

Abstract
TGFBR1*6A is a common hypomorphic variant of the type I transforming growth factor (TGF)-β receptor (TGFBR1), which transduces TGF-β growth inhibitory signals less effectively than TGFBR1. Recent studies suggest that TGFBR1*6A confers a selective growth advantage to both normal appearing and cancerous epithelial cells in the presence of TGF-β. We have previously shown that TGFBR1*6A is somatically acquired in head and neck and colon cancer (10). Using microdissected tissues, we show that TGFBR1*6A is somatically acquired by stromal and epithelial cells adjacent to colorectal and head and neck tumors. Somatic acquisition of the TGFBR1*6A allele is not accompanied by acquisition of other tumor-specific mutations. Furthermore, lymphocytes located within the stroma or the normal appearing epithelium do not have evidence of TGFBR1*6A acquisition. The highest TGFBR1*6A/TGFBR1 allelic ratio is observed at the tumor's edge, and traces of TGFBR1*6A are detected as far as 2 cm away from the tumor, which is suggestive of centrifugal spread of cells that harbor TGFBR1*6A. Assessment of CDH1 and CDH2 expression does not indicate epithelial–mesenchymal transformation. The results suggest that TGFBR1*6A somatic acquisition is a critical event in the early stages of cancer development that is associated with field cancerization. They also represent the first human report of somatically acquired altered stromal TGF-β signaling during oncogenesis and the first report of a concordant mutation in the stromal and epithelial compartments in colon cancer.
INTRODUCTION
Cancers arise frequently in epithelial tissues such as those of the aerodigestive tract where constant proliferation is necessary to provide a continued supply of newly differentiated cells. Replacement of the mature cells in these tissues is tightly regulated by a small population of self-renewing stem cells, which gives rise to proliferating progenitor cells that undergo limited rounds of mitotic division leading to differentiation and loss or further proliferating ability (1). The main signaling pathways regulating morphogenesis in early embryogenesis, like the Wnt, Notch, Hedgehog and TGF-β pathwaysm, are directly or indirectly involved in the development and progression of most carcinomas (2). For example, almost all colorectal cancer cases show aberrant activation of the Wnt pathway, predominantly because of inactivating mutations of the APC gene (3). Similarly, germline and somatically acquired mutations in several constitutive genes of the TGF-β signaling pathway have been implicated in the development and progression of various forms of aerodigestive cancer (4). Whether some of these genetic alterations are acquired by tissues other than epithelial tumor tissues is largely unknown.
TGFBR1*6A is a common variant of the type I transforming growth factor (TGF)-β receptor (TGFBR1) (5). Using mink lung epithelial cell lines that are devoid of endogenous type I TGF-β receptor, two groups of investigators have shown that mink cells transfected with TGFBR1*6A, compared with TGFBR1, are impaired as mediators of TGF-β antiproliferative signals (6,7). A meta-analysis of 13 113 cases and controls has shown that TGFBR1*6A carriers have an overall 22% increase in cancer risk. TGFBR1*6A predisposes to the development of breast, colorectal, ovarian and prostate cancer (8,9). This allele is emerging as a common tumor susceptible alleles as 14% of the general population carries at least one copy of TGFBR1*6A (8). Analysis of tumors and matched normal tissues has shown that TGFBR1*6A is somatically acquired in ∼2% of primary head and neck and colorectal tumors as well as in 30% of colorectal cancer metastases (10). However, the same study did not show evidence of TGFBR1*6A somatic acquisition in breast cancer, suggesting that this genetic event occurs only in certain tumor types. TGFBR1*6A somatic acquisition appears to be a stochastic event, which confers tumor cells that carry this allele a selective growth advantage in the presence of TGF-β as it switches TGF-β growth inhibitory signals into growth stimulatory signals (10). It is unknown whether TGFBR1*6A somatic acquisition is tumor-specific and whether TGFBR1*6A is acquired before the development of cancer or concomitantly with the emergence of cancer.
RESULTS
We obtained a series of unstained, histological sections from the tumor of three of the four previously identified patients with colon cancer and evidence of TGFBR1*6A somatic acquisition within the tumor tissue (10). The tumor of one patient had microsatellite instability (MSI-H), the other two did not (microsatellite stable, MSS) and both had germline TGFBR1/TGFBR1 genotype as assessed by genotyping DNA extracted from peripheral blood lymphocytes. Microdissection of stroma and normal epithelium adjacent to the colon tumors followed by DNA extraction and genotyping by direct sequencing showed that TGFBR1*6A alleles were present in both stromal and normal epithelial cells up to 2 cm away from the tumor (Fig. 1). The results were confirmed by repeated polymerase chain reaction (PCR) amplification followed by cloning and sequencing of the PCR products. To rule out the possibility of tumor tissue contaminating the microdissected samples of stroma and normal epithelium, we searched the microdissected tissues of the tumor for the tumor-specific frame shift poly(A) tract TGFBR2 mutations, a frequent occur-rence in MSI-H colorectal cancer (11). As expected, we found that the polyadenine tract was mutated in the tumor, but neither in the stroma nor in the normal epithelium adjacent to the tumor (Fig. 1). To more precisely identify which cell types surrounding the tumor had acquired TGFBR1*6A, we performed laser capture microdissection (LCM) of epithelial, stromal (fibroblasts) and lymphoid cells surrounding colorectal tumor tissue with evidence of TGFBR1*6A acquisition. Both epithelial and stromal cells (fibroblasts) had evidence of TGFBR1*6A acquisition, but lymphoid cells did not, confirming the presence of TGFBR1/TGFBR1 germline genotype (Fig. 1). To determine whether somatic acquisition of GCG repeats was specific to TGFBR1 as previously observed in colorectal and head and neck tumors, we searched the microdissected stromal and epithelial tissues for mutations of the GCG repeat sequences of three other genes: GDF11, STK39 and HLXB9 (10). None of the tissues analyzed had evidence of GCG repeat sequence mutation, demonstrating that TGFBR1*6A somatic acquisition by stromal and epithelial cells is not associated with a mutator phenotype, similar to what was observed in both head and neck and colon cancer epithelial tumor cells.
