

Abstract
LAU-0901, a novel platelet-activating factor (PAF) receptor antagonist, is highly neuroprotective in a rodent model of cerebral ischemia. This study was conducted to establish whether the neuroprotection induced by LAU-0901 persists with chronic survival. Male Sprague-Dawley rats were anesthetized with isoflurane and subjected to 2 h of temporary middle cerebral artery occlusion (MCAo) induced by means of a poly-L-lisine-coated intraluminal nylon suture. Animals were treated with either LAU-0901 (60 mg/kg) or vehicle (45% cyclodextran) administered i.p. at 2 h from onset of MCAo. They received neurobehavioral examinations during MCAo (60 min) and then at 1, 2, 3, 7, 14, 21 and 28 days followed by histopathology at 30 days. LAU-0901 significantly improved the behavior compared to the vehicle group, beginning on day 1 (by 29%, p=0.00007) and persisting throughout a 30-day survival period (42%, p=0.0001). Compared with vehicle treatment, LAU-0901 treatment significantly increased volume of non-infarcted brain tissue loss relative to the unlesioned hemisphere (16.3±4.6% vs. 46.0±10.3%, respectively). These results establish that LAU-0901 confers enduring ischemic neuroprotection.
Keywords: LAU-0901, PAF antagonist, Neuroprotection, Behavioral, Histopathology, Middle cerebral artery occlusion
1. Introduction
Platelet-activating factor (PAF) is a lipid mediator released by many types of cells – such as platelets, monocytes/macrophages, neutrophils and endothelial cells – that potently activates neutrophils, contributing to the pathogenesis of inflammation, endotoxic shock and lipopolysaccharide-mediated tissue injury (Bazan 2003). PAF in the nervous system serves a dual role. It modulates long-term potentiation (Kato K et al. 1994; Clark et al. 1992) and memory (Izquierdo et al. 1995). However, when overproduced, this phospholipid mediator becomes a potent pro-inflammatory mediator. During brain ischemia and in other pathologic conditions involving oxidative stress, PAF concentration increases and, in turn, it becomes a pro-inflammatory messenger and a mediator of neurotoxicity (Bazan and Allan, 1998). Excessive PAF production promotes neuronal damage; inhibition of this process plays a critical role in neuronal survival and prevention of ischemic brain injury (Aspey et al. 1997; Bozlu et al. 2007, Tian and Bazan 2005).
LAU-0901 (2,4,6-Trimethyl-1, 4-Dihydro-Pyridine-3, 5-Dicarboxylic Acid) is a highly potent and selective PAF receptor antagonist (Boetkjaer et al. 2007; Cortina et al. 2005; He and Bazan 2006). Recently, we have shown that LAU-0901 improved behavior and reduced brain infarction in focal cerebral ischemia in rats and mice (Belayev et al. 2008). The dose-response studies in rat and mouse models of transient middle cerebral artery occlusion (MCAo) showed that 60 mg/kg dosage was highly neuroprotective; thus, this dose was applied in this study (Belayev et al. 2008). The objective of the present study was to test the hypothesis that acute LAU-0901-induced neuroprotection endures in animals allowed to survive for several weeks after focal ischemic insult.
2. Results
2.1 Physiological variables
There were no significant differences with respect to rectal and cranial temperatures, arterial blood gases or plasma glucose between groups (Table 1). LAU-0901-treated rats significantly increased body weight from day 3, which persisted throughout the 4-week survival period (Table 1).
Table 1.
