

Abstract
In 2006, more than 55 000 patients died of colorectal cancer in the US, accounting for ∼10% of all cancer deaths. Despite significant progress in screening combined with the development of novel effective therapies, colorectal cancer ranks second to lung cancer as a cause of cancer death. Twin studies indicate that 35% of all colorectal cancers are inherited, but high-penetrance tumor susceptibility genes only account for ∼3–6% of all cases. The remainder of the unexplained familial risk is presumably due to other high-penetrance genes, but polygenic mechanisms and low-penetrance tumor susceptibility genes are likely to account for a greater proportion of familial colorectal cancers. In this regard, there is growing evidence that a common hypomorphic variant of the type I TGF-β receptor, TGFBR1*6A, may account for ∼3% of all colorectal cancer cases, a fraction higher than that attributable to mismatch repair genes MLH1, MSH2, MSH6 and PMS2. Furthermore, TGFBR1*6A is emerging as a potent modifier of colorectal cancer risk among individuals with a strong family of colorectal cancer. The TGF-β signaling pathway plays a central but paradoxical role in the predisposition and progression of colorectal cancer. TGF-β is a potent inhibitor of normal colonic epithelial cells acting as a tumor suppressor. However, TGF-β promotes the survival, invasion and metastasis of colorectal cancer cells, thereby acting as an oncogene. Understanding how selective alterations of the TGF-β signaling pathway contribute to colorectal cancer development and progression will likely permit the identification of an additional fraction of inherited colorectal cancer cases and provide novel opportunities for therapeutic intervention.
INTRODUCTION
Colorectal cancer is the third most common cancer and the second leading cause of cancer death in the US. In 2006, 148 810 new cases were diagnosed, of which 106 680 were colon and the remainder rectal cancers (1). Despite the increased use of screening strategies such as fecal occult blood testing, sigmoidoscopy and colonoscopy, more than one-third of patients with colorectal cancer will ultimately develop metastatic disease. Although several new treatment options have become available for metastatic colorectal cancer in the past decade, most patients will eventually die of uncontrolled metastatic spread within a few years of diagnosis, accounting for 55 000 deaths in 2006 (1). It is widely accepted that genetic factors play key roles in the predisposition to colorectal cancer development and progression, but the proportion of colorectal cancer cases attributable to inherited factors depends on the definition of familial colorectal cancer. More than 11% of patients with colorectal cancer have at least one first degree relative with the same disease (2), but twin studies suggest that ∼35% of colorectal cancers are inherited (3). One may therefore reasonably estimate that one-third of all colorectal cancer cases are caused by cancer susceptibility genes. Our current knowledge of cancer genetics is primarily based on the identification of high-penetrance tumor susceptibility genes such as APC, MLH1, MSH2, MSH6 and PMS2 (4). The high morbidity and mortality of colorectal cancer and the availability of screening and prevention strategies provide compelling reasons to search for additional colorectal cancer susceptibility genes. Indeed, identification and removal of early-stage colorectal cancers screened by fecal occult blood testing or sigmoidoscopy are feasible and effective methods to reduce mortality (5). Strategies consisting of colonoscopies every 1–3 years in individuals at high risk for colorectal cancer result in decreased colorectal cancer incidence (6), but such strategies cannot realistically be used in the general population. In this short review, we will focus on some of the recent developments in the study of the transforming growth factor-β (TGF-β) signaling pathway, as it relates to colorectal cancer susceptibility.
GENETICS OF COLORECTAL CANCER
Clues regarding important genetic targets in colorectal cancer have come from the study of two hereditary neoplastic syndromes: familial adenomatous polyposis (FAP) and Lynch syndrome, formerly named hereditary non-polyposis colorectal cancer (HNPCC). FAP is caused by inherited defects in the adenomatous polyposis coli (APC) gene, which lead to the development of multiple benign polyps throughout the colon. Additional mutations, including mutation of KRAS and TP53, and deletion on chromosome 18q are required for subsequent tumor progression (7). In contrast, Lynch syndrome is characterized by rapid progression of colorectal tumors, which is due to germline mutation of one of the DNA mismatch repair (MMR) genes: MLH1, MSH2, MSH6 or PMS2 (8). Interestingly, somatic inactivation of MMR genes, which is generally caused by hypermethylation in the promoter of MMR genes, has also been found in ∼15–20% of sporadic colorectal cancers (9). Cells with MMR gene inactivation are prone to insertion/deletion-type mutations during replication, which occur especially at sites of nucleotide repeat sequences known as microsatellites and result in microsatellite instability (MSI). Mutations in microsatellites of certain tumor-suppressor genes, such as the type II TGF-β receptor TGFBR2 (discussed in more detail in what follows), BAX, E2F4 and IGFR2, are frequently identified in MSI + colorectal tumors (9,10). Therefore, MSI may create a favorable state for accumulating mutations in genes that control cell growth and apoptosis, and these alterations ultimately lead to cancer progression.
Although the genetic mechanisms underlying FAP and Lynch syndrome are well understood, they only account for ∼0.2 (4) and 2% (11,12) of all colorectal cancers, respectively. Inherited variants of the MYH gene have been shown to cause MYH-associated polyposis and are thought to account for ∼1% of all colorectal cancers (13). Germline mutations of the STK11 gene underlie the Peutz–Jeghers syndrome (14), and mutations of SMAD4 (15) and BMPR1A (16) cause juvenile polyposis. Collectively, these syndromes account for 3–6% of all colorectal cancers (17). Much of the remaining familial colorectal cancers and a large proportion of sporadic cases are likely due to low-penetrance mutations, i.e. mutations that have low frequency of association with a specific phenotype (4). Several low-penetrance alleles that are associated with colorectal cancer have been identified, including TGFBR1*6A (discussed in what follows) and APC*I1307K (18).