Figure 1.
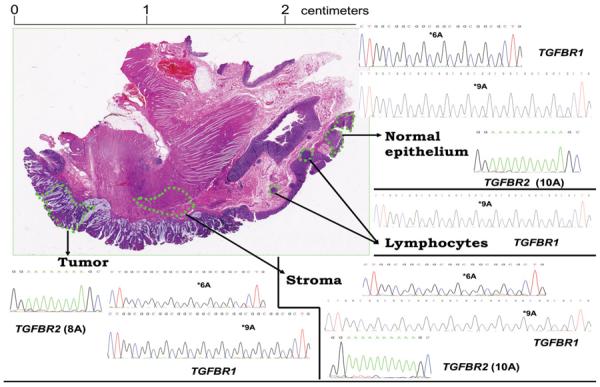
Hematoxylin and eosin stained colonic tumor block section from a patient with *9A/*9A germline genotype (peripheral blood lymphocytes) and evidence of *6A acquisition by microdissected normal epithelial cells, stromal cells and tumor cells. The patient's tumor exhibited microsatellite instability (MSI-H). Only *9A alleles were found in tissue lymphocytes (right middle quadrant) confirming the *9A/*9A germline genotype of the patient. The TGFBR2 polyadenine tract is mutated in the tumor (8A) (left lower quadrant) but neither in the stroma (10A) (right lower quadrant) nor in the normal colonic epithelium (10A) (right upper quadrant) showing the absence of tumor cell contamination in microdissected stroma and normal epithelium. The findings were confirmed by genotyping of DNA extracted by Laser Capture Microdissection from epithelial cells, fibroblasts and lymphoid cells. Genotyping results were confirmed by cloning of the PCR product and sequencing of at least ten clones. Each quadrant contains representative sequences of the clones.
We then obtained a series of unstained, histological sections from the tumor of three of the four previously identified patients with head and neck cancer and evidence of TGFBR1*6A somatic acquisition within the tumor tissue (10). We microdissected tumor cells (Fig. 2A and B), stromal cells (Fig. 2A and C) and histologically ‘normal’ epithelial cells adjacent to the tumor (Fig. 2D and E) from individual slides to avoid contamination. Additionally, normal epithelium (Fig. 2F and G) and stroma (Fig. 2F and H were isolated from the right true vocal cord located 1 cm lateral to the tumor edge to further exclude the possibility of microscopic contamination. Histopathological examination showed the absence of tumor cells in stroma as well as in normal squamous epithelium. PCR amplification followed by cloning of the PCR product and sequencing of the clones demonstrated the presence of TGFBR1*6A alleles in the tumor, immediately juxtaposed ‘normal’ squamous epithelium and stroma, as well as in adjacent true vocal cord epithelium and stroma. To again exclude the contamination of stroma and normal epithelium by tumor tissue, we searched for mutations of TP53, one of the most commonly targeted genes in head and neck cancer (12). We identified a TP53 747G>A silent substitution in the tumor of one of the three patients examined. As shown in Fig. 2I, this somatically acquired substitution was present only in the tumor of the patient, not in the distant or adjacent stroma, nor in the adjacent epithelium. Using three informative polymorphic markers (D17S796, D17S513, TP53 AC/GT), we also established that there was no TP53 LOH at 17p13, which is consistent with a neutral TP53 mutation that is not subject to selective pressure.
Figure 2.