Physiological variables
| Vehicle (n=11) | LAU-0901 (n=10) | |
|---|---|---|
| Before MCAo (15 min) | ||
| Rectal temperature (°C) | 36.8 ± 0.07 | 36.6 ± 0.10 |
| Cranial temperature (°C) | 36.7± 0.14 | 36.4 ± 0.11 |
| pH | 7.43 ± 0.01 | 7.42 ± 0.01 |
| PO2, mm Hg | 113 ± 6 | 122 ± 5 |
| PCO2, mm Hg | 40.5 ± 1.13 | 39 7 ± 0.91 |
| Plasma glucose, mg/dL | 167 ± 8 | 165 ± 10 |
| Body weight (g) | 294 ± 8 | 328 ± 7 |
| During MCAo (15 min) | ||
| Rectal temperature (°C) | 37.0 ± 0.10 | 36.8 ± 0.09 |
| Cranial temperature (°C) | 36.7 ± 0.15 | 36.6 ± 0.09 |
| pH | 7.42 ± 0.01 | 7.40 ± 0.01 |
| PO2, mm Hg | 99 ± 4 | 101 ± 3 |
| PCO2, mm Hg | 41.1 ± 0.97 | 43.0 ± 0.89 |
| Plasma glucose, mg/dL | 146 ± 6 | 150 ± 6 |
| After MCAo (2h) | ||
| Rectal temperature (°C) | 38.3 ± 0.25 | 38 0 ± 0.31 |
| Cranial temperature (°C) | 37.4 ± 0.24 | 36.9 ± 0.29 |
| During chronic survival | ||
| Rectal temperature (°C). | 38.0± 0.17 | 37.5 ± 0.61 |
| Body weight (g), 1 day | 269 ± 7 | 300 ± 8 |
| Rectal temperature (°C). | 37.1 ± 0.26 | 37.8 ± 0.13 |
| Body weight (g), 2 days | 261 ± 14 | 297 ± 10 |
| Rectal temperature (°C). | 37.2 ± 0.18 | 37.7 ± 0.16 |
| Body weight (g), 3 days | 259 ± 8 | 296 ± 10* |
| Rectal temperature (°C). | 37.5 ± 0.15 | 37.6 ± 0.13 |
| Body weight (g), 1 week | 286 ± 10 | 332 ± 10* |
| Rectal temperature (°C). | 37.5 ± 0.12 | 37.7 ± 0.10 |
| Body weight (g), 2 weeks | 328 ± 13 | 389 ± 8* |
| Rectal temperature (°C). | 37.2 ± 0.30 | 37.5 ± 0.18 |
| Body weight (g), 3 weeks | 370 ± 13 | 428 ± 9* |
| Rectal temperature (°C). | 37.7 ± 0.21 | 37.5 ± 0.12 |
| Body weight (g), 4 weeks | 397 ± 9 | 452 ± 9* |
Values are mean ± SEM
MCAo, middle cerebral artery occlusion
different from vehicle group (p<0.05, Student’s t-test).
2.2 Neurobehavioral assessment
Neurological score was normal (0) in all animals before MCAo. High-grade contralateral deficits (score, 10–11) were present at 60 min of MCAo in all rats (2); thus, no animals required exclusion based on inadequate ischemia. A significant improvement in neurological score was evident in LAU-0901-treated animals compared with vehicle-treated rats within 1 day of treatment, and the score was sustained at every observation point throughout the 30-day survival period (Figure 1). There were no adverse behavioral side effects observed with LAU-0901 administration.
Figure 1.

Total neurological score, which incorporates postural reflex and forelimb-placing tests (normal score=0, maximal score=12) during MCAo and at various times after treatment. At 60 min of MCAo, all animals had a score of 11 (of a possible 12). LAU-0901 (n=12) or vehicle (n=11) treatment was administered at 2 h after onset of ischemia. LAU-0901-treated rats had a significantly improved neurological score, compared to the corresponding vehicle group, throughout the 30-day survival period. Values shown are means ± SEM., *P<0.05, LAU-0901 vs. vehicle.
2.3 Histopathology
The volume of the left hemisphere contralateral to the infarction was the same in both treatment groups (LAU-0901, 543 ± 7 mm3; vehicle, 529 ± 7 mm3 p=N.S.); this allowed the data to be pooled for subsequent analysis. The rostrocaudal distribution of normal hemispheric tissue areas and the integrated normal hemisphere volumes are shown in Figure 2. Compared to vehicle treatment, LAU-0901 increased the non-infarcted tissue areas at six of nine coronal levels (Figure 2). Integrated right hemisphere volume was reduced compared with the contralateral hemisphere in both vehicle- and LAU-0901-treated groups (Figure 3). However, in LAU-0901-treated rats, the ratio of right/left hemisphere volume computed in individual animals averaged 0.88 ± 0.03 (mean ± SEM), whereas the corresponding ration in vehicle-treated rats was 0.72 ± 0.04. This intergroup difference was highly significant (p=0.001). Thus, LAU-0901 treatment, on average, resulted in a 20% relative increase in proportion of non-infarcted tissue. Treatment with LAU-0901 significantly reduced total lesion volume compared to the vehicle group (20 ± 6 vs. 68 ± 9 mm3, respectively, p=0.0003). Also, tissue loss and percent (relative to unlesion hemisphere) were more severe in vehicle- compared to LAU-0901-treated rats (149 ± 18 vs. 67 ± 14 mm3 and 43 ± 8 vs. 15 ± 4%, respectively, p<0.003). Figure 4 presents Nissl and GFAP brain sections of rats treated with vehicle (A and B) and LAU-0901 (C and D). Vehicle-treated rats showed ipsilateral-hemisphere tissue loss and extensive zones of cystic necrosis. In contrast, rats treated with LAU-0901 showed less extensive cortical and subcortical damage. After 1 month, a cavity had formed and was surrounded with the GFAP positive astrocytes. GFAP expression was more prominent in LAU-0901-treated compared to the vehicle-treated rats and was found in the boundary zone of the infarct (Figure 4B and D). Figure 5 presents GFAP and Nissl positive cell counts. Treatment with LAU-0901 significantly increased GFAP and Nissl positive cells compared to the saline-treated rats. The Nissl positive cells count was not different between the two groups in Cortex A, which was severely damaged in the vehicle group after 30 days; only a small number of neurons survived.
