

Abstract
E. coli AdhE has been reported to harbor three distinct enzymatic activities: alcohol dehydrogenase, acetaldehyde-CoA dehydrogenase, and pyruvate formate-lyase (PFL) deactivase. Herein we report on the cloning, expression, and purification of E. coli AdhE, and the re-investigation of its purported enzymatic activities. While both the alcohol dehydrogenase and acetaldehyde-CoA dehydrogenase activities were readily detectible, we were unable to obtain any evidence for catalytic deactivation of PFL by AdhE, regardless of whether the reported cofactors for deactivation (Fe(II), NAD, and CoA) were present. Our results demonstrate that AdhE is not a PFL deactivating enzyme. We have also examined the potential for deactivation of active PFL by small-molecule thiols. Both β-mercaptoethanol and dithiothreitol deactivate PFL efficiently, with the former providing quite rapid deactivation. PFL deactivated by these thiols can be reactivated, suggesting that this deactivation is non-destructive transfer of an H atom equivalent to quench the glycyl radical.
Keywords: pyruvate formate-lyase, PFL, AdhE, glycyl radical quenching, deactivation of glycyl radical enzyme
INTRODUCTION
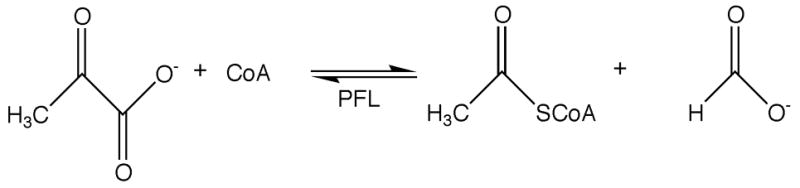
Pyruvate formate-lyase (PFL) is one of the central enzymes in anaerobic metabolism in Escherichia coli (E. coli), and is responsible for catalyzing the conversion of pyruvate and CoA to formate and acetyl-CoA by a nonoxidative route (Scheme 1). Consistent with its central role in anaerobic metabolism, pfl transcription and PFL biosynthesis increase 10- to 12-fold when E. coli are grown under anaerobic conditions [1, 2]. Induction of pfl gene expression under anaerobic conditions has been shown to involve both the FNR and the ArcA/ArcB regulatory proteins, which regulate gene expression in response to O2 and various metabolites, respectively [3, 4]. In addition to this transcriptional regulation, PFL requires post-translational activation by the pyruvate formate-lyase activating enzyme (PFL-AE) [5]. PFL-AE is constitutively expressed under both aerobic and anaerobic conditions [6], however in vitro studies have demonstrated that it is catalytically active only under anaerobic reducing conditions [7, 8]. There are, therefore, at least two levels of control for regulating PFL activity: transcriptional up-regulation and post-translational activation. A potential third level of control involving PFL deactivation is addressed herein.
Scheme 1.
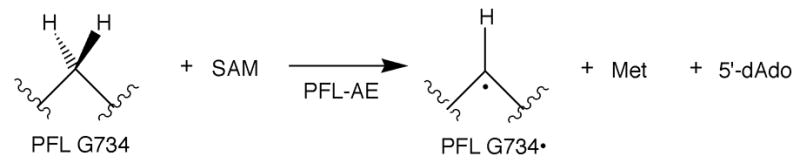
PFL-AE activates PFL by generation of a stable and catalytically essential radical at PFL G734 (Scheme 2) [9]. The activation reaction requires a cluster bound to PFL-AE [10], as well as S-adenosylmethionine (SAM or AdoMet) and a reductant, which in vivo is reduced flavodoxin [8]. These requirements place PFL-AE in the radical AdoMet superfamily [11]. The radical AdoMet enzymes use a reduced [4Fe-4S]+ cluster to reductively cleave AdoMet, producing methionine and a putative adenosyl radical intermediate; the adenosyl radical intermediate is believed to be responsible for H atom abstraction from substrate in all characterized members of the superfamily [12–15]. The radical AdoMet superfamily has hundreds of putative members spanning the phylogenetic kingdom; functions include cofactor biosynthesis, rearrangement reactions, DNA repair, and enzyme activation. In addition to PFL-AE, E. coli harbors another radical AdoMet enzyme that is central to its anaerobic metabolism, the anaerobic ribonucleotide reductase (ARR) activating enzyme (AE). ARR plays a central role in nucleotide metabolism in anaerobic bacteria; like PFL, it requires post-translational activation to generate its catalytically essential glycyl radical [16, 17].
Scheme 2.
E. coli thus utilizes post-translational glycyl radical generation as part of its regulatory mechanism for two of its most central metabolic functions: glucose and nucleotide metabolism. Once generated, the glycyl radicals in PFL [18] and ARR [19] are stable under anaerobic conditions; further, the radical is not consumed during catalysis but rather is a true cofactor. Thus once the respective activating enzymes activate PFL and ARR, the glycyl radicals are present and the enzymes are active indefinitely. The glycyl radicals in both PFL and ARR are, however, extremely sensitive to oxygen; oxygen reacts rapidly with the radical in vitro, resulting in protein cleavage at the site of the radical [9, 20]. This oxygen sensitivity raises the question of the fate of activated PFL and ARR when anaerobic E. coli are subjected to aerobic conditions, a situation that can occur routinely in facultative anaerobes such as E. coli.
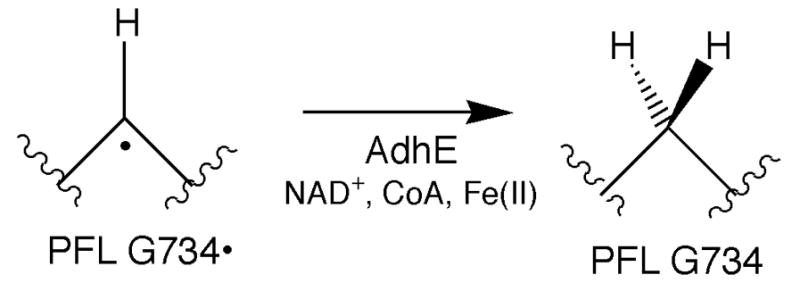
The report of PFL deactivation catalyzed by AdhE seemed to resolve the puzzle of the fate of this catalytically essential radical: rather than undergoing oxygenolytic cleavage, as occurs in vitro, the glycyl radical was reported to be catalytically deactivated, or quenched, by AdhE [21, 22]. This intriguing deactivation reaction resulted in the conversion of active PFL back to its activatable form, thereby circumventing the irreversible protein cleavage observed in vitro, and presumably allowing the protein to be re-activated upon return to anaerobic conditions. The deactivation of PFL by AdhE was reported to be dependent on Fe(II), NAD, and CoA, with both pyruvate and NADH acting as competitive inhibitors of deactivation (Scheme 3). The effects of NAD and NADH led the authors to propose that the intracellular NAD/NADH ratio controls the state of PFL in vivo, resulting in activation under anaerobic conditions and deactivation as conditions become more aerobic [22]. Their proposal was further supported by in vivo studies showing that PFL is deactivated by AdhE under microaerophilic conditions [22].
Scheme 3.