TGF-β SIGNALING PATHWAY IN COLORECTAL CANCER
The TGF-β signaling pathway is involved in the control of several biological processes, including cell proliferation, differentiation, migration and apoptosis (19). It is one of the most commonly altered cellular signaling pathways in human cancers (20). TGF-β signaling is initiated by the binding of TGF-β ligands to type II TGF-β receptors (TGFBR2). Three TGF-β isoforms (TGFB1, TGFB2 and TGFB3) are expressed in mammalian epithelium, and each is encoded by a unique gene and expressed in both a tissue-specific and developmentally regulated manner. TGFB1 is the most abundant and ubiquitously expressed isoform. Once bound to TGF-β, TGFBR2 recruits and phosphorylates the type I TGF-β receptor (TGFBR1), which stimulates TGFBR1 protein kinase activity. Activated TGFBR1 then phosphorylates two downstream transcription factors, SMAD2 and SMAD3, allowing them to bind to SMAD4. The resulting SMAD complexes translocate into the nucleus and interact with other transcription factors in a cell-specific manner to regulate the transcription of a multitude of TGF-β-responsive genes (21) (Fig. 1). It is increasingly apparent that TGF-β-related proteins initiate the activation not only of SMADs but also of other signaling pathways. These pathways regulate SMAD-mediated responses and also induce SMAD-independent responses (22).
Figure 1.
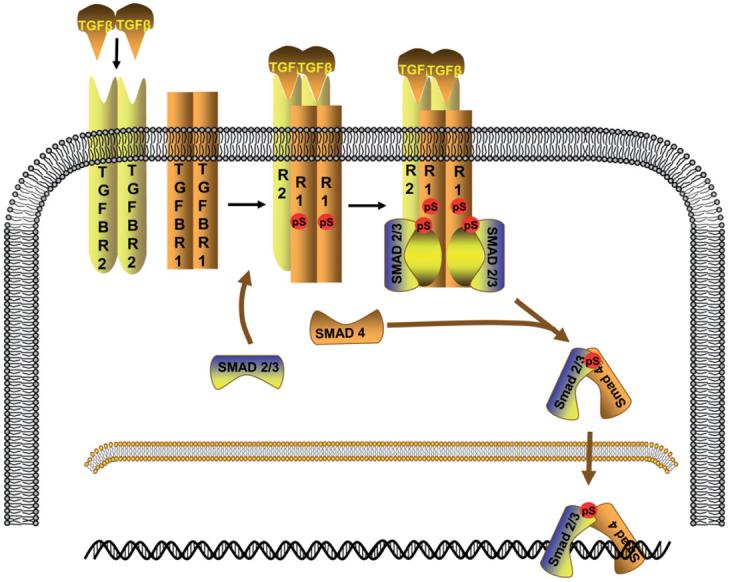
TGF-β/SMAD signaling pathway. TGF-β (TGFB1, TGFB2 or TGFB3) binds to the type II TGF-β receptor (TGFBR2: R2), which then binds to the type I TGF-β receptor (TGFBR1: R1) and forms a heterodimeric complex in which TGFBR1 kinase becomes activated. SMAD2 and SMAD3 are phosphorylated by activated TGFBR1. Once phosphorylated, they associate with SMAD4 and move to the nucleus where they act as transcription factors.
Some of the downstream targets of TGF-β signaling are important cell-cycle checkpoint genes, including CDKN1A (p21), CDKN1B (p27) and CDKN2B (p15), and their activation leads to growth arrest (19). Therefore, TGF-β serves as a tumor suppressor in the normal intestinal epithelium by inhibiting cell proliferation and inducing apoptosis. Many colorectal cancers escape the tumor-suppressor effects of TGF-β and are resistant to TGF-β-induced growth inhibition (23). However, during the late stages of colorectal carcinogenesis, TGF-β acts as a tumor promoter and is usually highly expressed. High levels of TGFB1 in the primary colorectal tumor are associated with advanced stages and a greater likelihood of recurrence and decreased survival (24,25). Experimentally, prolonged exposure to high levels of TGF-β promotes neoplastic transformation of intestinal epithelial cells (26), and TGF-β stimulates the proliferation and invasion of poorly differentiated and metastatic colon cancer cells (27).
Although the mechanism by which TGF-β switches its growth inhibitory effect into growth stimulatory effect is not well understood, TGF-β has been shown to increase the production of several mitogenic growth factors including TGF-α, FGF and EGF (21). In addition, TGF-β can activate SMAD-independent pathways, such as Ras/MAPK pathway, JNK pathway and PI3 kinase/Akt pathway (8,21). Thus, TGF-β may drive the proliferation of colorectal cancer cells in conjunction with these oncogenic pathways. TGF-β is also a potent regulator of cell adhesion, motility and the extracellular matrix composition, which are involved in tumor invasion and metastasis (21). In addition, TGF-β signaling promotes angiogenesis and immuno-suppression (21). Therefore, it is likely that cancer cells achieve resistance to the tumor-suppressor effects of TGF-β but remain responsive to the tumor-promoter effects of TGF-β via selective alterations of this signaling pathway.
TGFBR2 MUTATIONS IN COLORECTAL CANCER
The TGFBR2 gene contains an A (10) tract in exon 3 and GT (3) tracts in exon 5 and 7, which are microsatellite sequences prone to replication errors, especially in the presence of MMR gene inactivation (28). Frameshift mutations of TGFBR2 are found in >80% of MSI+ colorectal cancers (10). It is commonly speculated that colorectal cancers acquire partial TGF-β resistance largely because of TGFBR2 genetic alterations. In fact, the overall incidence of TGFBR2 mutations is close to 30% in colorectal cancer and is the most common mechanism identified so far that results in TGF-β signaling alteration (9,29). Interestingly, some colorectal cancer cell lines, which harbor homozygous mutation of TGFBR2, are growth-inhibited by TGFB1, which suggests that under certain circumstance, the cells can bypass TGFBR2 to maintain growth inhibition (28). Whether TGFBR2 mutations truly contribute to colorectal tumorigenesis or arise as bystander mutations in MSI+ tumors remains to be further studied. The fact that TGFBR2 missense mutations are identified in ∼15% of colon cancer cell lines that are microsatellite stable (29) provides some support for the notion that TGFBR2 serves as a tumor suppressor in colorectal cancer.