Figure 2.
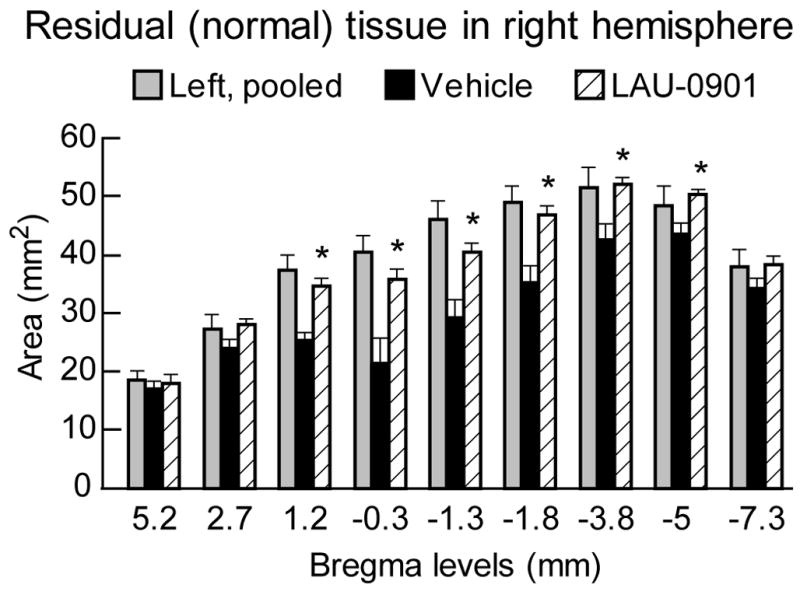
Areas of normal (i.e., non-infarcted, non-cystic) cerebral-hemisphere brain tissue at 30 days after MCAo, shown for nine rostrocaudal forebrain coronal levels in LAU-0901-and vehicle-treated rats. Left-hemisphere data for the vehicle and LAU-0901 groups were pooled since they were virtually identical. Values shown are means ± SEM, *P<0.05, LAU-0901 vs. vehicle.
Figure 3.
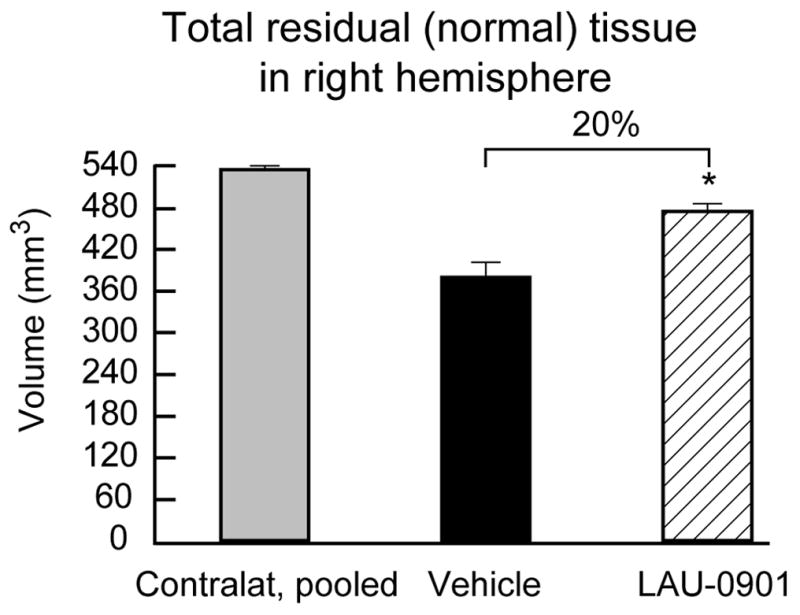
Integrated volumes of normal (i.e., non-infarcted, non-cystic) cerebral-hemisphere brain tissue at 30 days after MCAo in LAU-0901- (n=12) and vehicle- (n=11) treated rats. Left-hemisphere data for the vehicle and LAU-0901 groups were pooled since they were virtually identical. Values shown are means ± SEM., *, P< 0.05, LAU-0901 vs. vehicle.
Figure 4.