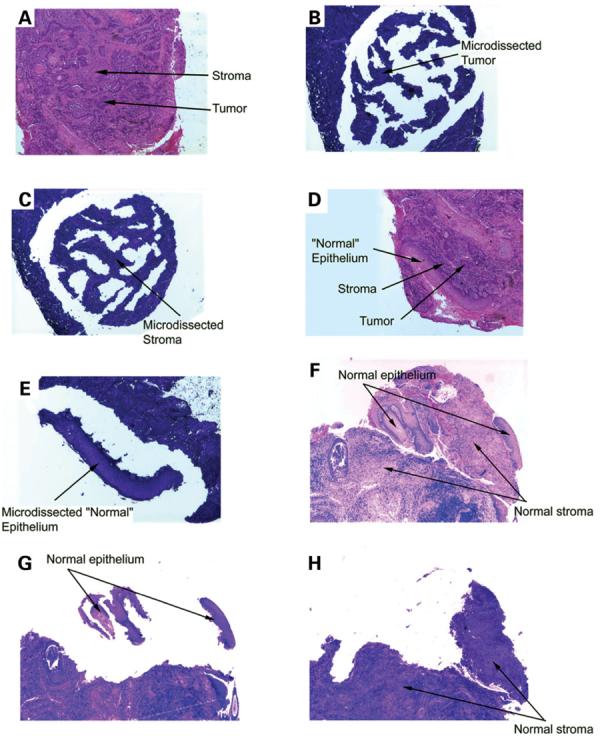
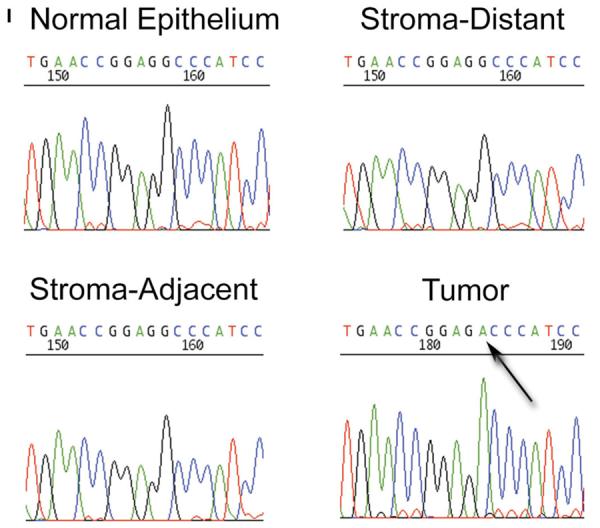
(A) Hematoxylin and eosin stained section used as histological control for tumor and stroma from a patient with *9A/*9A germline genotype (peripheral blood lymphocytes) and *9A/*6A tumor genotype. (B) Tumor cells: consecutive section stained with toluidine blue; stromal and inflammatory cells surrounding the tumor have been removed. (C) Stromal cells: consecutive section stained with toluidine blue; all tumor cells have been removed. (D) Hematoxylin and eosin stained section used as histological control for ‘normal’ epithelium directly adjacent to the tumor. (E) ‘Normal’ squamous epithelium: consecutive section stained with toluidine blue; tumor, stromal and inflammatory cells have been removed. (F) Hematoxylin and eosin stained section used as histological control for adjacent normal epithelium and stroma of the right true vocal cord located 1 cm lateral to the tumor. (G) Normal squamous epithelium of the right true vocal cord located 1 cm lateral to the tumor: consecutive section stained with toluidine blue; stromal and inflammatory cells have been removed. (H) Stromal cells of the right true vocal cord located 1 cm lateral to the tumor: consecutive section stained with toluidine blue; all normal squamous epithelium has been removed. (I)A TP53 747G>A silent substitution was identified in the tumor DNA, but not in the adjacent normal epithelium or stroma DNAs. Nucleotide sequence electropherograms representing a region of TP53 exon 7 amplified from isolated distant normal epithelium, distant stroma, or adjacent stroma DNA samples. The arrow points to a nucleotide substitution in TP53 at position 747 for the tumor DNA.
In a fourth patient with head and neck cancer and evidence of somatically acquired TGFBR1*6A, germline tissue had been obtained by biopsy of normal appearing epithelium more than 2 cm away from the tumor (10). Genotyping of DNA extracted from this tissue showed a TGFBR1/TGFBR1 genotype with a barely identifiable TGFBR1*6A peak. The TGFBR1*6A peak height was at least 40 times lower than that of TGFBR1. Upon sequencing of multiple clones of the PCR product, we identified traces of TGFBR1*6A alleles. This was in striking contrast to the near equivalent TGFBR1*6A and TGFBR1 peak heights identified in the tumor. This prompted us to quantify the ratios of TGFBR1*6A/TGFBR1 alleles in the various tissues examined. We therefore established a standard curve using various molar ratios of pCMV5–TGFBR1*6A–HA and pCMV5–TGFBR1–HA plasmids and amplified them by PCR with exonic primers covering TGFBR1 exon 1 GCG repeat sequence. We assessed the relative levels of TGFBR1*6A and TGFBR1 as peak heights. The correlation coefficient of expected (molar ratio) versus measured (peak height) amounts of TGFBR1*6A and TGFBR1 was 0.875, validating this measurement method for TGFBR1*6A/TGFBR1 ratios ranging from 0.2 to 5.0 (Fig. 3). Having demonstrated the feasibility of this approach to assess the relative proportion of TGFBR1*6A and TGFBR1 alleles, we investigated their ratios in patients' tissues. We found that germline TGFBR1*6A/TGFBR1 samples from peripheral blood specimens had comparable TGFBR1*6A/TGFBR1 molar ratio of ∼1.0, whereas TGFBR1*6A/TGFBR1*6A and TGFBR1/TGFBR1 samples had molar ratios at the upper end of the curve and close to zero, respectively (Fig. 3). Analysis of DNA extracted from the tissues obtained by LCM shown in Fig. 1d a ratio of TGFBR1*6A/TGFBR1 ranging from virtually 0 for lymphocytes, 0.13 for epithelial cells, 0.48 for fibroblasts and 0.72 for tumor cells (Fig. 3). Similarly, there were only traces of TGFBR1*6A in the predominantly TGFBR1/TGFBR1 germline genotype obtained more than 2 cm away from the tumor edge from one of the patient with head and neck cancer (Fig. 3). On the contrary, the tumor genotype of the same patient had a TGFBR1*6A/TGFBR1 ratio of 0.6 (Fig. 3). Additional samples of microdissected stromal, epithelial and tumor tissues from each of the other patients consistently showed higher TGFBR1*6A/TGFBR1 ratios in tumor tissues compared with stromal and epithelial tissues. Therefore, the presence of the somatically acquired TGFBR1*6A allele in normal epithelial and stromal cells surrounding the tumor appears to be inversely proportional to the distance from the primary tumor, suggestive of tumor-centered centrifugal growth.