AdhE, like PFL, is expressed under anaerobic conditions, as it plays an essential role in alcoholic fermentation. That AdhE should also function to deactivate PFL was a very intriguing proposal as it suggests that AdhE somehow acts as both a microaerophilic sensor and a deactivator of PFL, thus protecting it from oxygenolytic degradation. The proposed deactivation reaction, which involves the addition of a hydrogen atom equivalent to the glycyl radical of active PFL using the cofactors Fe(II), NAD, and CoA, is intriguing and unprecedented in biology. We therefore set out to investigate the PFL deactivase activity of AdhE in more detail, in order to gain some understanding of the catalytic mechanism. Described herein are our results showing that AdhE does not, in fact, catalyze the deactivation of PFL. We also provide data to show that some small-molecule thiols are quite efficient deactivators of PFL activity.
EXPERIMENTAL METHODS
Materials
Unless otherwise stated, all chemicals were obtained from commercial sources and used without further purification. Deazariboflavin was synthesized according to literature procedures.[23–25] Cloning vectors (PET-21a(+) and PET-Blue1) were obtained from Novagen. E. coli strains BL21(DE3)pLysS (Stratagen), Epicurian XL1 Blue (Novagen), NovaBlue (Novagen), and Tuner (DE3)pLacI (Novagen) were purchased from the indicated companies. Genomic E. coli (BL21(DE3)) DNA was purified using the Wizard genomic DNA extraction kit from Promega. Plasmid purification was performed using a purification kit from Stratagen. PCR primers were purchased from Integrated DNA Technologies, and PCR reactions were done using Pfu Turbo DNA polymerase (Stratagene). Immobilized metal ion affinity resin was purchased from Qiagen.
Cloning of His-tagged and Wild-type AdhE
For cloning of AdhE with a His6 affinity tag, PCR was performed using genomic E. coli BL21(DE3) DNA as the template with two primers (sense, 5′-CGCCGCCTCGAGAGCGGATTTTTTCGCTTT-3′ and antisense, 5′-CGCCGCGGATCCATGGCTGTTACTAATGTC-3′) that incorporated appropriate restriction sites (BamH1 and Xho, respectively) for cloning into pET-21(a)+. The PCR product and vector were digested with these enzymes and then ligated to produce pMN-1. Restriction digests verified the presence of a correctly-sized insert, and the resulting vector was subjected to DNA sequencing (MSU Biotechnology Core Facility) to verify the presence of adhE; this plasmid is hereafter referred to as pET-21(a)+/adhE. For cloning of wild type adhE, PCR was performed using genomic E. coli DNA as the template with two primers (sense, 5′-GCGCGCGGATCCATGGCTGTTACTAATGTCGCTGAACTTAACGCA-3′ and antisense, 5′-GCGCGCCTCGAGTTAAGCGGATTTTTTCGCTTTTTTCTCAGCTTT-3′). The resulting PCR product was subjected to an end-conversion reaction prior to ligation into pET-Blue-1 blunt vector. Isolation of plasmid with the correct insert was confirmed by restriction digest analysis and DNA sequencing; this plasmid is hereafter referred to as pET-Blue-1/adhE.
Purification of AdhE-His6
The plasmid PET-21a(+)/adhE was transformed into BL21(DE3)pLysS host strain. Cultures were grown in LB/Amp media to an OD600 of 0.5 to 0.7, and then induced by addition of isopropyl β-D-thiogalactopyranoside (IPTG) to a final concentration of 1 mM. After an additional 2 hours growth, the cells were harvested by centrifugation.
BL21(DE3)pLysS/PET-21a(+)/adhE cells were lysed in buffer (3 mL/g wet weight) containing 50 mM NaH2PO4, 10 mM imidazole, and 300 mM NaCl, pH 8, to which was added lysozyme (0.5 mg/mL), PMSF (1 mM), and trace quantities of DNAse and RNAse. After two hours at 4°C with gentle agitation, the suspension was centrifuged at 9,000 g for 30 minutes. The supernatant (7 mL) was loaded onto an immobilized metal affinity chromatography (IMAC) column (30 mL, QIAGEN) in 50 mM NaH2PO4, 10 mM imidazole, and 300 mM NaCl, pH 8. The column was washed with 50 mM NaH2PO4, 20 mM imidazole, and 300 mM NaCl, pH 8, and then the AdhE-His6 was eluted with 50 mM NaH2PO4, 250 mM imidazole, and 300 mM NaCl, pH 8. The purified protein was dialyzed into 20 mM Hepes pH 7.2 and stored at −80°C.
Purification of Wild Type AdhE
The plasmid PET-Blue-1/adhE was transformed into Tuner(DE3)pLacI cells. Using this vector/host combination, AdhE was found to be overexpressed constitutively, so growth and overexpression was carried out simply by growing a culture to its maximum optical density (OD600 ~ 2.5) prior to harvesting by centrifugation.
BL21(DE3)pLysS/PET-Blue-1/adhE cells were lysed at 4°C for 2.5 to 3 hours in lysis buffer (1.5 mL per gram of cells) containing 50 mM Tris-sulfate pH 7.5, 200 mM NaCl, 1% Triton X-100, 5% glycerol, 10 mM MgCl2, 1 mM DTT, 1 mM PMSF, 0.25 mg/mL lysozyme, and RNAse and DNAse (~0.1 mg each per 10 mL), prior to centrifugation to remove cell debris. The crude extract (30–40 mL) was loaded onto a Superdex 75 gel filtration column ( 5 x 60 cm) equilibrated with 50 mM Tris pH 7.5, and eluted with the same buffer. Fractions were analyzed using SDS-PAGE, and those containing >~80% total protein as AdhE were pooled, concentrated to 30–40 mL, and dialyzed against 20 mM Hepes pH 7.2. This protein was then loaded onto a Accell Plus QMA (Waters) ion exchange column (5 x 30 cm) equilibrated with 20 mM Hepes/1 mM DTT pH 7.2. After washing with the same buffer, AdhE was eluted using a gradient from the loading buffer to loading buffer + 200 mM NaCl. Fractions were analyzed by SDS-PAGE and those in which AdhE constituted >~90% of the total protein were pooled, concentrated, and flash-frozen for storage at −80°C.