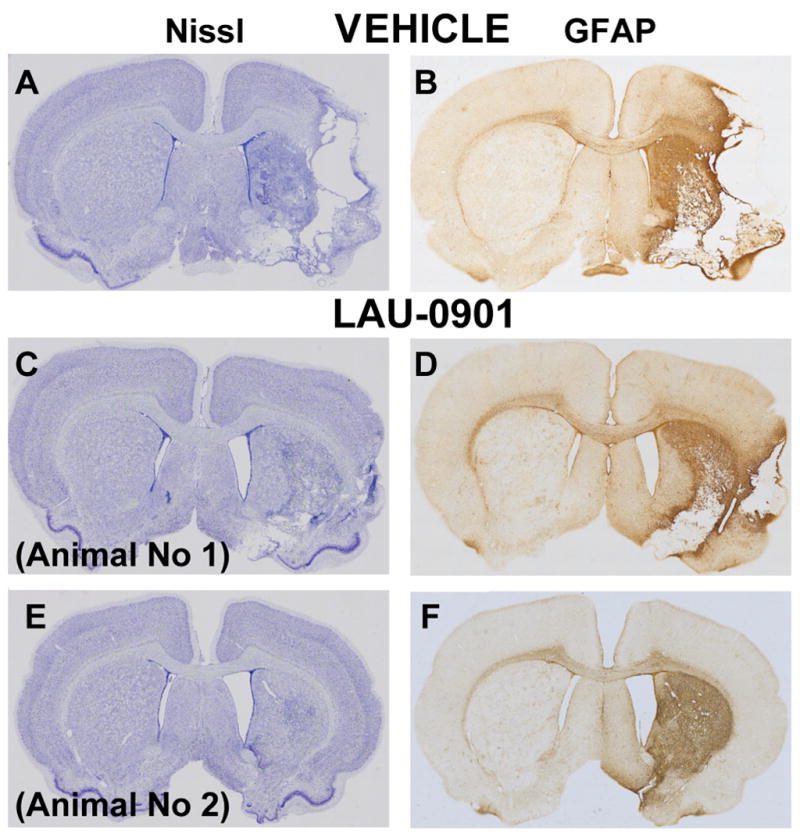
Computer generated MosaiX processed images (Carl Zeiss MicroImaging, Inc, Thornwood, NY) of Nissl (Panels A, C and E) and GFAP (Panels B, D and F) paraffin-embedded brain sections at coronal level (bregma +1.2 mm) from a rat treated with vehicle (Panels A and B) and two rats treated with LAU-0901 (Panels C–F). The saline-treated rat shows typical appearance of cystic necrosis, and pan-necrosis involves the entire neocortical thickness, extending to subjacent regions (Panels A–B). In contrast, the two rats treated with LAU-0901 show less extensive damage (Panels C–F).
Figure 5.
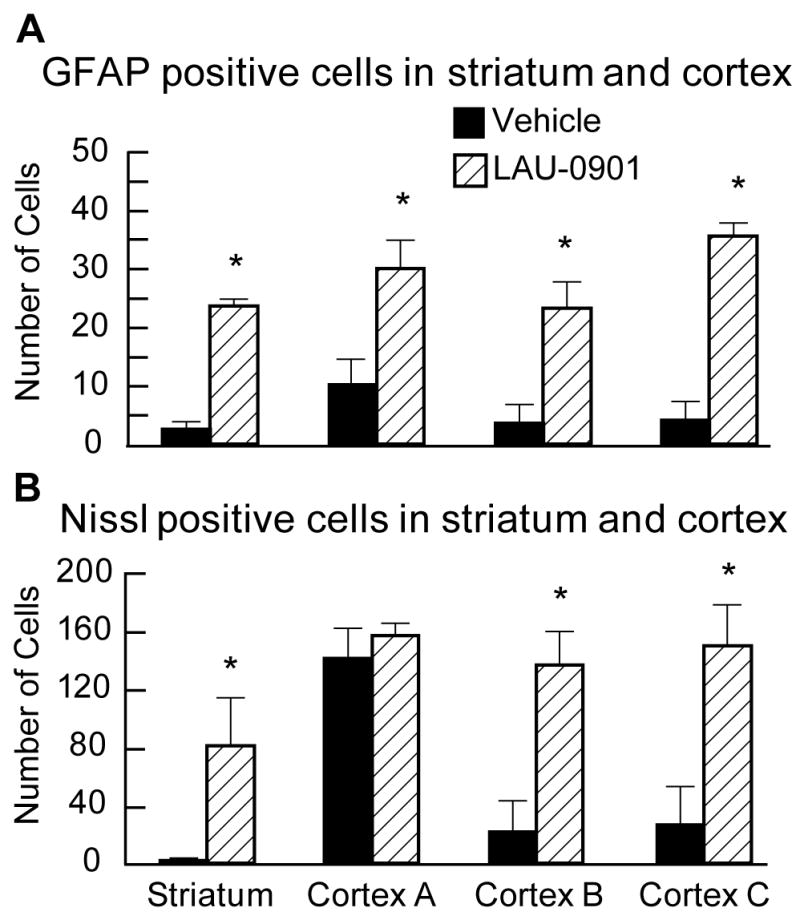
GFAP (Panel A) and Nissl (Panel B) positive cell counts. Treatment with LAU-0901 significantly increased GFAP and Nissl positive cells compared to the saline-treated rats. Values shown are means ± SEM., *, P< 0.05, LAU-0901 vs. vehicle.