Figure 3.
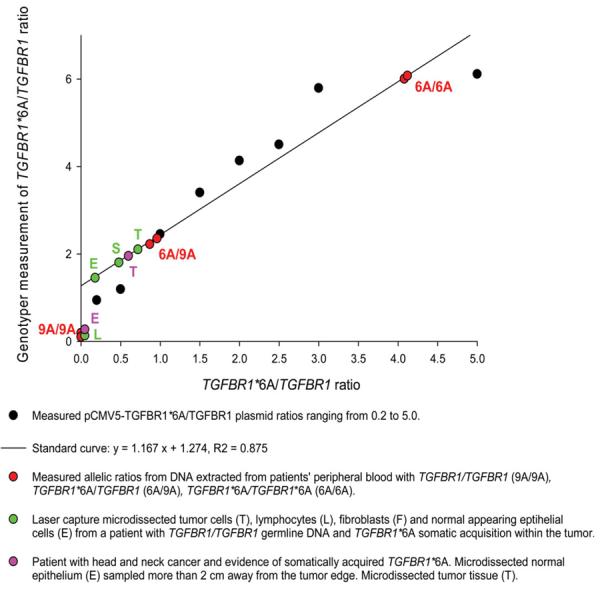
Various molar ratios (0.2, 0.5, 1.0, 1.5, 2.0, 2.5, 3.0 and 5.0) of pCMV5-TGFBR1*6A-HA and pCMV5-TGFBR1-HA constructs were amplified by PCR. Using a genotype TGFBR1*6A and TGFBR1, amplicons were separated based on their size (107 bp for TGFBR1*6A and 115 bp for TGFBR1) and the measured TGFBR1*6A/TGFBR1 peak height ratios (y-axis) were plotted against the expected peak heights (x-axis): black circles. The correlation coefficient (R2) was 0.875. DNA extracted from patients' tissues was analyzed in the same manner and the resulting TGFBR1*6A/TGFBR1 ratios plotted using the formula derived from the standard curve: y = 1.167x + 1.274, R2 = 0.875.
Epithelial–mesenchymal transformation (EMT) is a process whereby epithelial cell layers lose polarity and cell–cell contacts and undergo a dramatic remodeling of the cytoskeleton (13). EMT occurs during critical phases of embryonic development and many parallels are found between this process in embryonic development, in tissue culture and in tumors. For example, in colorectal adenocarcinoma, invasion involves the release of single cells by a process that involves EMT in the zone of the tumor that is progressing within the stroma (14,15). Several member of the TGF-β family of growth factors can initiate and maintain EMT in various biological systems through activation of several signaling pathways and transcriptional factors (16). For example the TGF-β signaling pathway synergizes with Ras to induce EMT in culture and metastasis in mouse models indicating the pivotal role of the TGF-β signaling pathway in these phenomena (17). Two of the hallmarks of EMT are loss of E-cadherin (CDH1) and overexpression of N-cadherin (CDH2). To test the hypothesis that TGFBR1*6A somatic acquisition by stromal and epithelial cells is associated with EMT, we assessed CDH1 and CDH2 expression in the tissues of four patients with evidence of TGFBR1*6A somatic acquisition, two with colorectal cancer and two with head and neck cancer. As shown in Fig. 4, CDH1 was overexpressed in both tumor types. On the contrary, we did not find any evidence of CDH2 overexpression (Fig. 4). These findings strongly suggest that the acquisition of TGFBR1*6A by normal appearing epithelial and stromal cells in colorectal as well as head and neck cancer is not caused by EMT.
Figure 4.