Purification of PFL
Pyruvate formate-lyase (PFL) was overexpressed using E. coli BL21(DE3)pLysS transformed with pKK/pfl. An overnight culture (50 mL) grown in LB containing 50 μg/mL Amp (LB/Amp) was used to inoculate 10 L of LB/Amp in a benchtop fermenter. The culture was grown at 37°C to an optical density at 600 nm of 0.7, at which time IPTG was added to a final concentration of 1 mM. Two hours later, the cells were harvested by centrifugation, the supernatant was decanted, and the pellets stored at −80°C until needed for purification. Cells were lysed in buffer (~2 mL per g of cell paste) containing 20 mM HEPES pH 7.2, 1 mM DTT, 1% Triton X-100, 5% glycerol, 10 mM MgCl2, 8 mg lysozyme, 1 mM PMSF, and trace quantities of DNAse and RNAse. The cell suspension was thoroughly mixed and then homogenized continuously with a magnetic stirrer at 4°C for 2 hours. The lysed cells were centrifuged at 15,000 rpm (Sorvall SS34) for 30 min at 4°C. Crude lysate (50 mL) was loaded onto an Accell Plus QMA (Waters) ion exchange column (5 x 30 cm) previously equilibrated with a low-salt buffer (Buffer A, 20 mM HEPES pH 7.2/1 mM DTT). The column was then washed with 180 mL of Buffer A before eluting the protein with a 540 mL gradient from Buffer A to Buffer B (20 mM HEPES pH 7.2/1 mM DTT/500 mM NaCl), followed by 180 mL isocratic at 100% Buffer B. Under these conditions, PFL elutes in the final quarter of the gradient. Fractions were analyzed by SDS-PAGE, and those containing PFL as >~75% of total protein were pooled, concentrated, and dialyzed against Buffer C (40 mM HEPES pH 7.2, 1 mM DTT, 1 M ammonium sulfate). The dialyzed protein was loaded onto a Hi-Load (Pharmacia) phenyl-sepharose column (1.6 x 10 cm) equilibrated with Buffer C. After washing the column with 120 mL of Buffer C, PFL was eluted with a 100 mL gradient from Buffer C to Buffer D (40 mM HEPES pH 7.2, 1 mM DTT) followed by isocratic elution with Buffer D for 80 mL. Under these conditions, PFL elutes in the last third of the gradient. Fractions containing PFL as >95% of total protein, as judged by SDS-PAGE, were pooled, concentrated, and dialyzed against Buffer A. Protein was flash-frozen and stored at −80°C.
Activation of PFL
PFL activation reactions were performed in an anaerobic chamber (Mbraun) maintained at < 2 ppm O2. All reagents were thoroughly degassed prior to beginning the reaction. A typical PFL activation mix contained 150 mM Tris-Cl pH 7.6, 0.1 M KCl, 10 mM oxamate, 8 mM DTT, 0.03–15 μM PFL-AE, 2 – 150 μM PFL, 0.2 mM AdoMet, and 50 μM 5-deazariboflavin, added in the order given. After mixing, activation was initiated by illuminating the sample with a 300 W halogen lamp. The activation sample was kept at approximately 5 cm from the lamp in a water bath to which ice was added periodically to keep the temperature at approximately 20–23 °C. Illumination was typically carried out for 30–45 minutes, until maximal PFL activity was obtained.
PFL Activity Assay
The PFL activity assay was carried out in an anaerobic chamber (Mbraun) maintained at < 2 ppm O2 and in sealed quartz cuvettes. PFL activity was measured by using a modification of the coupled enzymatic assay previously described [7]. The coupling assay mix contained 150 mM Tris-Cl pH 8.5, 3 mM NAD+, 55 μM CoA, 0.1 mg BSA, 10 mM pyruvate, 10 mM malate, 6 units citrate synthase, and 14 units malic dehydrogenase in a total volume of 700 μL. PFL activity was assayed by adding a small volume of the activated PFL (typically 1–5 μL) to the coupling assay mix, and monitoring the rate of production of NADH at 340 nm.
AdhE Deactivation Activity Assays
The ability of AdhE to deactivate active PFL was investigated using a modification of previously reported assays; these assays were carried out in an anaerobic chamber and sealed cuvettes. To activated PFL (typically approximately 200 μM final concentration) was added AdhE (generally to 10–15 μM final concentration) NAD+ (100–200 μM final), CoA (50–100 μM final), and Fe(II) (0.5–1 mM final). Some assays also included 5 mM MgSO4 and 2 mM NAD. The mixture was incubated under anaerobic conditions (in the dark unless stated otherwise, in order to prevent competing re-activation by PFL-AE during deactivation experiments), and aliquots were removed periodically to assay for residual PFL activity. For some assays, one or more of the cofactors or AdhE was left out of the mixture, in order to determine the effect of each component individually.
Other AdhE Activity Assays
In order to assay alcohol dehydrogenase activity, an aliquot of AdhE was added to a solution containing 300 mM K2CO3 (pH 10), 0.25 mM NAD+, and 170 mM ethanol. The production of NADH was monitored at 340 nm. To assay acetaldehyde reductase activity, an aliquot of AdhE (generally 2–4 μM final concentration) was added to a solution containing 50 mM Tris-Cl pH 7.6, 2.5 μM NADH, and 0.2–0.7 M acetaldehyde, and NADH oxidation was monitored at 340 nm. To assay acetyl-CoA reductase activity, an aliquot of AdhE (1–10 μM) was added to a solution containing 50 mM CHES pH 9.5, 0.2 mM dithiothreitol, 0.075 mM NAD, 0.1 mM CoA, and 10 mM acetaldehyde, and the absorbance at 340 nm was monitored. For each of the activities of AdhE, the specific activity is defined as 1 U = 1 μmol NADH/min.
Other PFL Deactivation Assays
The ability of a series of small molecules to inactivate active PFL was examined by adding the small molecules at a range of concentrations to active PFL in Tris-Cl buffer pH 8.5. The residual PFL activity was measured using the coupling assay as described previously.
Electron Microscopy
Electron microscopy was performed using a Zeiss 100 CA microscope and a LEO 912 AB Zeiss microscope. AdhE (50 μg/mL) was in either 50 mM anaerobic MOPS/KOH pH 7.5, or in the same buffer containing 3mM DTT, 3 mM MgSO4, and 0.3 mM Fe(NH4)2(SO4)2, or in the same buffer containing 3mM DTT, 3 mM MgSO4, and 0.3 mM Fe(NH4)2(SO4)2, 5 mM NAD and 5 □M CoA. Samples were adsorbed on carbon film supported by a 200 mesh copper grid, and then stained by 1 % uranyl acetate, pH 4.5.
PFL Cleavage as a Function of Redox Potential
BL21(DE3)pLysS cells containing the PFL or AdhE expression vectors were grown in 50 mL of Luria Broth media at 37ºC overnight. A 5 mL aliquot of each of the overnight cultures was transferred into 850 mL of LB media or a defined minimal medium [26] with glucose as the carbon source. The cultures were grown aerobically to an OD600 of 0.9, and then induced by addition of IPTG to 1 mM. After 75 (LB) or 120 (minimal media) minutes of induction, both the AdhE and PFL cultures were purged with N2 gas for 2 hours, and then pooled into centrifuge bottles in a Coy anaerobic chamber. The cells were harvested at 8,000 rpm, 4ºC for 10 minutes. The PFL pellets were split in half; half was lysed alone, the other was lysed together with AdhE pellets. The pellets were lysed in the anaerobic chamber in 50 mL of lysis buffer containing 20 mM HEPES, 200 mM NaCl, 10 mM MgCl2, 1% Triton X-100, 5% glycerol, 1 mM PMSF, 0.5 mg of DNAse and RNAse. After 30 minutes of lysis, the redox potential was measured to be −360 (LB) or −346 (minimal media) mV (vs. SHE) by using an Orion 9678BNWP electrode and an Orion 3 Star meter; a 1 mL aliquot was taken out, mixed with 100 □L glycerol, and frozen in liquid N2. The potential of the lysis was then adjusted by injecting air; at the potentials −30, +49, and +102 mV (LB cells) or −30, +49, and +99 mV (minimal media cells), aliquots were taken and mixed with glycerol and frozen in liquid N2. Each aliquot was thawed under air, and purged with air thoroughly by using a 1 mL pipet. Each sample was then prepared and run on a 12% Tris-HCl SDS-PAGE gel in order to examine the state of the PFL protein.