Five animals died during the experiments: four rats in the vehicle group (died on days 1, 2, 6 and 7) and one rat in the LAU-0901 group (died on day 1). These animals were eliminated from the neurobehavioral and histopathological analysis.
3. Discussion
The results of this study establish that systemic administration of LAU-0901, a novel PAF receptor antagonist, confers marked and enduring neuroprotection, as assessed by both neurobehavioral and histological methods, in animals followed for 30 days after focal cerebral ischemia. Neurobehavioral improvement began within 1 day after insult and endured for 30 days (Figure 1). The histological results in LAU-0901-treated animals were concordant with the neurological outcome: namely, a 20% increase in preserved brain tissue at 30 days (Figures 3), a striking reduction in the incidence of extensive cystic-necrotic lesions (Figure 3), and markedly improved microscopic histopathology with increased GFAP and Nissl positive cells (Figure 5). These results are very encouraging, because the neuroprotective efficacy shown with short survival periods may not necessarily persist during longer survival periods (Dietrich et al. 1993).
The beneficial effect of LAU-0901 has been shown in a well-controlled animal model of MCAo in rats, which produces consistent cortical and subcortical infarcts that closely resemble the large hemispheric infarcts resulting from proximal MCAo in patients (Belayev et al. 1996). We also closely monitored and controlled physiological parameters, including body and cranial temperatures. Cranial temperature control is extremely important, as it is essential for determining the extent of ischemic brain injury (Ginsberg and Busto 1998).
It has been established that PAF is one of the key brain damage factors following cerebral ischemia reperfusion, and its biological effect is produced via the PAF receptor (Bazan 2001). PAF receptor is present functionally in brain tissue that regulates several events. It activates intracellular signaling cascades, including arachidonic acid metabolism, intracellular calcium changes, increase of the tyrosine phosphorylation and immediate-early gene expression, blood-brain barrier damage, edema formation and neuronal cell death (Kato et al. 1994). The tight regulation of the balance between synthesis (via phospholipases) and degradation (via acetylhydrolases) of PAF modulates the functions of this lipid messenger. During ischemia, seizures, and in other pathological conditions involving oxidative stress, the rates of PAF synthesis and degradation no longer maintain a modulated PAF pool size; consequently, the concentration of PAF increases and it becomes a pro-inflammatory messenger and a mediator of neurotoxicity (Bazan 2005). PAF produces cerebral damage by increasing intracellular calcium concentrations, disrupting the blood brain barrier, reducing cerebral blood flow, and stimulating leucocytes in damaged neuronal tissue (Bazan and Allan 1998).
PAF receptor antagonists reduce experimental brain dysfunction with improvement in neuron survival and function (Bazan 2005). Recently, we discovered a novel platelet-activating factor receptor antagonist, LAU-0901, which has been shown to inhibit apoptosis, to repress the chemotaxis of inflammatory cells, to protect photoreceptors from light-induced oxidative stress, and to inhibit inflammatory responses in retinal, corneal and neural cells (Boetkjaer et al. 2007; Cortina et al. 2005; He and Bazan 2006) (Bazan, N.G., Sunkel, C., Marcheselli, V.L., Builla, G.J., 2003. Assignee Louisiana State University, 2,4,6-trimethyl-1,4-dihydro-pyridine-3,5-dicarboxylic acid esters as neuroprotective drugs. US Patent 6,566,359 B1).
LAU-0901 was also evaluated in animal models of focal cerebral ischemia in rats and mice and showed marked infarct volume reduction and neurobehavioral improvement at seven days of survival (Belayev et al. 2008). LAU-0901 treatment (30, 60 and 90 mg/kg) significantly reduced total corrected infarct volume compared to vehicle rats by 76, 88 and 90%, respectively. Mice treated with LAU-0901 (30 and 60 mg/kg) reduced total infarction by 29 and 66%, respectively. All doses of LAU-0901 significantly improved the neurological score compared with vehicle-treated rats at 24, 48, 72 h and 7 days (Belayev et al, 2008).
The remarkable effect of LAU-0901 is not completely understood. Recently we showed that the recovery of LCBF after 1 h of MCAo was remarkable in LAU-0901-treated mice (Belayev et al, 2008). In contrast to the vehicle group, LCBF continually increased in mice treated with LAU-0901 (by 77%) of baseline at 6 h. Cerebral blood flow improvement is therefore a likely explanation for the smaller infarction in the cortical area and, especially, the subcortical area, which is usually very resistant to any therapeutic intervention. Further studies are needed to reveal the exact mechanism by which LAU-0901 protects the brain after focal cerebral ischemia.