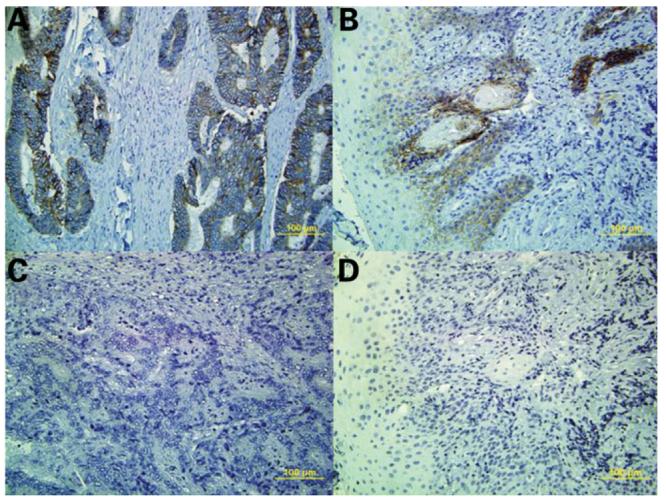
CDH1 expression was assessed by immunohistochemistry in colorectal cancer tissue (upper left panel) and head and neck cancer (upper right panel) with evidence of TGFBR1*6A somatic acquisition. The brown colored areas reflect CDH1 expression. CDH2 expression was assessed by immunohistochemistry in the same tissues: colorectal cancer (left lower panel) and head and neck cancer (right lower panel). There was no evidence of CDH2 overexpression.
DISCUSSION
Recent molecular findings support a multiphase carcinogenesis model in which the development of ‘a field’ with genetically altered cells plays a central role (18). In the initial phase, a stem cell acquires genetic alterations and forms a ‘patch’, a clonal unit of altered daughter cells. These patches can be recognized on the basis of somatically acquired mutations, loss of heterozygosity, microsatellite alterations and chromosomal instability. This phenomenon termed as ‘field cancerization’ has been reported for several types of cancers, including head and neck (19,20), breast (21,22), esophagus (23) and colon (24). Conversion of a patch into an expanding field is the next critical step in epithelial carcinogenesis. Additional genetic alterations are required for this step, and by virtue of its growth advantage, a proliferating field gradually displaces the normal mucosa. A field lesion has a monoclonal origin, and does not show invasive growth and invasive behavior, the hallmark criteria of cancer. It has been shown that genetically altered cells in a field show a high proliferative capacity, as determined with Ki-67 staining (25). Such fields have been detected with dimensions of up to 7 cm in diameter in the mucosa of the head and neck as well as the esophagus (26,27).
The data presented in this report indicate that TGFBR1*6A is somatically acquired in a small proportion of primary colorectal and head and neck cancers by normal appearing epithelial cells. The absence of cancer-specific mutations in normal epithelial cells and stromal cells corroborated by LCM experiments virtually exclude the possibility of contamination as an explanation for our findings. The findings also strongly suggest that TGFBR1*6A acquisition by surrounding fibroblasts is not the result of EMT but rather because of the growth advantage in the presence of TGF-β bestowed to epithelial and stromal cells as previously shown in mink lung epithelial (7), breast cancer and colon cancer cells (10). Hence, our results are consistent with the novel concept that TGFBR1*6A somatic acquisition is an early genetic event, associated with field cancerization in both colorectal cancer and head and neck cancer.
Concordant allelic losses and mutations have been previously described in epithelial and stromal compartments in breast and head and neck cancer (20,22,28). One report suggests that somatically acquired genomic alterations are rather common in breast cancer and may portend a poor prognosis (22). With respect to colorectal cancer, our findings provide support for the model proposed by Brabletz et al. (1) and suggest that a common cancer stem cell may give rise to epithelial carcinoma cells, stromal cells as well as normal appearing epithelial cells, which spread in a centrifugal fashion from the tumor site. To the best of our knowledge, our findings also constitute the first report of a concordant mutation in the stromal and epithelial compartments in colorectal cancer. A potentially crucial role for altered stromal TGF-β signaling during oncogenesis has been highlighted by the discovery that mice that harbor an inactivated type II TGF-β receptor (Tgfbr2) develop intraepithelial neoplasia of the prostate and invasive squamous cell carcinoma of the forestomach (29). A recent report has further highlighted the relevance of altered stromal TGF-β signaling with respect to colorectal cancer development by demonstrating that stromal inactivation of the type II bone morphogenetic protein receptor (BMPR2), a member of the TGF-β signaling pathway family, leads to colorectal polyp overgrowth and polyp formation (30).
One report has shown concordant regions of loss of heterozygosity but discordant TP53 mutations in the epithelial and stromal compartment of colon cancer (31) and other reports have shown somatically acquired stromal genetic alterations in juvenile polyposis (32) as well as stromal and epithelial genetic alterations in ulcerative colitis (33). Our results constitute the first report of a somatically acquired TGF-β pathway mutation by stromal cells in cancer and provide a strong support for the notion that TGF-β signaling pathway alterations within the tumor microenvironment contribute to cancer development and progression. They provide the first evidence in humans that hypomorphic TGF-β signaling within the stroma may be involved in colon cancer development. The overall frequency of TGFBR1*6A somatic acquisition in primary tumors is low (1.8–2.5%), but appears to be much higher in colorectal cancer liver metastases (30%). Six out of six tumors with evidence of TGFBR1*6A somatic acquisition also had evidence of acquisition of TGFBR1*6A by stromal and normal appearing epithelial cells. This provides strong preliminary evidence that TGFBR1*6A somatic acquisition is predominantly not tumor specific. Additional studies of primary and metastatic colorectal and head and neck tumors are needed to confirm our findings and to determine whether TGFBR1*6A somatic acquisition correlates with prognosis and metastatic progression. These results also provide a strong rationale to study the role of TGFBR1*6A-mediated signaling in stromal cells as it relates to the development of cancer.