In a second set of experiments, the potential of the growth culture, rather than that of the lysis, was adjusted. The BL21(DE3)pLysS cells overexpressing PFL or AdhE were grown in 50 mL of Luria Broth or a defined minimal medium with glucose as the carbon source aerobically. The cultures were induced at an OD600 of 0.5 by addition of IPTG to 1 mM. After 60 minutes (LB cultures) or 120 (minimal media cultures) min, both the cultures were purged by N2 gas for 2 hours. The redox potential was −68 (LB culture) or +12 (minimal media culture) mV (vs. SHE) at this point, as measured by an Orion 9678BNWP electrode and an Orion 3 Star meter. A 10 mL aliquot of each culture was taken out, mixed with 1 mL of glycerol, and frozen in liquid N2. The potential was then adjusted to −32, +52, and +99 mV (LB cultures) or to 50, 102, and 201 mV (minimal media culture) by purging with air; at each potential, an additional 1 mL aliquot of each culture was taken out, mixed with 100 μL glycerol, and frozen in liquid N2. The cells were thawed in the anaerobic chamber and harvested by centrifugation. Each pellet was lysed in 20 μL of the lysis buffer described previously. The PFL cells taken before or after potential adjustment were lysed together with AdhE cells taken at different potentials. After 30 minutes, the lysates were exposed to air, and purged with air thoroughly. The samples were then run on an SDS-PAGE gel to examine the extent of PFL cleavage.
Other Methods
Protein concentrations were determined by the method of Bradford, using dye reagent purchased from Bio-Rad. Iron content was determined using a modification of the procedure previously reported by Beinert [27]. Purified AdhE-His6 was subjected to matrix-assisted laser desorption ionization mass spectrometry (MS-MALDI) at the Michigan State University Macromolecular Facility.
RESULTS
Overexpression and Purification
Both AdhE and AdhE-His6 were overexpressed efficiently using the constructs and host cells described in Materials and Methods. The AdhE-His6 overexpressed at a higher level and was easier to purify due to the presence of the His tag. Typical yields were 25 mg purified AdhE-His6 per liter of culture. The untagged AdhE was partially purified using gel filtration and ion exchange chromatography and was used primarily to confirm results obtained with the AdhE-His6. Yields of purified wt-AdhE were typically on the order of 9 mg per liter of culture. PFL overexpressed efficiently and could be purified in reasonably large quantities (generally 120–160 mg purified PFL per L of culture).
Alcohol Dehydrogenase Activities
AdhE catalyzes the generation of ethanol from acetyl-CoA by two sequential NADH-dependent reactions, as shown in Scheme 4. Each of the reactions shown in Scheme 4, the acetyl-CoA reductase and the acetaldehyde reductase reactions, can be assayed independently in either the forward or reverse direction by applying the appropriate substrates. Both activities have been identified in our purified AdhE, both the wild type and the hexahistidine-tagged versions, with specific activities similar to those reported in the literature. For example, we have found the specific activity of AdhE for the ethanol dehydrogenase reaction to be 9.5 U/mg (for both wild type and histidine-tagged enzyme), as compared to values of 10 U/mg reported previously for wild-type AdhE.[21] All other specific activities were also similar to those reported in the literature, thereby verifying that we have isolated AdhE in its fully functional form.
Scheme 4.

Deactivation of PFL by AdhE
AdhE has been reported to harbor PFL deactivation as a third enzymatic activity, with a requirement for NAD, Fe2+, and CoA for full PFL deactivation activity. We have re-investigated this report through a series of detailed experiments as outlined below. In general, our approach was to fully activate PFL using PFL activating enzyme and the procedures described in the Materials and Methods. Then, under strictly anaerobic conditions (due to the extreme air sensitivity of the glycyl radical present in active PFL), we incubated PFL alone or with AdhE and/or a range of possible cofactors. Residual PFL activity was monitored as a function of time in order to probe the rate of deactivation, if any, resulting from the added components.
Initial experiments involved incubating PFL with AdhE alone, with the putative cofactors NAD, Fe2+, and CoA, or with both AdhE and the putative cofactors. The results of representative experiments are shown in Figure 1. Over the time course of the experiment, the activity of the PFL incubated with AdhE alone increased slightly and then remained essentially constant, suggesting that AdhE in the absence of any added cofactors is unable to deactivate PFL. PFL incubated alone under identical conditions exhibited identical behavior (data not shown). It is noteworthy that the PFL activity is stable over 8 hours in this experiment, as previous reports estimate the half-life of the PFL glycyl radical to be on the order of several hours at 4°C; the half-life under our current conditions and at 25°C is clearly significantly longer than several hours, which is quite remarkable for a protein backbone-centered radical. PFL incubated with AdhE and the putative cofactors (NAD, Fe2+, and CoA), however, gradually loses activity over the course of the experiment, which at first glance would be consistent with prior reports of AdhE-catalyzed PFL deactivation. However, the results of control reactions containing PFL and the putative cofactors but no AdhE showed identical results, demonstrating that the gradual loss of PFL activity was independent of the presence of AdhE. Furthermore, in contrast to the report of Kessler and Knappe, the presence of Mg2+ had no effect on the observed results, nor did varying concentrations of NAD nor varying ratios of NADH to NAD (data not shown). We were initially concerned about artifacts in these experiments, particularly since the PFL glycyl radical is very oxygen-sensitive and therefore prone to inactivation by trace oxygen. However, repeated experiments under stringently anaerobic conditions provided results consistent with those shown in Figure 1. The inactivation of PFL observed in Fig. 1 is attributed to the CoA and Fe(II) present, as each individually was correlated with loss of PFL activity in separate experiments (data not shown).
Figure 1.
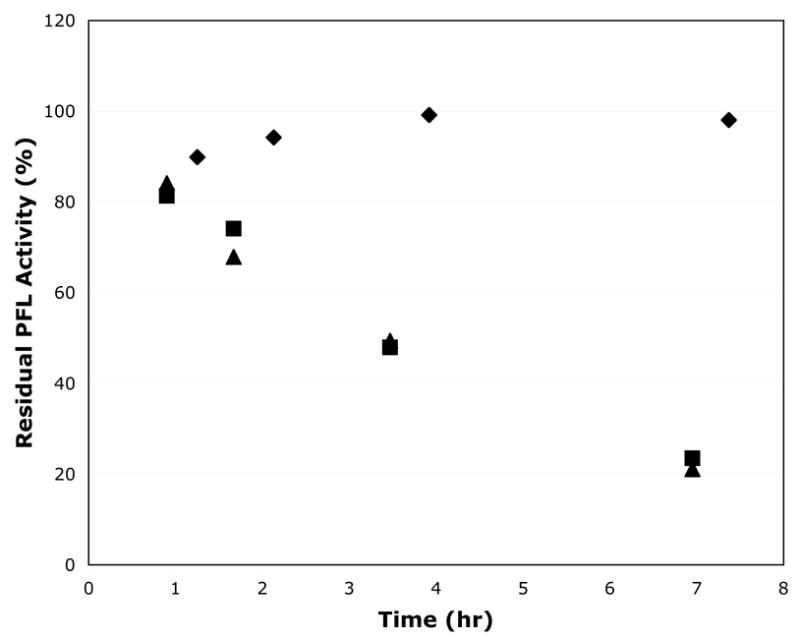
Lack of deactivation of PFL by AdhE. Activated PFL (193 μM) was incubated with AdhE (3 μM), with AdhE and its reported “PFL deactivase” cofactors NAD+ (0.2 mM), CoA (0.1 mM), and Fe(II) (1 mM), with the cofactors alone, or with no additives; experiments were performed in the dark. The results clearly demonstrate that AdhE has no effect on the deactivation of PFL, although some of the cofactors appear to display a modest effect. ♦ PFL + AdhE; ▪ PFL + AdhE, NAD, CoA, and Fe(II); ▴ PFL + NAD, CoA, and Fe(II).