To determine the pathogenic relevance of TGFBR2 inactivation in colorectal cancer, a conditional mouse model that was null for Tgfbr2 in the colonic epithelium was generated and treated with azoxymethane to induce colorectal cancer formation (30). Mice that lack Tgfbr2 in the colon epithelium developed more colorectal adenomas and adenocarcinomas and had increased neoplastic proliferation when compared with mice with intact Tgfbr2 (30). The increase in cellular proliferation in the absence of TGFBR2 is likely due to the failure to inactivate cdk4 expression in TGFBR2-deficient cells, as cdk4 expression is upregulated in MSI+ cancers (31). In addition, reconstitution of TGFBR2 in an MSI+ colorectal cancer cell line resulted in a decrease in cdk4 expression and activity, accompanied with a decrease in cellular proliferation (31). These studies suggest that TGFBR2 inactivation may be a factor contributing to the transformation of colorectal cancers.
TGFBR1 MUTATIONS AND POLYMORPHISMS IN COLORECTAL CANCER
Mutations in TGFBR1 have been identified in human colorectal cancer cell lines but are not common (32). However, decreased TGFBR1 expression levels are frequently observed. In such cells, reconstitution of TGFBR1 expression has been shown to decrease tumorigenicity (33). Our search for TGFBR1 tumor-specific mutations led us to the discovery of a polymorphic allele of the type I receptor, which was first named TβR-Iβ (6A), that has a deletion of three alanines within a 9-alanine stretch of exon 1 coding sequence (34). In 2003, this variant was renamed TGFBR1*6A (*6A) to be consistent with the HUGO nomenclature (35). Other rare polyalanine stretch variants such as *10A, *8A and so on have also been described (36). The fact that a significantly higher *6A allelic frequency was found among patients with a diagnosis of cancer than among healthy controls prompted us to postulate that *6A may act functionally as a tumor susceptibility allele (36). Over the past few years, some studies have confirmed an association between *6A and cancer, but others have failed to establish any correlation (37-41). To resolve these discrepancies, we carried out a meta-analysis of all published case–control studies on *6A, which included 2438 patients with a diagnosis of cancer and 1846 healthy controls. The combined analysis of seven studies showed that *6A allelic frequency was 28.8% higher among all cancer cases (0.0935) combined than among controls (0.0726) (P < 0.001) (35). A second updated meta-analysis of 12 case–control studies that included 7850 individuals confirmed these original findings (42). A third meta-analysis that included 6968 cases and 6145 controls was published in 2005 by Zhang et al. (43). The combined analysis of those 17 case–control studies that included more than 13 000 cases and controls showed that TGFBR1*6A allelic frequency was 44% higher among all cancer cases (0.082) than among controls (0.057) (P < 0.0001). Overall, *6A carriers have a 22% (OR 1.22; 95% CI 1.12–1.34) increased risk of cancer at any site. The overall cancer risk is significantly increased for both *6A heterozygotes (OR 1.13; 95% CI 1.03–1.24) and homozygotes (OR 2.04; 95% CI 1.49–2.78). A combined analysis of the six studies assessing *6A in colon cancer cases and controls indicates that TGFBR1*6A carriers are at increased risk of developing colorectal cancer (OR 1.20; 95% CI 1.01–1.43) (42). Estimates based on these numbers predict that TGFBR1*6A population attributable risk is 3.2% (0.6–5.8%).
Low-penetrance genes, such as TGFBR1*6A, may also account for a proportion of familial colorectal cancer cases. To test this hypothesis, we determined whether TGFBR1*6A contributes to a proportion of mismatch repair gene mutation-negative (MMR−) HNPCC cases. In that study, 208 index patients with HNPCC meeting the Amsterdam criteria were examined for mutations and genomic rearrangements in the MLH1, MSH2 and MSH6 genes and genotyped for TGFBR1*6A; 144 patients (69.2%) carried a deleterious mutation and were classified as mismatch repair gene mutation-positive (MMR+), i.e. having Lynch syndrome; 64 patients (30.8%) had no evidence of mutations and were classified as MMR−. TGFBR1*6A allelic frequency was significantly higher among MMR − patients (0.195) than among MMR+ patients (0.104) (P = 0.011). The proportion of TGFBR1*6A homozygotes was 9-fold higher among MMR − (6.3%) than among MMR+ patients (0.7%) (P = 0.032) (44). We found that MMR − *6A homozygotes have a 10-fold higher colorectal cancer risk than MMR − that do not carry the *6A allele (Table 1). Our calculations based on published reports suggest that overall 7% of all HNPCC cases and 13% of MMR − HNPCC cases may be associated with *6A. These results are in agreement with the recent findings of Kemp et al. (45), who showed that *6A is not a major colon cancer susceptibility gene with a dominant pattern of inheritance, co-segregating with cancer in HNPCC families. Rather, they suggest that *6A may act as a significant modifier of colorectal cancer susceptibility in concert with other, yet to be discovered genes.
Table 1.
Colorectal cancer risk among HNPCC patients based on TGFBR1*6A status: adjusted ORs for the associations between MMR mutation status and TGFBR1 genotype
| TGFBR1 genotype | MMR mutation status (N = 208) N (positive/negative) |
Crude ORs for MMR – (95% Cl)a |
|
|---|---|---|---|
| Dominant model | |||
| 9A/9A | 115/43 | 1.00 | |
| 9A/6A or 6A/6A | 29/21 | 1.94 (1.00–3.75) | |
| Additive model | |||
| 9A/9A | 115/43 | 1.00 | |
| 9A/6A | 28/17 | 1.62 (0.81–3.26) | |
| 6A/6A | 1/4 | 10.70 (1.16–98.4) | |
| Recessive model | |||
| 9A/9A or 9A/6A | 143/60 | 1.00 | |
| 6A/6A | 1/4 | 9.53 (1.04–87.1) | |
ORs were adjusted for age at diagnosis and gender. Nine subjects with unknown age or gender were excluded from the analysis.
The molecular mechanism by which TGFBR1*6A contributes to cancer development is still under investigation. Expression of TGFBR1*6A in a TGFBR1-deficient mink lung epithelium cell line results in reduced TGF-β-mediated antiproliferative response in comparison with cells that express the wild-type TGFBR1 (36,46). However, the same experiments have shown that TGFBR*10A, another rare variant, transduces TGF-β growth-inhibitory signals as effectively as TGFBR1 (36), which suggests that the 6 Ala repeat of *6A has specific biological properties. In addition, when we expressed *6A in the MCF-7 breast carcinoma cells, we observed that the cells became growth stimulated upon exposure to TGFB1, whereas cells transfected with the wild-type form remained growth inhibited (47). More interestingly, one colorectal cancer cell line, DLD-1, which harbors one *6A allele, was also growth stimulated by TGFB1 (47). These results suggest that TGFBR1*6A may contribute to tumorigenesis by switching TGF-β growth inhibitory signals into growth stimulatory signals.