MATERIALS AND METHODS
Sample acquisition
All tumor tissue samples were obtained from patients enrolled in Investigation Review Board (IRB) approved protocols at Northwestern Memorial Hospital in Chicago and The James Cancer Hospital and Solove Research Institute in Columbus as described previously (10).
Microdissection, LCM and DNA extraction
Microdissection and DNA extraction
Sections of formalinfixed, paraffin-embedded head and neck and colorectal tissues were cut at 7 μm and DNA prepared following tissue microdissection from the slides as previously described (34). Pure populations of normal appearing epithelial cells, stromal cells and lymphoid cells were obtained by LCM using a Pixcell II LCM (Arcturus Bioscience Inc., Mountain View, CA, USA) and DNA extracted according to the manufacturer's instructions (35).
TGFBR1, TGFBR2, TP53 genotyping and TP53 loss of heterozygosity assessment
TGFBR1 exon 1 was amplified by PCR using the Advantage-GC genomic polymerase mix (BD Biosciences Clontech, San Jose, CA) and the following primers: 5′- GAGGCGAGGTTTGCTGGGGTGAGGCA-3′ and 5′-CATGTTTGAGAAAGAGCAGGAGCGAG-3′. TP53 exon 7 was amplified using the following primers: 5′-CCTCATCTTGGGCCTGTGTT-3′ and 5′-GTGGCAAGTGGCTCCTGAC-3′. TGFBR2 exon 3 that contains the (A)10 repeat was amplified using the following primers: 5′-CCTCGCTTCCAATGAATCTC-3′ and 5′-TTGGCACAGATCTCAGGTCC-3′ for TGFBR2. PCR amplification was carried out in standard buffer, 2.0 mmol/l MgCl2, 0.25 mmol/l dNTPs, 25 pmol of each primers, 1 unit of Taq polymerase and 100 ng DNA per 25 μl reaction. TP53 LOH analysis was performed using four dinucleotide polymorphic markers, two markers mapped to 17p13 (D17S796 and D17S513), and two markers spanning the TP53 gene (TP53 AC/GT and TP53 D1/D2). A standard PCR containing gamma [32P]-ATP end-labeled forward primer, analysis of PCR products on denaturing polyacrylamide sequencing gels, and scoring of LOH on autoradiograms was performed as described previously (34). Data were collected using an ABI 377 automated DNA sequencer, analyzed with Genescan software, and allelic imbalance determined using GeneScan/Genotyper software (Applied Biosystems, Foster City, CA). Either the oligonucleotide sense primer for each amplimer set was synthesized with 5′-FAM, -TET or -HEX fluorescent labels, or unlabeled primers were utilized and products were labeled during amplification with appropriate fluorescent nucleotides. A TAMRA-labeled fluorescent internal size standard was included with each experimental sample.
GCG-rich genes
To investigate whether the deletion of GCG repeats within TGFBR1 was gene specific or associated with generalized genomic instability, we identified and sequenced other genes containing a similar sequence as described previously (10).
Cloning and sequencing
PCR products were cloned into the pCR 2.1 vector (Clontech). Automated sequencing of ten clones was performed to determine the presence/absence of TGFBR1*6A and TP53 747G>A as well as the length of the polyadenine tract of TGFBR2. Microsatellite instability assessment was performed as described previously (10).
Plasmid construction
TGFBR1-HA and TGFBR1*6A-HA were cloned into the pGEM1 plasmid (Promega, Madison, WI) with the initiator ATG codon in the context of a ‘Kozak consensus’ sequence as described previously (10).
ACKNOWLEDGEMENTS
The authors wish to thank Wendy Frankel and Heather Hampel from the Ohio State University Comprehensive Cancer Center, Columbus, OH, USA for help and advice, and Albert de la Chapelle for access to patient samples.
FUNDING
This work was supported by grants CA112520 and CA108741 from the NCI, the Jeannik M. Littlefield-AACR Grant in Metastatic Colon Cancer Research and the Walter S. Mander Foundation, Chicago, IL. The authors wish to thank Albert de la Chapelle (grants CA67941 and CA16058 from the NCI).
Footnotes
Conflict of Interest statement. None declared.