Kessler and Knappe previously showed that in E. coli K12 cells, protection of PFL against oxygenolytic cleavage was dependent on both the presence of AdhE and the redox potential of the culture, such that deactivation was turned on only under microaerophilic conditions (+100 mV). Therefore we examined the PFL deactivation activity of our overexpressed AdhE as a function of culture and lysate redox potential. Briefly, anaerobic cultures expressing AdhE and PFL separately were adjusted to a range of redox potentials and then lysed together prior to exposure to oxygen to cleave any remaining active PFL. Alternatively, anaerobic cultures expressing AdhE and PFL separately were lysed alone or in combination prior to adjusting the redox potential of the lysate; the lysate was then exposed to oxygen to cleave any remaining active PFL. In all experiments, complete cleavage of PFL is observed, as evidenced by the equal intensity bands corresponding to the uncleaved PFL polypeptide (85 kDa) and the large fragment of the oxygenolytically cleaved polypeptide (82 kDa), as seen in Figure 2. The cleavage of one-half of all PFL polypeptides is considered complete PFL cleavage due to the half-of-sites reactivity of PFL. It should be noted that even if our cloned AdhE was somehow defective in PFL deactivation, the wt AdhE of the host cells would be induced to fairly high levels (3 x 104 copies/cell) [28] under the anaerobic growth conditions used in these experiments. Thus the redox shifting should have resulted in PFL deactivation (even in the absence of our overexpressed AdhE). No evidence for inactivation of PFL by wt AdhE is observed, however, providing further support for our conclusion that AdhE is not a PFL deactivating enzyme.
Figure 2.
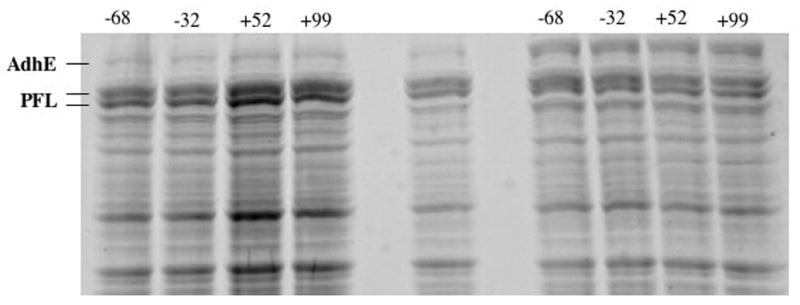
Effect of redox potential shifting on PFL cleavage. The potential of the growth cultures were adjusted to the indicated values by air purge prior to lysis and exposure to air. Lanes 1–4: cultures overexpressing PFL. Lane 5: Cells overexpressing PFL exposed to air after N2 gas purge (without redox potential adjustment). Lane 6–9: PFL cells (at anaerobic potential of −68 mV) were lysed with AdhE cells at the indicated potentials. In all cases complete cleavage of PFL is observed.
Oligomerization Properties of Overexpressed AdhE
Kessler and Knappe were the first to observe the unusual oligomerization properties of AdhE, which forms rod-like oligomeric particles that change conformation, as observed by electron microscopy, upon addition of Fe2+, CoA, and NAD. In order to further address the similarities between our overexpressed AdhE and the wt protein characterized by Kessler and Knappe, we performed electron microscopy on our purified AdhE in the absence and presence of cofactors (data not shown). Similar to the observations of Kessler and Knappe, we see rodlike particles (average length 56 nm and average width of 13 nm) for the protein in MOPS buffer with or without Fe2+. In the presence of NAD and CoA, further aggregation occurs, and the rods change shape (average length 100 nm and average width 13 nm).
Inactivation of PFL by Small Molecule Reductants
An observation made consistently in these deactivation experiments was that reactions that included either Fe2+ or CoA exhibited faster rates of loss of PFL activity than the control reactions, regardless of other components present in the mix (data not shown). These observations suggested that reducing agents such as thiols or reduced metals might be able to slowly quench the glycyl radical of PFL. To investigate this further, we have examined the inactivation of active PFL by small-molecule thiols and other reducing agents. Both β-mercaptoethanol (βME) and dithiothreitol (DTT) were observed to inactivate PFL, with the former doing so at a much faster rate (Figure 3). βME inactivation is quite rapid, with complete loss of activity observed in just over one hour under the conditions of this experiment. DTT inactivation is almost imperceptible over the same time period, but is complete within 7 hours under these conditions. Loss of PFL activity is also accompanied by a loss of the EPR signal due to the PFL glycyl radical (data not shown), demonstrating that these thiols inactivate PFL by destroying the glycyl radical found in active PFL. That these two thiols, commonly used in biological buffers, should inactivate PFL is curious, since both thiols have been used in the past in various stages of PFL purifications and activations reported in the literature. In fact, it has been observed in our laboratory that PFL must be treated with DTT prior to activation in order to achieve maximal activity (unpublished observation). However since DTT inactivation is so slow, it presumably would not have interfered with previous experiments in which PFL activation was carried out for one hour or less.
Figure 3.
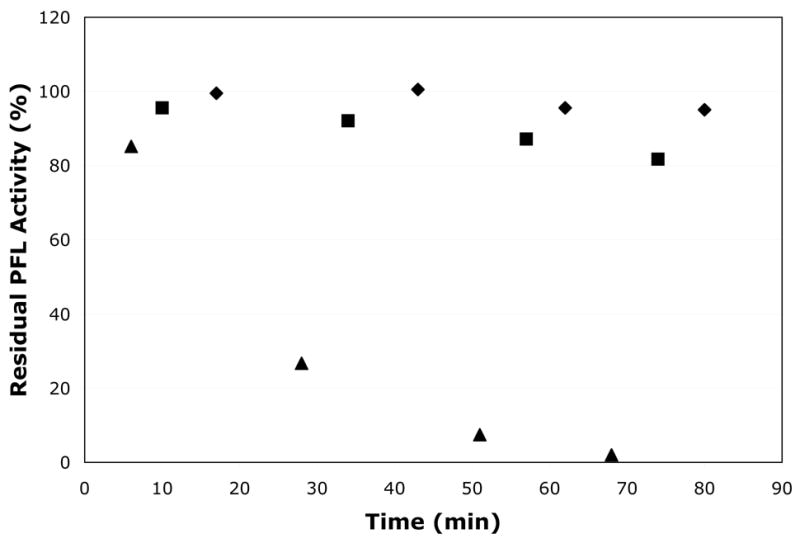
Inactivation of active PFL by DTT and β-ME. Activated PFL (220 μM) was incubated with DTT or β-ME (14 mM each) under anaerobic conditions in the dark, and residual PFL activity was measured periodically. β-ME was found to rapidly inactivate PFL, while DTT inactivated PFL much more slowly. Experiments were carried out in the dark. Data shown is for PFL alone (♦), PFL + DTT (▪), and PFL + βME (▴).