The 9-alanine repeat sequence is part of TGFBR1 signal peptide (47) and is responsible for targeting the receptor to the membrane. We found that the signal sequences of wild-type TGFBR1 and TGFBR1*6A were cleaved at the same site, resulting in identical mature receptors (47) (Fig. 2). Furthermore, expression of *6A with abolished kinase activity stimulates MCF-7 cell growth in response to TGF-β (47). Therefore, the biological effects of *6A are mediated by the signal sequence rather than by the mature receptor. After cleavage, the signal sequence remains in the cytoplasm, where it is likely to modulate specific gene expression or have other cellular functions (Fig. 2). Hence, *6A is the first example of signal sequence secondary signaling in cancer.
Fig. 2.
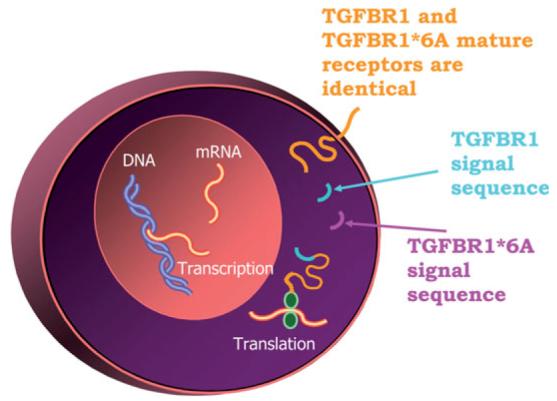
Secondary signaling effects of TGFBR1*6A. Amino terminus sequencing has shown that the polyalanine tract that contains the 3-Ala deletion, which differentiates TGFBR1*6A from TGFBR1, is part of the signal sequence. Given the fact that TGFBR1*6A and TGFBR1 mature receptors are identical, the biological actions of TGFBR1*6A are likely due to its signal sequence secondary signaling effects.
SMAD MUTATIONS AND ISOFORMS IN COLORECTAL CANCER
TGF-β signaling can also be impaired by deletions or mutations in the SMAD genes, which encode for proteins that act downstream of the receptors. SMAD4, originally identified as a tumor-suppressor gene lost in pancreatic cancers called DPC4 (deleted in pancreatic cancer 4) (48), is mutated in 16–25% of colorectal cancer cases, and alterations of SMAD2 have been identified in ∼6% of colorectal cancers (10). In fact, SMAD2 and SMAD4 are both localized at chromosome 18q, a region commonly deleted in colon adenocarcinomas (9). Germline mutations of SMAD4 cause juvenile polyposis (15) as well as hamartomatous polyposis (49). Although both Smad2-null and Smad4-null mice die in utero, mice with heterozygous deletion of Smad4 are predisposed to gastrointestinal polyps that can develop into tumors at later stage (50), which suggests that Smad4 functions as a tumor suppressor in colorectal cancer progression. In agreement with these findings, it has recently been shown that TGF-β-induced epithelial–mesenchymal transition is not dependent on SMAD4, and SMAD4 mainly mediates the antiproliferative response to TGF-β (51). A recent study showed that mice with selective loss of Smad4-dependent signaling in T-cells develop spontaneous epithelial cancers throughout the gastrointestinal tract, including colon and rectum. Interestingly, mice with epithelial-specific deletion of the Smad4 gene do not have the same phenotype (52). These results suggest that SMAD4 mediates crosstalk between stromal and epithelial cells, and interference with this process may eventually lead to cancer.
SMAD3 mutations in human colorectal cancer are rare (32) and their true pathogenic role has yet to be defined. Interestingly, mice with loss of Smad3 develop a high frequency of metastatic colon carcinoma by 6 months of age (53). However, two additional lines of Smad3-deficient mice do not develop colonic adenocarcinoma (54,55). Recently, it has been reported that chronic inflammation may explain this discrepancy (56). Smad3-null mice maintained in a bacteria-free environment did not develop colon cancer for up to 9 months, although infection of these mice with the Gram-negative enterohepatic bacteria Helicobacter triggered colon cancer in more than half of the animals, which suggests that bacteria infection plays an important role in triggering colorectal cancer in the context of gene mutation in the TGF-β signaling pathway (56).
Smad3 has two phosphoisoforms: one phosphorylated at the C-terminal region (pSmad3C) by TGFBR1, and the other phosphorylated at the linker region (pSmad3L) by JNK (57). The pSmad3C is expressed in normal colorectal epithelial cells and inhibits their growth, whereas pSmad3L is highly expressed in late-stage sporadic colorectal tumors and mediates mesenchymal cell invasion (57). It has been suggested that during sporadic colorectal carcinogenesis, tumor cells gain growth advantage through a shift from TGFBR1/pSmad3C-mediated to JNK/pSmad3L-mediated signaling (58,59).
CROSSTALK BETWEEN TGF-β AND WNT SIGNALING PATHWAYS IN COLORECTAL CANCER
The APC gene, which encodes a Wnt signaling molecule, is frequently mutated in patients with FAP and some sporadic colorectal cancer cases. Several mice models carrying different Apc mutations have been generated and tumors that develop in these mice are similar in histology but vary in terms of age of onset, number and location (60). Recent studies suggest that TGF-β and Wnt pathways synergistically promote colorectal tumorigenesis. For example, compound heterozygous ApcΔ716/+, Smad4+/− mice develop large and invasive intestinal carcinomas not seen in the ApcΔ716/+ mice (61). Smad4 heterozygosity also increased tumor multiplicity of the Apc+/N1638 mice (62). Smad2 haploinsufficiency accelerated tumor progression in the Apc580D mice but had no effect in the ApcΔ716/+ mice (63,64). Another compound mouse model was generated by crossbreeding Apcmin/+ mice with mice null for Smad3, where Smad-mediated TGF-β signaling was completely abolished (65). Interestingly, Smad3 deficiency promoted tumorigenesis only in the distal colon of the Apcmin/+ mice, whereas the proximal colon was not affected. This supports the notion that proximal and distal diseases in human sporadic colorectal cancer have different etiologies and respond differently to TGF-β signaling (65). More recently, Grady and colleagues developed a mouse model by mating Apc+/N1638 mice with mice that are null for Tgfbr2 in the intestinal epithelium (66). They observed that Tgfbr2 inactivation in the intestinal epithelial cells promoted the transformation and invasion of tumors initiated by Apc mutation in a cell-autonomous manner (66). In summary, the results from these mouse models confirm that deregulation of both TGF-β and Wnt signaling cooperate to drive tumor initiation and progression in vivo. A recent whole-genome analysis of mutated genes confirms that these two pathways are targeted in colorectal cancer (67).