REFERENCES
- 1.Brabletz T, Hlubek F, Spaderna S, Schmalhofer O, Hiendlmeyer E, Jung A, Kirchner T. Invasion and metastasis in colorectal cancer: epithelial-mesenchymal transition, mesenchymal-epithelial transition, stem cells and beta-catenin. Cells Tissues Organs. 2005;179:56–65. doi: 10.1159/000084509. [DOI] [PubMed] [Google Scholar]
- 2.Radtke F, Clevers H, Riccio O. From gut homeostasis to cancer. Curr. Mol. Med. 2006;6:275–289. doi: 10.2174/156652406776894527. [DOI] [PubMed] [Google Scholar]
- 3.Morin PJ, Sparks AB, Korinek V, Barker N, Clevers H, Vogelstein B, Kinzler KW. Activation of beta-catenin-Tcf signaling in colon cancer by mutations in beta-catenin or APC. Science. 1997;275:1787–1790. doi: 10.1126/science.275.5307.1787. [DOI] [PubMed] [Google Scholar]
- 4.Akhurst RJ. TGF beta signaling in health and disease. Nat. Genet. 2004;36:790–792. doi: 10.1038/ng0804-790. [DOI] [PubMed] [Google Scholar]
- 5.Pasche B, Luo Y, Rao PH, Nimer SD, Dmitrovsky E, Caron P, Luzzatto L, Offit K, Cordon-Cardo C, Renault B, et al. Type I transforming growth factor beta receptor maps to 9q22 and exhibits a polymorphism and a rare variant within a polyalanine tract. Cancer Res. 1998;58:2727–2732. [PubMed] [Google Scholar]
- 6.Chen T, de Vries EG, Hollema H, Yegen HA, Vellucci VF, Strickler HD, Hildesheim A, Reiss M. Structural alterations of transforming growth factor-beta receptor genes in human cervical carcinoma. Int. J. Cancer. 1999;82:43–51. doi: 10.1002/(sici)1097-0215(19990702)82:1<43::aid-ijc9>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 7.Pasche B, Kolachana P, Nafa K, Satagopan J, Chen YG, Lo RS, Brener D, Yang D, Kirstein L, Oddoux C, et al. T beta R-I(6A) is a candidate tumor susceptibility allele. Cancer Res. 1999;59:5678–5682. [PubMed] [Google Scholar]
- 8.Zhang HT, Zhao J, Zheng SY, Chen XF. Is TGFBR1*6A really associated with increased risk of cancer? J. Clin. Oncol. 2005;23:7743–7744. doi: 10.1200/JCO.2005.02.9108. [DOI] [PubMed] [Google Scholar]
- 9.Xu Y, Pasche B. TGF-beta signaling alterations and susceptibility to colorectal cancer. Hum. Mol. Genet. 2007;16:R14–R20. doi: 10.1093/hmg/ddl486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pasche B, Knobloch TJ, Bian Y, Liu J, Phukan S, Rosman D, Kaklamani V, Baddi L, Siddiqui FS, Frankel W, et al. Somatic acquisition and signaling of TGFBR1*6A in cancer. JAMA. 2005;294:1634–1646. doi: 10.1001/jama.294.13.1634. [DOI] [PubMed] [Google Scholar]
- 11.Markowitz S, Wang J, Myeroff L, Parsons R, Sun L, Lutterbaugh J, Fan RS, Zborowska E, Kinzler KW, Vogelstein B. Inactivation of the type II TGF-beta receptor in colon cancer cells with microsatellite instability. Science. 1995;268:1336–1338. doi: 10.1126/science.7761852. [DOI] [PubMed] [Google Scholar]
- 12.Forastiere A, Koch W, Trotti A, Sidransky D. Head and neck cancer. N. Engl. J. Med. 2001;345:1890–1900. doi: 10.1056/NEJMra001375. [DOI] [PubMed] [Google Scholar]
- 13.Thiery JP. Epithelial-mesenchymal transitions in tumour progression. Nat. Rev. Cancer. 2002;2:442–454. doi: 10.1038/nrc822. [DOI] [PubMed] [Google Scholar]
- 14.Hlubek F, Jung A, Kotzor N, Kirchner T, Brabletz T. Expression of the invasion factor laminin gamma2 in colorectal carcinomas is regulated by beta-catenin. Cancer Res. 2001;61:8089–8093. [PubMed] [Google Scholar]
- 15.Brabletz T, Herrmann K, Jung A, Faller G, Kirchner T. Expression of nuclear beta-catenin and c-myc is correlated with tumor size but not with proliferative activity of colorectal adenomas. Am. J. Pathol. 2000;156:865–870. doi: 10.1016/s0002-9440(10)64955-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zavadil J, Bottinger EP. TGF-[beta] and epithelial-to-mesenchymal transitions. Oncogene. 2004;24:5764–5774. doi: 10.1038/sj.onc.1208927. [DOI] [PubMed] [Google Scholar]
- 17.Kang Y, Massague J. Epithelial–mesenchymal transitions: twist in development and metastasis. Cell. 2004;118:277–279. doi: 10.1016/j.cell.2004.07.011. [DOI] [PubMed] [Google Scholar]
- 18.Braakhuis BJM, Tabor MP, Kummer JA, Leemans CR, Brakenhoff RH. A genetic explanation of Slaughter's concept of field cancerization: evidence and clinical implications. Cancer Res. 2003;63:1727. [PubMed] [Google Scholar]