Other thiols, including ethanethiol and cysteine, were also observed to inactivate PFL (Figure 4). Ethanethiol, like βME, inactivates PFL rapidly, while cysteine caused much slower inactivation. In contrast, larger thiols (such as homocysteine and glutathione) or non-thiolic compounds (such as methionine) showed no detectable inactivation. The results suggest that thiols must access the active site glycyl radical directly in order to efficiently promote inactivation, since the smallest thiols provide the most efficient inactivation. Non-thiolic reductants, such as dithionite and ascorbic acid, also inactivate PFL, with dithionite showing a rate similar to that of DTT and ascorbate being significantly slower (data not shown).
Figure 4.
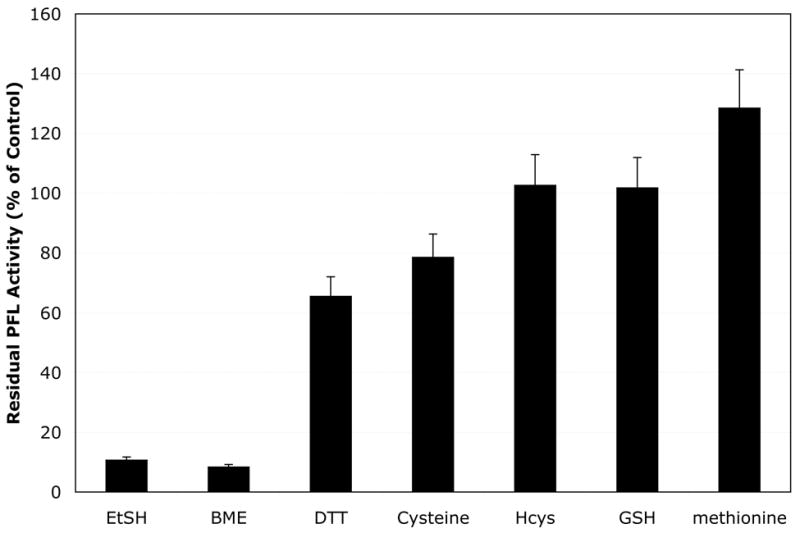
Inactivation of active PFL by thiols. Activated PFL (370 μM) was incubated with the indicated reagents (4 mM) under anaerobic conditions in the dark, and residual PFL activity was monitored after 4 hours.
Reversibility of Inactivation
Observation of a loss of PFL activity over time could be due to deactivation, i.e. conversion of the glycyl radical to a non-radical glycyl residue without damaging the protein, or to inactivation, in which the glycyl radical is quenched in a destructive manner. The former process is reversible and would allow re-generation of the glycyl radical, while the latter process involves irreversible destruction of the protein. The latter process, inactivation, is commonly observed when working with active PFL. For example, exposure of active PFL to even trace quantities of oxygen in vitro results in oxygenolytic cleavage at the site of the glycyl radical. The former process, deactivation, was previously ascribed only to AdhE, and was proposed to be the means by which PFL is conserved in vivo when E. coli cycle between anaerobic and aerobic conditions. However in our hands AdhE does not bring about a loss of PFL activity. Since small molecule thiols and other reductants do bring about a loss of PFL activity, we proceeded to investigate whether the loss of activity mediated by thiols was reversible, i.e. whether these thiols were mediating deactivation.
Active PFL was incubated with β-ME as previously described, and essentially complete loss of activity was observed, while essentially no loss of activity was observed for the control reaction containing no βME (Figure 5). Additional PFL activating enzyme, 5-deazariboflavin, and AdoMet were added to both reactions at this point, and both reactions were exposed to a 300 W halogen lamp for 30 minutes. As can be seen in Figure 5, this treatment had essentially no effect on the control (-βME) reaction, other than a slight (~ 5–10%) increase in PFL activity; this is consistent with the fact that the PFL used in the experiment had been essentially completely activated prior to beginning the experiment. The addition of PFL-AE, 5-deazariboflavin, and AdoMet and exposure to light did, however, have a dramatic effect on the +βME reaction, in which the PFL activity returned to that of the control. Removal of the light source again leads to loss of PFL activity in the +βME reaction, while the activity of the control again remains constant. That full PFL activity was restored to the βME-treated PFL clearly demonstrates that the βME-mediated deactivation is a reversible process that does not destroy G734 or the PFL protein structure. That is, βME inactivates PFL by the simple addition of a hydrogen atom equivalent to the glycyl radical. Similar experiments performed with DTT, ethanethiol, and other thiols showed similar results, in which PFL inactivation by these thiols was completely reversible (data not shown).
Figure 5.
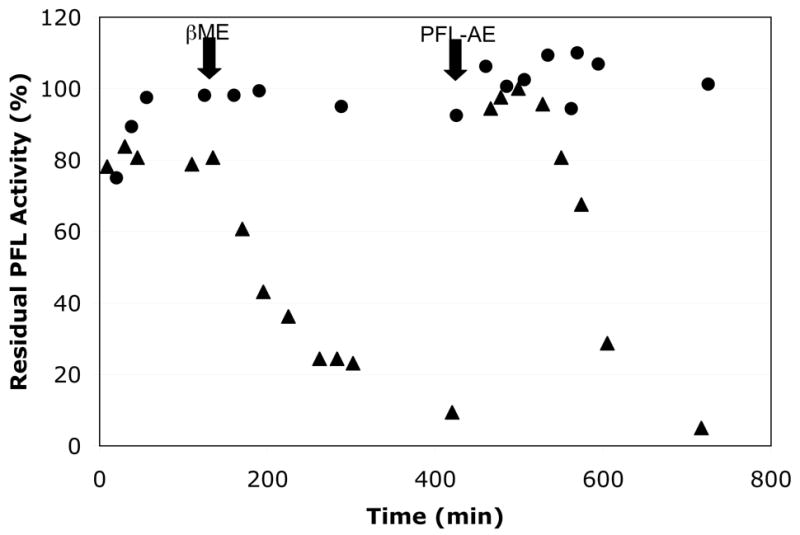
Reversibility of β-ME-mediated inactivation of PFL. Activated PFL (containing 125 μM PFL, 20 μM PFL-AE, 1 mM SAM, 10 μM 5-deazariboflavin, 300 mM TrisCl pH 8.5, 1 mM oxamate) was incubated alone (▪) or with 1.4 mM □-ME (●; □-ME added at 130 min) under anaerobic conditions in the dark. After essentially complete inactivation of the sample containing β-ME, additional PFL-AE (to 40 μM final concentration) and 5-deazariboflavin (to 20 μM final concentration) was added to both samples. The samples were then subjected to illumination with a 300 W halogen lamp for 30 min. This treatment had no effect on the control (-β-ME) reaction, but resulted in complete restoration of full PFL activity in the +β-ME reaction. Subsequent incubation in the dark resulted in re-inactivation of the PFL in the +β-ME reaction.