CONCLUSION
The role of TGF-β in colorectal tumorigenesis has been recognized over the past decade. There is growing evidence that TGF-β signaling alterations mediated by mutations or polymorphisms of TGF-β receptors or SMADs contribute to colon cancer development and progression. However, there are still many unanswered questions. Why is SMAD4 more frequently mutated in colorectal cancer than SMAD2 and SMAD3? What are the molecular causes accounting for differences in TGF-β response between normal and transformed intestinal epithelial cells? What are the downstream targets of TGFBR1*6A? Ongoing advances in understanding the involvement of each TGF-β signaling component in colorectal cancer will provide novel opportunities to target this pathway for the prevention and treatment of colorectal cancer.
ACKNOWLEDGEMENTS
This work was supported by grants from the NCI (R01 CA112520, R01 CA108741), the AACR (Jeannik M. Littlefield-AACR Grant in Metastatic Colon Cancer Research) and the Walter S. Mander Foundation (Chicago, IL). The authors would like to thank Drs Habibul Ahsan, University of Chicago and Dr Yu Chen, New York University for the Odds Ratios presented in Table 1.
Footnotes
Conflict of Interest statement. None declared.
REFERENCES
- 1.Jemal A, Siegel R, Ward E, Murray T, Xu J, Smigal C, Thun MJ. Cancer statistics, 2006. CA Cancer J. Clin. 2006;56:106–130. doi: 10.3322/canjclin.56.2.106. [DOI] [PubMed] [Google Scholar]
- 2.Olsson L, Lindblom A. Family history of colorectal cancer in a Sweden county. Fam. Cancer. 2003;2:87–93. doi: 10.1023/a:1025734200635. [DOI] [PubMed] [Google Scholar]
- 3.Lichtenstein P, Holm NV, Verkasalo PK, Iliadou A, Kaprio J, Koskenvuo M, Pukkala E, Skytthe A, Hemminki K. Environmental and heritable factors in the causation of cancer—analyses of cohorts of twins from Sweden, Denmark, and Finland. N. Engl. J. Med. 2000;343:78–85. doi: 10.1056/NEJM200007133430201. [DOI] [PubMed] [Google Scholar]
- 4.de la Chapelle A. Genetic predisposition to colorectal cancer. Nat. Rev. Cancer. 2004;4:769–780. doi: 10.1038/nrc1453. [DOI] [PubMed] [Google Scholar]
- 5.Walsh JM, Terdiman JP. Colorectal cancer screening: scientific review. JAMA. 2003;289:1288–1296. doi: 10.1001/jama.289.10.1288. [DOI] [PubMed] [Google Scholar]
- 6.Mecklin JP, Jarvinen HJ. Surveillance in Lynch syndrome. Fam. Cancer. 2005;4:267–271. doi: 10.1007/s10689-005-1475-x. [DOI] [PubMed] [Google Scholar]
- 7.Kinzler KW, Vogelstein B. Lessons from hereditary colorectal cancer. Cell. 1996;87:159–170. doi: 10.1016/s0092-8674(00)81333-1. [DOI] [PubMed] [Google Scholar]
- 8.Roman C, Saha D, Beauchamp R. TGF-beta and colorectal carcinogenesis. Microsc. Res. Tech. 2001;52:450–457. doi: 10.1002/1097-0029(20010215)52:4<450::AID-JEMT1030>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 9.Grady WM, Markowitz SD. Genetic and epigenetic alterations in colon cancer. Annu. Rev. Genomics Hum. Genet. 2002;3:101–128. doi: 10.1146/annurev.genom.3.022502.103043. [DOI] [PubMed] [Google Scholar]
- 10.Takayama T, Miyanishi K, Hayashi T, Sato Y, Niitsu Y. Colorectal cancer: genetics of development and metastasis. J. Gastroenterol. 2006;41:185–192. doi: 10.1007/s00535-006-1801-6. [DOI] [PubMed] [Google Scholar]
- 11.Pinol V, Castells A, Andreu M, Castellvi-Bel S, Alenda C, Llor X, Xicola RM, Rodriguez-Moranta F, Paya A, Jover R, et al. Accuracy of revised Bethesda guidelines, microsatellite instability, and immunohistochemistry for the identification of patients with hereditary nonpolyposis colorectal cancer. JAMA. 2005;293:1986–1994. doi: 10.1001/jama.293.16.1986. [DOI] [PubMed] [Google Scholar]
- 12.Hampel H, Frankel WL, Martin E, Arnold M, Khanduja K, Kuebler P, Nakagawa H, Sotamaa K, Prior TW, Westman J, et al. Screening for the Lynch syndrome (hereditary nonpolyposis colorectal cancer) N. Engl. J. Med. 2005;352:1851–1860. doi: 10.1056/NEJMoa043146. [DOI] [PubMed] [Google Scholar]
- 13.Al-Tassan N, Chmiel NH, Maynard J, Fleming N, Livingston AL, Williams GT, Hodges AK, Davies DR, David SS, Sampson JR, et al. Inherited variants of MYH associated with somatic G:C– > T:A mutations in colorectal tumors. Nat. Genet. 2002;30:227–232. doi: 10.1038/ng828. [DOI] [PubMed] [Google Scholar]
- 14.Hemminki A, Markie D, Tomlinson I, Avizienyte E, Roth S, Loukola A, Bignell G, Warren W, Aminoff M, Hoglund P, et al. A serine/threonine kinase gene defective in Peutz–Jeghers syndrome. Nature. 1998;391:184–187. doi: 10.1038/34432. [DOI] [PubMed] [Google Scholar]
- 15.Howe JR, Roth S, Ringold JC, Summers RW, Jarvinen HJ, Sistonen P, Tomlinson IP, Houlston RS, Bevan S, Mitros FA, et al. Mutations in the SMAD4/DPC4 gene in juvenile polyposis. Science. 1998;280:1086–1088. doi: 10.1126/science.280.5366.1086. [DOI] [PubMed] [Google Scholar]