Our data show that after 30 days of MCAo: (1) GFAP expression was found in the boundary zone to the infarct or in the areas of selective incomplete ischemic necrosis; (2) GFAP expression was localized to the same areas where neurons are destined to survive the ischemic insult (detected by Nissl positive cells count). Glial cells are activated after brain ischemia. It has been a general conviction that microglia secretes neurotoxic agents and astrocytes produce neurotrophic factors (Eddleston and Mucke 1993). GFAP reactivity may be associated with neuronal resistance to ischemic damage. The contributions of astrocytes to normal brain physiology have been demonstrated by many reports (Kimelberg and Norenberg 1989; Tower 1992). Astrocytes can release a number of growth factors for neurons (Kimelberg and Norenberg 1989). Some of these, such as nerve growth factor, may stimulate the neuron as a whole as well as promote neurite growth. Thus, GFAP expression may be related to a possible “protective” effect on the adjacent neurons. Our data may be interpreted as evidence for the engagement of protective mechanisms in astrocytes and neurons within regions of the brain that remain viable following focal cerebral ischemia.
4. Conclusions
In summary, the present results establish that LAU-0901, a novel platelet-activating factor receptor antagonist, confers enduring neuroprotection in an in vivo model of temporary focal cerebral ischemia compared to the vehicle treatment. Therefore, a pharmacological agent such as LAU-0901 may have potential use in treating focal ischemic stroke in the clinical setting.
5. Experimental procedures
5.1 Protocols and animal care
Experimental protocols were approved by the Institutional Animal Care and Use Committee of the Louisiana State University Health Sciences Center, New Orleans.
5.2 Focal cerebral ischemia
Twenty-eight adult male Sprague–Dawley rats (260 to 329 grams; Charles River Laboratories, Wilmington, Mass) were fasted overnight but allowed free access to water. Anesthesia was induced with 3.5% halothane in a mixture of 70% nitrous oxide and 30% oxygen. Rats were orally intubated, immobilized with pancuronium bromide (0.6 mg/kg, intravenous), mechanically ventilated, and inserted with femoral arterial and venous catheters for blood sampling and drug infusion. Rectal and cranial (left temporalis muscle) temperatures were separately monitored and held at normothermic levels (36–37°C). Arterial blood gases, pH and glucose were measured 15 min before, during, and 15 min after MCAo. The right middle cerebral artery (MCA) was occluded for 2 h by the intraluminal-filament method using a poly-L-lysine-coated suture as previously reported (Belayev et al. 1996). After 2 h of MCAo, rats were re-anesthetized and the intraluminal suture was carefully removed. The neck incisions were closed with silk sutures, and the animals were allowed to survive for 30 days with free access to food and water.
5.3 Treatment
The agents (LAU-0901; 60 mg/kg; n=12) or vehicle (45% cyclodextran, 1 ml/kg; n=11) were administered i.p. at the time of reperfusion, i.e., 2 h from onset of MCAo in rats.
5.4 Behavioral evaluation
A standardized battery of behavioral tests was used to quantify sensorimotor neurological function during MCAo (60 min) and then at 1, 2, 3, 7, 14, 21 and 28 days after MCAo (Belayev et al. 1996). The battery, which incorporates postural reflex and forelimb-placing tests, yields a 12-point score (normal=0, maximal=12) (Belayev et al. 1996). Tests were conducted by an observer blinded to the treatment group.
5.5 Histopathology
Following a 30-day survival period, animals were deeply anesthetized with isoflurane and perfused transcardially with isotonic saline, followed by a perfusion with paraformaldehyde (4% in phosphate buffer). Then brains were removed and brain blocks were embedded in paraffin. Twelve-micron-thick sections were cut in the coronal plane and stained with thionine (Nissl), and then adjacent sections were used for glial fibrilliary acidic protein (GFAP) immunostaining (Kokubo et al. 2002). Brain sections were then digitized (MCID™ Core imaging software, InterFocus Imaging Ltd, Linton, Cambridge, UK) at nine standardized coronal levels (bregma levels: +5.2, +2.7, +1.2, −0.3, −1.3, −1.8, −3.8, −5.0 and −7.3 mm) (Konig and Klippel 1963). Image analysis was conducted by an operator blinded to the treatment group assignment. Since chronic histopathology of untreated focal ischemic infarction involves extensive tissue loss with residual cavitation, cystic alterations, and ventricular dilatation, image analysis consisted of outlining the areas of the lesion (which were clearly demarcated), left and right ventricles, and the left- (contralateral) and right- (MCAo) hemisphere contours at each level. The following analysis was conducted: (A) Lesion volume was calculated as the product of the cross-sectional area for all sections, and the distance between the sections was determined using Simpson’s method (Carnevale 1986). (B) Residual (normal) tissue in the right hemisphere (mm3) was calculated applying the following formula: (right hemisphere volume - right ventricle - lesion volume). (C) Tissue loss was calculated as a difference in the amount of histologically-intact residual tissue between the lesioned and the unlesioned hemispheres. (D) Percent [relative to unlesioned (left) hemisphere volume] was calculated applying the following formula: (Tissue loss × 100/Residual tissue in left hemisphere). GFAP and Nissl positive cell counts were conducted in the cortex (A, B and C) and striatum (S) at the level of the central lesion (bregma level −0.3 mm; Figure 6). Data were expressed as numbers of GFAP and Nissl positive cells per high-power microscopic field (magnification × 40).