The nature of PFL inactivation by βME was further investigated by examining the effect of dioxygen on βME-inactivated PFL. Oxygenolytic cleavage of activated PFL can be readily observed by SDS-PAGE (Figure 6). Unactivated PFL migrates as an 85 kDa band on SDS-PAGE. If activated PFL is fully inactivated by exposure to oxygen, the resulting PFL migrates as two observed bands of equal intensity, one at 85 kDa and one at 82 kDa (Figure 6, lane 1). The latter band is the larger fragment resulting from oxygenolytic cleavage at G734; the smaller 3 kDa fragment is not observed. Observation of both 85 and 82 kDa fragments at equal intensities is consistent with the previously reported half-of-sites reactivity for the PFL dimer; one glycyl radical per dimer results in oxygenolytic cleavage of exactly half of the PFL molecules. When activated PFL is deactivated with βME and then exposed to oxygen, no oxygenolytic cleavage is observed (Figure 6, lane 2), consistent with the quenching of the glycyl radical by βME.
Figure 6.
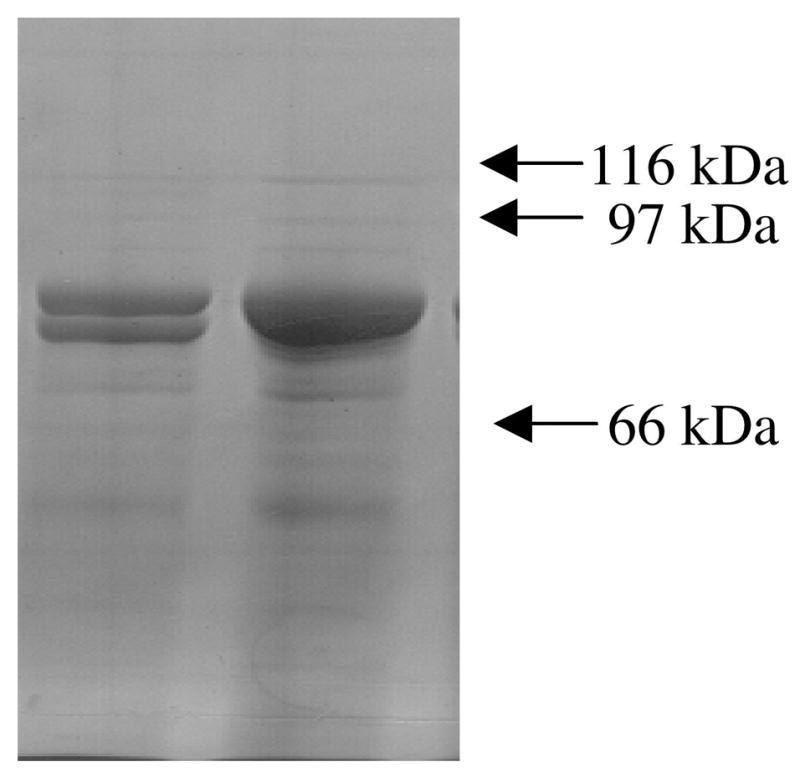
SDS-PAGE gel showing effect of βME on oxygenolytic cleavage of activated PFL. The lanes show (1) activated PFL exposed to oxygen, and (2) □ME-inactivated PFL exposed to oxygen. The amount of PFL loaded in each lane is 12 μg.
PFL Activation using Ambient Light
We observed during these experiments that the rate of inactivation could be perceptibly slowed if the reactions were not protected from ambient light; in fact, if PFL had not been fully activated prior to deactivation experiments, we could observe activation of PFL, particularly certain controls, during the deactivation experiments (Figure 7). We surmised that this slowing of deactivation, or further activation, of PFL was due to a competition between deactivation and re-activation (by the trace PFL-AE, SAM, and the photoreductant 5-deazariboflavin, which was not removed from PFL prior to these inactivation experiments). In order for this to be true, the relatively dim ambient lighting in the anaerobic chamber would have to be sufficient to promote the 5-deazariboflavin-mediated photoreduction of PFL-AE. This possibility was independently tested by incubating inactive PFL with PFL-AE, AdoMet, and 5-deazariboflavin in the ambient lighting in the anaerobic chamber. Although activation was slower than that observed using the intense 300 W halogen lamp as described in Materials and Methods, full activation could be achieved with ambient lighting (data not shown). Activation using ambient lighting was found to be preferable in many instances for these experiments, as it avoided the temperature-control problems associated with intense illumination. In addition, the PFL activity appeared to be more reproducible and more stable when activation was done with ambient lighting.
Figure 7.
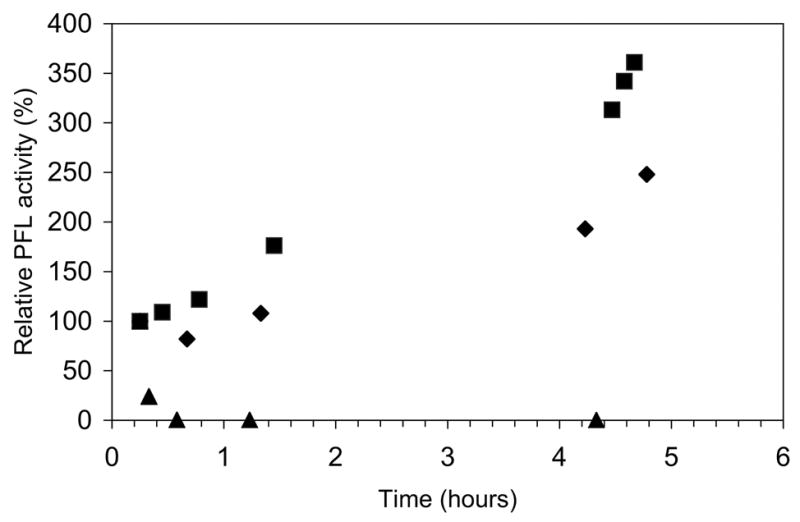
PFL inactivation experiments performed under ambient lighting. Active PFL (98 μM) in 300 mM Tris-Cl pH 8.5 was incubated alone (▪) or with βME (▴) or DTT (♦) (10 mM each) under anaerobic conditions in the ambient lighting in the glove box. Also present in the reactions were PFL-AE (4.5 μM), SAM (0.7 mM), and deazariboflavin (10 μM) left over from the activation of PFL. Under ambient lighting conditions, further activation of PFL by the activation components is competing with deactivation by the thiols.
DISCUSSION
Our laboratory has a long-standing interest in the mechanism of radical generation by which PFL is activated, therefore we were naturally intrigued by the report of an enzyme that catalytically deactivates PFL. Although several other glycyl radical enzymes and their corresponding activating enzymes are known, no potential deactivases (other than AdhE) have been identified. Despite this, the possibility of an enzymatic means by which to deactivate PFL (or other anaerobic glycyl radical enzymes such as the anaerobic ribonucleotide reductase) during cycling between anaerobic and aerobic conditions is compelling, as oxygenolytic cleavage upon transition to aerobic conditions followed by de novo protein synthesis and activation would be energetically costly and inefficient.