- 16.Howe JR, Bair JL, Sayed MG, Anderson ME, Mitros FA, Petersen GM, Velculescu VE, Traverso G, Vogelstein B. Germline mutations of the gene encoding bone morphogenetic protein receptor 1A in juvenile polyposis. Nat. Genet. 2001;28:184–187. doi: 10.1038/88919. [DOI] [PubMed] [Google Scholar]
- 17.Kemp Z, Thirlwell C, Sieber O, Silver A, Tomlinson I. An update on the genetics of colorectal cancer. Hum. Mol. Genet. 2004;13(Spec no 2):R177–R185. doi: 10.1093/hmg/ddh247. [DOI] [PubMed] [Google Scholar]
- 18.Laken SJ, Petersen GM, Gruber SB, Oddoux C, Ostrer H, Giardiello FM, Hamilton SR, Hampel H, Markowitz A, Klimstra D, et al. Familial colorectal cancer in Ashkenazim due to a hypermutable tract in APC. Nat. Genet. 1997;17:79–83. doi: 10.1038/ng0997-79. [DOI] [PubMed] [Google Scholar]
- 19.Massague J, Blain SW, Lo RS. TGFβ signaling in growth control, cancer, and heritable disorders. Cell. 2000;103:295–309. doi: 10.1016/s0092-8674(00)00121-5. [DOI] [PubMed] [Google Scholar]
- 20.Akhurst RJ. TGF beta signaling in health and disease. Nat. Genet. 2004;36:790–792. doi: 10.1038/ng0804-790. [DOI] [PubMed] [Google Scholar]
- 21.Elliott RL, Blobe GC. Role of transforming growth factor beta in human cancer. J. Clin. Oncol. 2005;23:2078–2093. doi: 10.1200/JCO.2005.02.047. [DOI] [PubMed] [Google Scholar]
- 22.Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature. 2003;425:577–584. doi: 10.1038/nature02006. [DOI] [PubMed] [Google Scholar]
- 23.Hoosein NM, McKnight MK, Levine AE, Mulder KM, Childress KE, Brattain DE, Brattain MG. Differential sensitivity of subclasses of human colon carcinoma cell lines to the growth inhibitory effects of transforming growth factor-beta 1. Exp. Cell Res. 1989;181:442–453. doi: 10.1016/0014-4827(89)90101-8. [DOI] [PubMed] [Google Scholar]
- 24.Friedman E, Gold LI, Klimstra D, Zeng ZS, Winawer S, Cohen A. High levels of transforming growth factor beta 1 correlate with disease progression in human colon cancer. Cancer Epidemiol. Biomarkers Prev. 1995;4:549–554. [PubMed] [Google Scholar]
- 25.Robson H, Anderson E, James RD, Schofield PF. Transforming growth factor beta 1 expression in human colorectal tumours: an independent prognostic marker in a subgroup of poor prognosis patients. Br. J. Cancer. 1996;74:753–758. doi: 10.1038/bjc.1996.432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sheng H, Shao J, O'Mahony CA, Lamps L, Albo D, Isakson PC, Berger DH, DuBois RN, Beauchamp RD. Transformation of intestinal epithelial cells by chronic TGF-beta1 treatment results in downregulation of the type II TGF-beta receptor and induction of cyclooxygenase-2. Oncogene. 1999;18:855–867. doi: 10.1038/sj.onc.1202397. [DOI] [PubMed] [Google Scholar]
- 27.Schroy P, Rifkin J, Coffey RJ, Winawer S, Friedman E. Role of transforming growth factor beta 1 in induction of colon carcinoma differentiation by hexamethylene bisacetamide. Cancer Res. 1990;50:261–265. [PubMed] [Google Scholar]
- 28.Ilyas M, Efstathiou JA, Straub J, Kim HC, Bodmer WF. Transforming growth factor beta stimulation of colorectal cancer cell lines: type II receptor bypass and changes in adhesion molecule expression. Proc. Natl Acad. Sci. USA. 1999;96:3087–3091. doi: 10.1073/pnas.96.6.3087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Grady WM, Myeroff LL, Swinler SE, Rajput A, Thiagalingam S, Lutterbaugh JD, Neumann A, Brattain MG, Chang J, Kim SJ, et al. Mutational inactivation of transforming growth factor beta receptor type II in microsatellite stable colon cancers. Cancer Res. 1999;59:320–324. [PubMed] [Google Scholar]
- 30.Biswas S, Chytil A, Washington K, Romero-Gallo J, Gorska AE, Wirth PS, Gautam S, Moses HL, Grady WM. Transforming growth factor beta receptor type II inactivation promotes the establishment and progression of colon cancer. Cancer Res. 2004;64:4687–4692. doi: 10.1158/0008-5472.CAN-03-3255. [DOI] [PubMed] [Google Scholar]
- 31.Grady WM, Willis JE, Trobridge P, Romero-Gallo J, Munoz N, Olechnowicz J, Ferguson K, Gautam S, Markowitz SD. Proliferation and Cdk4 expression in microsatellite unstable colon cancers with TGFBR2 mutations. Int. J. Cancer. 2006;118:600–608. doi: 10.1002/ijc.21399. [DOI] [PubMed] [Google Scholar]
- 32.Ku JL, Park SH, Yoon KA, Shin YK, Kim KH, Choi JS, Kang HC, Kim IJ, Han IO, Park JG. Genetic alterations of the TGF-beta signaling pathway in colorectal cancer cell lines: a novel mutation in Smad3 associated with the inactivation of TGF-beta-induced transcriptional activation. Cancer Lett. 2006 doi: 10.1016/j.canlet.2006.05.008. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 33.Wang J, Han W, Zborowska E, Liang J, Wang X, Willson JK, Sun L, Brattain MG. Reduced expression of transforming growth factor beta type I receptor contributes to the malignancy of human colon carcinoma cells. J. Biol. Chem. 1996;271:17366–17371. doi: 10.1074/jbc.271.29.17366. [DOI] [PubMed] [Google Scholar]