Figure 6.
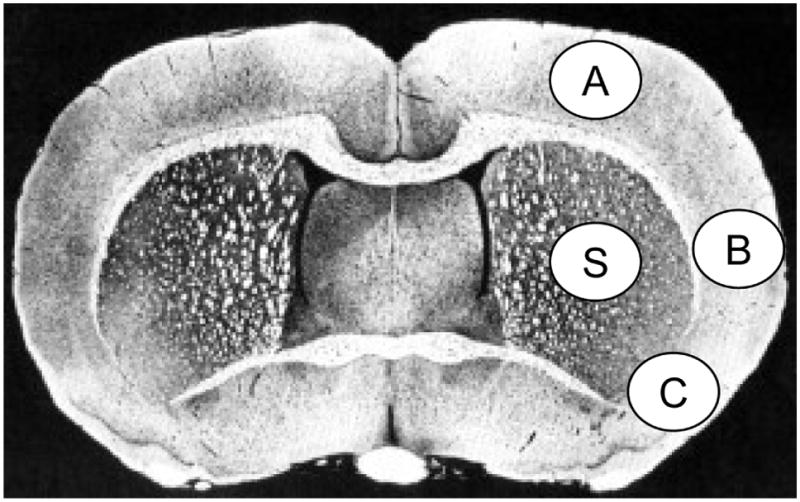
Schematic brain diagram showing locations of regions for GFAP and Nissl positive cell counts in the cortex (A, B and C) and striatum (S).
5.6 Statistical analysis
Data were presented as mean values ± SEM. Neurobehavioral scores and infarct size data were analyzed by repeated-measures analysis of variance (ANOVA) with post hoc Banferroni tests. Physiological variables were compared using Student t tests. Differences at P<0.05 were considered statistically significant.
Acknowledgments
This investigation was supported by NIH Grant NS023002 (NGB). The authors thank Neuroscience Associates, Inc., for histology service.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aspey BS, Alp MS, Patel Y, Harrison MJ. Effects of combined glutamate and platelet-activating factor inhibition on the outcome of focal cerebral ischemia - an initial screening study. Metab Brain Dis. 1997;12:237–249. [PubMed] [Google Scholar]
- Bazan NG. COX-2 as a multifunctional neuronal modulator. Nat Med. 2001;7:414–415. doi: 10.1038/86477. [DOI] [PubMed] [Google Scholar]
- Bazan NG. Lipid signaling in neural plasticity, brain repair, and neuroprotection. Mol Neurobiol. 2005;32:89–103. doi: 10.1385/MN:32:1:089. [DOI] [PubMed] [Google Scholar]
- Bazan NG. Synaptic lipid signaling: significance of polyunsaturated fatty acids and platelet-activating factor. J Lipid Res. 2003;44:2221–2233. doi: 10.1194/jlr.R300013-JLR200. [DOI] [PubMed] [Google Scholar]
- Bazan NG, Allan G. Platelet-activating factor and other bioactive lipids. In: Ginsberg MD, Bogousslavsky J, editors. Cerebrovascular Disease, Pathophysiology, Diagnosis and Management. Blackwell Science Publishers; Malden, Massachusetts: 1998. pp. 532–555. [Google Scholar]
- Belayev L, Alonso OF, Busto R, Zhao W, Ginsberg MD. Middle cerebral artery occlusion in the rat by intraluminal suture. Neurological and pathological evaluation of an improved model. Stroke. 1996;27:1616–1622. doi: 10.1161/01.str.27.9.1616. [DOI] [PubMed] [Google Scholar]
- Belayev L, Khoutorova L, Atkins K, Gordon WC, Alvarez-Builla J, Bazan NG. LAU-0901, a novel platelet-activating factor antagonist, is highly neuroprotective in cerebral ischemia. Exp Neurol. 2008 doi: 10.1016/j.expneurol.2008.08.009. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boetkjaer A, Boedker M, Cui JG, Zhao Y, Lukiw WJ. Synergism in the repression of COX-2- and TNFalpha-induction in platelet activating factor-stressed human neural cells. Neurosci Lett. 2007;426:59–63. doi: 10.1016/j.neulet.2007.08.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bozlu G, Atici A, Turhan AH, Polat A, Nayci A, Okuyaz C, Taskinlar H. Platelet-activating factor antagonist (ABT-491) decreases neuronal apoptosis in neonatal rat model of hypoxic ischemic brain injury. Brain Res. 2007;1143:193–198. doi: 10.1016/j.brainres.2007.01.094. [DOI] [PubMed] [Google Scholar]
- Carnevale NT. Integration of data obtained at fixed intervals. Brain Res Bull. 1986;16:137–142. doi: 10.1016/0361-9230(86)90022-5. [DOI] [PubMed] [Google Scholar]
