes partially solvent-accessible. This noncanonical binding results in a frameshift in subsite interaction: Thr-P3 penetrates into the S2 pocket and Asn-P5 binds at the S4 subsite. We note that the P2 residue specificity of SARS-CoV Mpro is less stringent than that of other CoV Mpros, which, with few exceptions, are restricted to leucine (9). The unexpected binding mode of the substrate-like inhibitor may well provide the structural basis for the more relaxed requirements of the enzyme.
Fig. 4.

Molecular recognition interactions in the substrate-analogue hexapetidyl CMK inhibitor (Cbz-Val-Asn-Ser-Thr-Leu-Gln-CMK) complexed with SARS Mpro. (A) A stereoview of the substrate-binding pocket (green) in protomer A of the CMK inhibitor complex. The inhibitor molecule (red) is shown in the 2.5-Å original Fo - Fc difference electron-density map (1.5 σ). Hydrogen bonds are shown as dashed lines. The Gln-P1 is bound to the S1 substrate-specificity subsite, but Leu-P2 fails to bind at the S2 subsite (near Asp-A187), which is instead occupied by Thr-P3. The amino acid residues of the protein are labeled in single letters; for example, H163A stands for His-163 of monomer A (i.e., His-A163). (B) A stereoview of the substrate-binding pocket (green) in protomer B of the CMK inhibitor complex. The inhibitor molecule (red) is shown in the original Fo - Fc difference electron-density map (1.5 σ). The Gln-P1 does not bind to the partly collapsed S1 subsite in this protomer, but Leu-P2 and Ser-P4 are in their canonical binding sites. See text for further details.
The inhibitor also binds to the inactive protomer, but in a different mode. Promoter B remains in the inactive conformation, and the inhibitor does not open the occluded S1 pocket to allow the Gln-P1 to enter the specificity site (Fig. 4B). Instead, the Gln-P1 points out of the substrate-binding cleft and toward bulk solvent. On the other hand, Leu-P2 and Ser-P4 bind to the appropriate specificity pockets that have been observed in the structure of TGEV Mpro (18), whereas Thr-P3 is oriented toward bulk solvent.
Conclusion
In summary, we have determined the crystal structures of SARS-CoV Mpro at different pH values, and with a bound substrate-analogue inhibitor. Our structural data illustrate that SARS-CoV Mpro shares common features with other CoV proteases, such as dimerization and the S1 substrate-specificity pocket. We also observed a pH-dependent activation switch, a unique feature of SARS-CoV Mpro, which results in large, cooperative movements of the side chains of Glu-166, Phe-140, Leu-141, and Tyr-118, and the N terminus of the partner protomer in the dimer. The unexpected binding mode of the substrate-analogue inhibitor provides a structural explanation for the lower P2-specificity of the SARS enzyme compared with those of other CoVs. These structures provide essential structural data of the dimerization and substrate-binding sites to underpin the design and screening of inhibitors with anti-SARS activity.
Acknowledgments
We thank Xuemei Li, Shentao Li, Yinghua Jin, Xueqing Han, Fei Sun, Feng Gao, Xiaodong Zhao, and Xiuhua Dong from Z.R.'s laboratory for technical assistance; Hualiang Jiang, Luhua Lai, Song Li, and Parvesh Wadwani for supplying inhibitors; and Luet Wong, Robert Sim, Ian Jones, John Ziebuhr, Po Tien, Dongcai Liang, and Zhu Chen for discussion and advice. M.Y. is a Ph.D. student from Peking Union Medical College, Beijing, under the supervision of Professor Linfang Wang. G.F.G. is at the Nuffield Department of Clinical Medicine, University of Oxford, Oxford, and is a visiting professor at the Institute of Biophysics, Chinese Academy of Sciences. The stay of R.H. at Tsinghua University was supported by the Ministry of Science and Technology and the Sino-German Center for the Promotion of Science. This work was supported by Ministry of Science and Technology, Chinese Academy of Sciences, and Natural Science Foundation of China Grants 2002BA711A12, 2001AA233011, and G1999075600 (to Z.R.) for the ”Human Structural Genomics Initiative.” R.H. also acknowledges support by Deutsche Forschungsgemeinschaft Grant Hi611/2-2 and the Fonds der Chemischen Industrie.
Abbreviations: SARS, severe acute respiratory syndrome; CoV, coronavirus; HcoV, human CoV; Mpro, main protease; TGEV, porcine transmissible gastroenteritis virus; PEG, polyethylene glycol; CMK, chloromethyl ketone; N-finger, N-terminal residues 1-7.