- 34.Pasche B, Luo Y, Rao PH, Nimer SD, Dmitrovsky E, Caron P, Luzzatto L, Offit K, Cordon-Cardo C, Renault B, et al. Type I transforming growth factor beta receptor maps to 9q22 and exhibits a polymorphism and a rare variant within a polyalanine tract. Cancer Res. 1998;58:2727–2732. [PubMed] [Google Scholar]
- 35.Kaklamani VG, Hou N, Bian Y, Reich J, Offit K, Michel LS, Rubinstein WS, Rademaker A, Pasche B. TGFBR1*6A and cancer risk: a meta-analysis of seven case–control studies. J. Clin. Oncol. 2003;21:3236–3243. doi: 10.1200/JCO.2003.11.524. [DOI] [PubMed] [Google Scholar]
- 36.Pasche B, Kolachana P, Nafa K, Satagopan J, Chen YG, Lo RS, Brener D, Yang D, Kirstein L, Oddoux C, et al. TβR-I(6A) is a candidate tumor susceptibility allele. Cancer Res. 1999;59:5678–5682. [PubMed] [Google Scholar]
- 37.Samowitz WS, Curtin K, Leppert MF, Slattery ML. Uncommon TGFBRI allele is not associated with increased susceptibility to colon cancer. Genes Chromosomes Cancer. 2001;32:381–383. doi: 10.1002/gcc.1203. [DOI] [PubMed] [Google Scholar]
- 38.van Tilborg AA, de Vries A, Zwarthoff EC. The chromosome 9q genes TGFBR1, TSC1 and ZNF189 are rarely mutated in bladder cancer. J. Pathol. 2001;194:76–80. doi: 10.1002/path.860. [DOI] [PubMed] [Google Scholar]
- 39.Pasche B, Bian YS, Reich J, Rademaker A, Kolachana P, Offit K. TβR-I(6A) in colorectal cancer: a new twist? Cancer Res. 2001;61:8351. [Google Scholar]
- 40.Stefanovska AM, Efremov GD, Dimovski AJ, Jasar D, Zografski G, Josifovski T, Panovski M, Jankova R, Spiroski M. TβR-I(6A) polymorphism is not a tumor susceptibility allele in Macedonian colorectal cancer patients. Cancer Res. 2001;61:8351–8352. Correspondence re: B. Pasche et al. (1998) Type I TβR-I(6A) is a candidate tumor susceptibility allele. Cancer Res., 58, 2727–2732. [PubMed] [Google Scholar]
- 41.Baxter SW, Choong DY, Eccles DM, Campbell IG. Transforming growth factor beta receptor 1 polyalanine polymorphism and exon 5 mutation analysis in breast and ovarian cancer. Cancer Epidemiol. Biomarkers Prev. 2002;11:211–214. [PubMed] [Google Scholar]
- 42.Pasche B, Kaklamani V, Hou N, Young T, Rademaker A, Peterlongo P, Ellis N, Offit K, Caldes T, Reiss M, et al. TGFBR1*6A and cancer: a meta-analysis of 12 case–control studies. J. Clin. Oncol. 2004;22:756–758. doi: 10.1200/JCO.2004.99.271. [DOI] [PubMed] [Google Scholar]
- 43.Zhang HT, Zhao J, Zheng SY, Chen XF. Is TGFBR1*6A really associated with increased risk of cancer? J. Clin. Oncol. 2005;23:7743–7744. doi: 10.1200/JCO.2005.02.9108. Author reply 7744–7746. [DOI] [PubMed] [Google Scholar]
- 44.Bian Y, Caldes T, Wijnen J, Franken P, Vasen H, Kaklamani V, Nafa K, Peterlongo P, Ellis N, Baron JA, et al. TGFBR1*6A may contribute to hereditary colorectal cancer. J. Clin. Oncol. 2005;23:3074–3078. doi: 10.1200/JCO.2005.00.281. [DOI] [PubMed] [Google Scholar]
- 45.Kemp ZE, Carvajal-Carmona LG, Barclay E, Gorman M, Martin L, Wood W, Rowan A, Donohue C, Spain S, Jaeger E, et al. Evidence of linkage to chromosome 9q22.33 in colorectal cancer kindreds from the United Kingdom. Cancer Res. 2006;66:5003–5006. doi: 10.1158/0008-5472.CAN-05-4074. [DOI] [PubMed] [Google Scholar]
- 46.Chen T, de Vries EG, Hollema H, Yegen HA, Vellucci VF, Strickler HD, Hildesheim A, Reiss M. Structural alterations of transforming growth factor-beta receptor genes in human cervical carcinoma. Int. J. Cancer. 1999;82:43–51. doi: 10.1002/(sici)1097-0215(19990702)82:1<43::aid-ijc9>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 47.Pasche B, Knobloch TJ, Bian Y, Liu J, Phukan S, Rosman D, Kaklamani V, Baddi L, Siddiqui FS, Frankel W, et al. Somatic acquisition and signaling of TGFBR1*6A in cancer. JAMA. 2005;294:1634–1646. doi: 10.1001/jama.294.13.1634. [DOI] [PubMed] [Google Scholar]
- 48.Hahn SA, Schutte M, Hoque AT, Moskaluk CA, da Costa LT, Rozenblum E, Weinstein CL, Fischer A, Yeo CJ, Hruban RH, et al. DPC4, a candidate tumor suppressor gene at human chromosome 18q21.1. Science. 1996;271:350–353. doi: 10.1126/science.271.5247.350. [DOI] [PubMed] [Google Scholar]
- 49.Sweet K, Willis J, Zhou XP, Gallione C, Sawada T, Alhopuro P, Khoo SK, Patocs A, Martin C, Bridgeman S, et al. Molecular classification of patients with unexplained hamartomatous and hyperplastic polyposis. JAMA. 2005;294:2465–2473. doi: 10.1001/jama.294.19.2465. [DOI] [PubMed] [Google Scholar]
- 50.Mishra L, Shetty K, Tang Y, Stuart A, Byers SW. The role of TGF-beta and Wnt signaling in gastrointestinal stem cells and cancer. Oncogene. 2005;24:5775–5789. doi: 10.1038/sj.onc.1208924. [DOI] [PubMed] [Google Scholar]