- Clark GD, Happel LT, Zorumski CF, Bazan NG. Enhancement of hippocampal excitatory synaptic transmission by platelet-activating factor. Neuron. 1992;9:1211–1216. doi: 10.1016/0896-6273(92)90078-r. [DOI] [PubMed] [Google Scholar]
- Cortina MS, Gordon WC, Lukiw WJ, Bazan NG. Oxidative stress-induced retinal damage up-regulates DNA polymerase gamma and 8-oxoguanine-DNA-glycosylase in photoreceptor synaptic mitochondria. Exp Eye Res. 2005;81:742–750. doi: 10.1016/j.exer.2005.04.017. [DOI] [PubMed] [Google Scholar]
- Dietrich WD, Dewanjee S, Prado R, Watson BD, Dewanjee MK. Transient platelet accumulation in the rat brain after common carotid artery thrombosis. An 111 In-labeled platelet study. Stroke. 1993;24:1534–1540. doi: 10.1161/01.str.24.10.1534. [DOI] [PubMed] [Google Scholar]
- Eddleston M, Mucke L. Molecular profile of reactive astrocytes--implications for their role in neurologic disease. Neuroscience. 1993;54:15–36. doi: 10.1016/0306-4522(93)90380-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginsberg MD, Busto R. Combating hyperthermia in acute stroke: a significant clinical concern. Stroke. 1998;29:529–534. doi: 10.1161/01.str.29.2.529. [DOI] [PubMed] [Google Scholar]
- He J, Bazan HE. Synergistic effect of platelet-activating factor and tumor necrosis factor-alpha on corneal myofibroblast apoptosis. Invest Ophthalmol Vis Sci. 2006;47:883–891. doi: 10.1167/iovs.05-0581. [DOI] [PubMed] [Google Scholar]
- Izquierdo I, Fin C, Schmitz PK, Da Silva RC, Jerusalinsky D, Quillfeldt JA, Ferreira MB, Medina JH, Bazan NG. Memory enhancement by intrahippocampal, intraamygdala, or intraentorhinal infusion of platelet-activating factor measured in an inhibitory avoidance task. Proc Natl Acad Sci U S A. 1995;92:5047–5051. doi: 10.1073/pnas.92.11.5047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato K, Clark GD, Bazan NG, Zorumski CF. Platelet-activating factor as a potential retrograde messenger in CA1 hippocampal long-term potentiation. Nature. 1994;367:175–179. doi: 10.1038/367175a0. [DOI] [PubMed] [Google Scholar]
- Kimelberg HK, Norenberg MD. Astrocytes. Sci Am. 1989;260:66–72. 74–76. doi: 10.1038/scientificamerican0489-66. [DOI] [PubMed] [Google Scholar]
- Kokubo Y, Matson GB, Liu J, Mancuso A, Kayama T, Sharp FR, Weinstein PR. Correlation between changes in apparent diffusion coefficient and induction of heat shock protein, cell-specific injury marker expression, and protein synthesis reduction on diffusion-weighted magnetic resonance images after temporary focal cerebral ischemia in rats. J Neurosurg. 2002;96:1084–1093. doi: 10.3171/jns.2002.96.6.1084. [DOI] [PubMed] [Google Scholar]
- Konig JFR, Klippel RA. The Rat Brain: A Stereotaxic Atlas of the Forebrain and Lower Parts of the Brain Stem. Lippincott Williams and Wilkins; Baltimore: 1963. [Google Scholar]
- Tian X, Bazan NG. Neuroprotection by platelet-activating factor antagonism. Ann N Y Acad Sci. 2005;1053:455–456. doi: 10.1111/j.1749-6632.2005.tb00054.x. [DOI] [PubMed] [Google Scholar]
- Tower DB. A century of neuronal and neuroglial interactions, and their pathological implications: an overview. Prog Brain Res. 1992;94:3–17. doi: 10.1016/s0079-6123(08)61735-5. [DOI] [PubMed] [Google Scholar]