- 19.Copper MP, Braakhuis BJ, de Vries N, van Dongen GA, Nauta JJ, Snow GB. A panel of biomarkers of carcinogenesis of the upper aerodigestive tract as potential intermediate endpoints in chemoprevention trials. Cancer. 1993;71:825–830. doi: 10.1002/1097-0142(19930201)71:3<825::aid-cncr2820710327>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 20.Weber F, Xu Y, Zhang L, Patocs A, Shen L, Platzer P, Eng C. Microenvironmental genomic alterations and clinicopathological behavior in head and neck squamous cell carcinoma. JAMA. 2007;297:187–195. doi: 10.1001/jama.297.2.187. [DOI] [PubMed] [Google Scholar]
- 21.Forsti A, Louhelainen J, Soderberg M, Wijkstrom H, Hemminki K. Loss of heterozygosity in tumour-adjacent normal tissue of breast and bladder cancer. Eur. J. Cancer. 2001;37:1372–1380. doi: 10.1016/s0959-8049(01)00118-6. [DOI] [PubMed] [Google Scholar]
- 22.Fukino K, Shen L, Patocs A, Mutter GL, Eng C. Genomic instability within tumor stroma and clinicopathological characteristics of sporadic primary invasive breast carcinoma. JAMA. 2007;297:2103–2111. doi: 10.1001/jama.297.19.2103. [DOI] [PubMed] [Google Scholar]
- 23.Prevo LJ, Sanchez CA, Galipeau PC, Reid BJ. p53-mutant clones and field effects in Barrett's esophagus. Cancer Res. 1999;59:4784–4787. [PubMed] [Google Scholar]
- 24.Jothy S, Slesak B, Harlozinska A, Lapinska J, Adamiak J, Rabczynski J. Field effect of human colon carcinoma on normal mucosa: relevance of carcinoembryonic antigen expression. Tumour Biol. 1996;17:58–64. doi: 10.1159/000217967. [DOI] [PubMed] [Google Scholar]
- 25.Tabor MP, Braakhuis BJ, van der Wal JE, van Diest PJ, Leemans CR, Brakenhoff RH, Kummer JA. Comparative molecular and histological grading of epithelial dysplasia of the oral cavity and the oropharynx. J. Pathol. 2003;199:354–360. doi: 10.1002/path.1285. [DOI] [PubMed] [Google Scholar]
- 26.Tabor MP, Brakenhoff RH, Ruijter-Schippers HJ, van der Wal JE, Snow GB, Leemans CR, Braakhuis BJ. Multiple head and neck tumors frequently originate from a single preneoplastic lesion. Am. J. Pathol. 2002;161:1051–1060. doi: 10.1016/S0002-9440(10)64266-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Barrett MT, Sanchez CA, Prevo LJ, Wong DJ, Galipeau PC, Paulson TG, Rabinovitch PS, Reid BJ. Evolution of neoplastic cell lineages in Barrett oesophagus. Nat. Genet. 1999;22:106–109. doi: 10.1038/8816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kurose K, Gilley K, Matsumoto S, Watson PH, Zhou XP, Eng C. Frequent somatic mutations in PTEN and TP53 are mutually exclusive in the stroma of breast carcinomas. Nat. Genet. 2002;32:355–357. doi: 10.1038/ng1013. [DOI] [PubMed] [Google Scholar]
- 29.Bhowmick NA, Chytil A, Plieth D, Gorska AE, Dumont N, Shappell S, Washington MK, Neilson EG, Moses HL. TGF-fbetag signaling in fibroblasts modulates the oncogenic potential of adjacent epithelia. Science. 2004;303:848–851. doi: 10.1126/science.1090922. [DOI] [PubMed] [Google Scholar]
- 30.Beppu H, Mwizerwa ON, Beppu Y, Dattwyler MP, Lauwers GY, Bloch KD, Goldstein AM. Stromal inactivation of BMPRII leads to colorectal epithelial overgrowth and polyp formation. Oncogene. 2007 Aug 13; doi: 10.1038/sj.onc.1210720. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 31.Wernert N, Locherbach C, Wellmann A, Behrens P, Hugel A. Presence of genetic alterations in microdissected stroma of human colon and breast cancers. Anticancer Res. 2001;21:2259–2264. [PubMed] [Google Scholar]
- 32.Jacoby RF, Schlack S, Cole CE, Skarbek M, Harris C, Meisner LF. A juvenile polyposis tumor suppressor locus at 10q22 is deleted from nonepithelial cells in the lamina propria. Gastroenterology. 1997;112:1398–1403. doi: 10.1016/s0016-5085(97)70156-2. [DOI] [PubMed] [Google Scholar]
- 33.Matsumoto N, Yoshida T, Okayasu I. High epithelial and stromal genetic instability of chromosome 17 in ulcerative colitis-associated carcinogenesis. Cancer Res. 2003;63:6158–6161. [PubMed] [Google Scholar]
- 34.Baisse B, Bian YS, Benhattar J. Microdissection by exclusion and DNA extraction for multiple PCR analyses from archival tissue sections. Biotechniques. 2000;28:856–858, 860, 862. [PubMed] [Google Scholar]
- 35.Eltoum IA, Siegal GP, Frost AR. Microdissection of histologic sections: past, present, and future. Adv. Anat. Pathol. 2002;9:316–322. doi: 10.1097/00125480-200209000-00006. [DOI] [PubMed] [Google Scholar]