- 51.Levy L, Hill CS. Smad4 dependency defines two classes of transforming growth factor β (TGF-β) target genes and distinguishes TGF-β-induced epithelial–mesenchymal transition from its antiproliferative and migratory responses. Mol. Cell. Biol. 2005;25:8108–8125. doi: 10.1128/MCB.25.18.8108-8125.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kim BG, Li C, Qiao W, Mamura M, Kasperczak B, Anver M, Wolfraim L, Hong S, Mushinski E, Potter M, et al. Smad4 signalling in T cells is required for suppression of gastrointestinal cancer. Nature. 2006;441:1015–1019. doi: 10.1038/nature04846. [DOI] [PubMed] [Google Scholar]
- 53.Zhu Y, Richardson JA, Parada LF, Graff JM. Smad3 mutant mice develop metastatic colorectal cancer. Cell. 1998;94:703–714. doi: 10.1016/s0092-8674(00)81730-4. [DOI] [PubMed] [Google Scholar]
- 54.Yang X, Letterio JJ, Lechleider RJ, Chen L, Hayman R, Gu H, Roberts AB, Deng C. Targeted disruption of SMAD3 results in impaired mucosal immunity and diminished T cell responsiveness to TGF-beta. EMBO J. 1999;18:1280–1291. doi: 10.1093/emboj/18.5.1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Datto MB, Li Y, Panus JF, Howe DJ, Xiong Y, Wang XF. Transforming growth factor beta induces the cyclin-dependent kinase inhibitor p21 through a p53-independent mechanism. Proc. Natl Acad. Sci. USA. 1995;92:5545–5549. doi: 10.1073/pnas.92.12.5545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Maggio-Price L, Treuting P, Zeng W, Tsang M, Bielefeldt-Ohmann H, Iritani BM. Helicobacter infection is required for inflammation and colon cancer in SMAD3-deficient mice. Cancer Res. 2006;66:828–838. doi: 10.1158/0008-5472.CAN-05-2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yamagata H, Matsuzaki K, Mori S, Yoshida K, Tahashi Y, Furukawa F, Sekimoto G, Watanabe T, Uemura Y, Sakaida N, et al. Acceleration of Smad2 and Smad3 phosphorylation via c-Jun NH(2)-terminal kinase during human colorectal carcinogenesis. Cancer Res. 2005;65:157–165. [PubMed] [Google Scholar]
- 58.Matsuzaki K. Smad3 phosphoisoform-mediated signaling during sporadic human colorectal carcinogenesis. Histol. Histopathol. 2006;21:645–662. doi: 10.14670/HH-21.645. [DOI] [PubMed] [Google Scholar]
- 59.Matsuzaki K, Seki T, Okazaki K. TGF-beta during human colorectal carcinogenesis: the shift from epithelial to mesenchymal signaling. Inflammopharmacology. 2006 doi: 10.1007/s10787-006-1536-2. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 60.Boivin GP, Washington K, Yang K, Ward JM, Pretlow TP, Russell R, Besselsen DG, Godfrey VL, Doetschman T, Dove WF, et al. Pathology of mouse models of intestinal cancer: consensus report and recommendations. Gastroenterology. 2003;124:762–777. doi: 10.1053/gast.2003.50094. [DOI] [PubMed] [Google Scholar]
- 61.Takaku K, Oshima M, Miyoshi H, Matsui M, Seldin MF, Taketo MM. Intestinal tumorigenesis in compound mutant mice of both Dpc4 (Smad4) and Apc genes. Cell. 1998;92:645–656. doi: 10.1016/s0092-8674(00)81132-0. [DOI] [PubMed] [Google Scholar]
- 62.Alberici P, Jagmohan-Changur S, De Pater E, Van Der Valk M, Smits R, Hohenstein P, Fodde R. Smad4 haploinsufficiency in mouse models for intestinal cancer. Oncogene. 2006;25:1841–1851. doi: 10.1038/sj.onc.1209226. [DOI] [PubMed] [Google Scholar]
- 63.Hamamoto T, Beppu H, Okada H, Kawabata M, Kitamura T, Miyazono K, Kato M. Compound disruption of smad2 accelerates malignant progression of intestinal tumors in apc knockout mice. Cancer Res. 2002;62:5955–5961. [PubMed] [Google Scholar]
- 64.Takaku K, Wrana JL, Robertson EJ, Taketo MM. No effects of Smad2 (madh2) null mutation on malignant progression of intestinal polyps in Apc(delta716) knockout mice. Cancer Res. 2002;62:4558–4561. [PubMed] [Google Scholar]
- 65.Sodir NM, Chen X, Park R, Nickel AE, Conti PS, Moats R, Bading JR, Shibata D, Laird PW. Smad3 deficiency promotes tumorigenesis in the distal colon of ApcMin/+ mice. Cancer Res. 2006;66:8430–8438. doi: 10.1158/0008-5472.CAN-06-1437. [DOI] [PubMed] [Google Scholar]
- 66.Munoz NM, Upton M, Rojas A, Washington MK, Lin L, Chytil A, Sozmen EG, Madison BB, Pozzi A, Moon RT, et al. Transforming growth factor beta receptor type II inactivation induces the malignant transformation of intestinal neoplasms initiated by Apc mutation. Cancer Res. 2006;66:9837–9844. doi: 10.1158/0008-5472.CAN-06-0890. [DOI] [PubMed] [Google Scholar]
- 67.Sjoblom T, Jones S, Wood LD, Parsons DW, Lin J, Barber TD, Mandelker D, Leary RJ, Ptak J, Silliman N, et al. The consensus coding sequences of human breast and colorectal cancers. Science. 2006;314:268–274. doi: 10.1126/science.1133427. [DOI] [PubMed] [Google Scholar]