Data deposition: The atomic coordinates and structure factors have been deposited in the Protein Data Bank, www.rcsb.org [PDB ID codes 1UJ1, 1UK3, 1UK2 (SARS-CoV Mpro, apoenzyme at pH 6.0, 7.6, and 8.0, respectively), and 1UK4 (SARS-CoV Mpro in complex with the hexapeptidyl CMK inhibitor].
Footnotes
World Health Organization press release, August 15, 2003, www.who.int/crs/sars/country/2003_08_15/en.
References
- 1.Drosten, C., Gunther, S., Preiser, W., van der Werf, S., Brodt, H. R., Becker, S., Rabenau, H., Panning, M., Kolesnikova, L., Fouchier, R. A., et al. (2003) N. Engl. J. Med. 348, 1967-1976. [DOI] [PubMed] [Google Scholar]
- 2.Fouchier, R. A., Kuiken, T., Schutten, M., van Amerongen, G., van Doornum, G. J., van den Hoogen, B. G., Peiris, M., Lim, W., Stohr, K. & Osterhaus, A. D. (2003) Nature 423, 240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ksiazek, T. G., Erdman, D., Goldsmith, C. S., Zaki, S. R., Peret, T., Emery, S., Tong, S., Urbani, C., Comer, J. A., Lim, W., et al. (2003) N. Engl. J. Med. 348, 1953-1966. [DOI] [PubMed] [Google Scholar]
- 4.Peiris, J. S., Lai, S. T., Poon, L. L., Guan, Y., Yam, L. Y., Lim, W., Nicholls, J., Yee, W. K., Yan, W. W., Cheung, M. T., et al. (2003) Lancet 361, 1319-1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Marra, M. A., Jones, S. J., Astell, C. R., Holt, R. A., Brooks-Wilson, A., Butterfield, Y. S., Khattra, J., Asano, J. K., Barber, S. A., Chan, S. Y., et al. (2003) Science 300, 1399-1404. [DOI] [PubMed] [Google Scholar]
- 6.Rota, P. A., Oberste, M. S., Monroe, S. S., Nix, W. A., Campagnoli, R., Icenogle, J. P., Penaranda, S., Bankamp, B., Maher, K., Chen, M. H., et al. (2003) Science 300, 1394-1399. [DOI] [PubMed] [Google Scholar]
- 7.Ruan, Y. J., Wei, C. L., Ee, A. L., Vega, V. B., Thoreau, H., Su, S. T., Chia, J. M., Ng, P., Chiu, K. P., Lim, L., et al. (2003) Lancet 361, 1779-1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Thiel, V., Herold, J., Schelle, B. & Siddell, S. G. (2001) J. Virol. 75, 6676-6681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Anand, K., Ziebuhr, J., Wadhwani, P., Mesters, J. R. & Hilgenfeld, R. (2003) Science 300, 1763-1767. [DOI] [PubMed] [Google Scholar]
- 10.Ziebuhr, J., Snijder, E. J. & Gorbalenya, A. E. (2000) J. Gen. Virol. 81, 853-879. [DOI] [PubMed] [Google Scholar]
- 11.Zhu, J., Li, P., Wu, T., Gao, F., Ding, Y., Zhang, C. W., Rao, Z., Gao, G. F. & Tien, P. (2003) Protein Eng. 16, 373-379. [DOI] [PubMed] [Google Scholar]
- 12.Otwinowski, Z. & Minor, W. (1997) in Macromolecular Crystallography, Part A, ed. Sweet, R. M. (Academic, New York), Vol. 276, pp. 307-326. [Google Scholar]
- 13.Matthews, B. W. (1968) J. Mol. Biol. 33, 491-497. [DOI] [PubMed] [Google Scholar]
- 14.Laskowski, R. A., MacArthur, M. W., Moss, D. S. & Thornton, J. M. (1993) J. Appl. Crystallogr. 26, 283-291. [Google Scholar]
- 15.Brunger, A. T., Adams, P. D., Clore, G. M., DeLano, W. L., Gros, P., Grosse-Kunstleve, R. W., Jiang, J. S., Kuszewski, J., Nilges, M., Pannu, N. S., et al. (1998) Acta Crystallogr. D 54, 905-921. [DOI] [PubMed] [Google Scholar]
- 16.Jones, T. A., Zou, J. Y., Cowan, S. W. & Kjeldgaard, M. (1991) Acta Crystallogr. A 47, 110-119. [DOI] [PubMed] [Google Scholar]
- 17.Ziebuhr, J., Heusipp, G. & Siddell, S. G. (1997) J. Virol. 71, 3992-3997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Anand, K., Palm, G. J., Mesters, J. R., Siddell, S. G., Ziebuhr, J. & Hilgenfeld, R. (2002) EMBO J. 21, 3213-3224. [DOI] [PMC free article] [PubMed] [Google Scholar]