The results presented herein, however, demonstrate that AdhE and AdhE-His6 purified from overexpressing cultures do not catalyze the deactivation of active PFL, despite previous reports to the contrary for the wt enzyme [21, 22]. Repeated deactivation experiments in the presence of the purported cofactors Fe(II), CoA, and NAD+ show that AdhE has no effect on the rate of deactivation of PFL, although both Fe(II) and CoA appear to increase the rate of deactivation. Small-molecule thiols were found to have an even more dramatic effect on the rate of loss of PFL activity, with βME causing essentially complete inactivation within one hour. Thiol-dependent inactivation of PFL was shown to be a nondestructive process that allowed regeneration of the active, glycyl radical-containing PFL by incubation with the activation components.
In contrast to the results of Kessler and Knappe, redox shifting of overexpressing cultures or lysates did not have an effect on the amount of activated PFL, as evidenced by SDS-PAGE gels showing fully cleaved PFL in all cases, regardless of the presence of overexpressed AdhE or the potential of the culture or lysate. It should be noted that in the cultures overexpressing PFL, anaerobic incubation of the culture allows the wt PFL-AE present in the host cells to fully activate the overexpressed PFL, such that subsequent aerobic lysis of those cells results in full PFL cleavage. Likewise, it is expected that the wt AdhE present in the host cells would be able to deactivate some of the overexpressed PFL during redox-potential shifting of the PFL cultures, as previously reported by Knappe and Kessler; however no evidence for deactivation is observed by SDS-PAGE (Fig. 2). The overexpressed AdhE, when combined with the PFL at different potentials, also provides no hint of PFL deactivation.
We are unable at this time to resolve the discrepancies between our results and the AdhE-catalyzed deactivation reported by Kessler and Knappe [21, 22]. Our data suggests that we have isolated fully functional AdhE; this is evidenced by the presence of the other two AdhE activities (alcohol dehydrogenase and acetaldehyde-CoA dehydrogenase) and by our ability to reproduce the cofactor-mediated change in oligomeric state of AdhE. Yet in hundreds of experiments in our laboratory, no evidence for AdhE-catalyzed PFL deactivation can be found. Our results are, however, consistent with a recent report on YfiD, a PFL homolog in E. coli that can be activated to generate a glycyl radical by PFL-AE, but cannot be deactivated by AdhE [29].
Acknowledgments
This work was supported by a grant from the NIH (GM54608).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Sawers G, Bock A. Anaerobic regulation of pyruvate formate-lyase from Escherichia coli K12. J Bacteriol. 1988;170:5330–5336. doi: 10.1128/jb.170.11.5330-5336.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sawers G, Bock A. Novel transcriptional control of the pyruvate formate-lyase gene: upstream regulatory sequences and multiple promoters regulate anaerobic expression. J Bacteriol. 1989;171:2485–2498. doi: 10.1128/jb.171.5.2485-2498.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sawers G, Summpann B. Anaerobic induction of pyruvate formate-lyase gene expression is mediated by the ArcA and FNR proteins. J Bacteriol. 1992;174:3474–3478. doi: 10.1128/jb.174.11.3474-3478.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sawers G. The aerobic/anaerobic interface. Curr Op Microbiol. 1999;2:181–187. doi: 10.1016/S1369-5274(99)80032-0. [DOI] [PubMed] [Google Scholar]
- 5.Conradt H, Hohmann-Berger M, Hohmann HP, Blaschkowski HP, Knappe J. Pyruvate Formate-Lyase (Inactive Form) and Pyruvate Formate-Lyase Activating Enzyme of Escherichia coli: Isolation and Structural Properties. Arch Biochem Biophys. 1984;228:133–142. doi: 10.1016/0003-9861(84)90054-7. [DOI] [PubMed] [Google Scholar]
- 6.Sauter M, Sawers RG. Transcriptional analysis of the gene encoding pyruvate formate-lyase-activating enzyme of Escherichia coli. Mol Microbiol. 1990;4:355–363. doi: 10.1111/j.1365-2958.1990.tb00603.x. [DOI] [PubMed] [Google Scholar]
- 7.Knappe J, Blaschkowski HP, Gröbner P, Schmitt T. Pyruvate formate-lyase of Escherichia coli: the acetyl-enzyme intermediate. Eur J Biochem. 1974;50:253–263. doi: 10.1111/j.1432-1033.1974.tb03894.x. [DOI] [PubMed] [Google Scholar]
- 8.Knappe J, Blaschkowski HP. Pyruvate formate-lyase from Escherichia coli and its activation system. Meth Enzymol. 1975;41 doi: 10.1016/s0076-6879(75)41107-7. [DOI] [PubMed] [Google Scholar]
- 9.Wagner AFV, Frey M, Neugebauer FA, Schäfer W, Knappe J. The Free Radical in Pyruvate Formate-Lyase is Located on Glycine-734. Proc Natl Acad Sci USA. 1992;89:996–1000. doi: 10.1073/pnas.89.3.996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Henshaw TF, Cheek J, Broderick JB. The [4Fe-4S]+ cluster of pyruvate formate-lyase activating enzyme generates the glycyl radical on pyruvate formate-lyase: EPR-detected single turnover. J Am Chem Soc. 2000;122:8331–8332. [Google Scholar]
- 11.Sofia HJ, Chen G, Hetzler BG, Reyes-Spindola JF, Miller NE. Radical SAM, a novel protein superfamily linking unresolved steps in familiar biosynthetic pathways with radical mechanisms: functional characterization using new analysis and information visualization methods. Nucleic Acids Res. 2001;29:1097–1106. doi: 10.1093/nar/29.5.1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cheek J, Broderick JB. Adenosylmethionine-dependent iron-sulfur enzymes: versatile clusters in a radical new role. J Biol Inorg Chem. 2001;6:209–226. doi: 10.1007/s007750100210. [DOI] [PubMed] [Google Scholar]
- 13.Fontecave M, Mulliez E, Ollagnier-de-Choudens S. Adenosylmethionine as a source of 5′-deoxyadenosyl radicals. Curr Op Chem Biol. 2001;5:506–511. doi: 10.1016/s1367-5931(00)00237-4. [DOI] [PubMed] [Google Scholar]
- 14.Broderick JB. In: Iron-sulfur clusters in enzyme catalysis, in Comprehensive Coordination Chemistry II. McCleverty JA, Meyer TJ, editors. Elsevier Ltd.; Oxford, UK: 2004. pp. 739–757. [Google Scholar]
- 15.Frey PA, Magnusson OT. S-Adenosylmethionine: A wolf in sheep's clothing, or a rich man's adenosylmethionine? Chem Rev. 2003;103:2129–2148. doi: 10.1021/cr020422m. [DOI] [PubMed] [Google Scholar]
- 16.Ollagnier S, Mulliez E, Schmidt PP, Eliasson R, Gaillard J, Deronzier C, Bergman T, Gräslund A, Reichard P, Fontecave M. Activation of the Anaerobic Ribonucleotide Reductase from Escherichia coli. The essential role of the iron-sulfur center for S-adenosylmethionine reduction. J Biol Chem. 1997;272:24216–24223. doi: 10.1074/jbc.272.39.24216. [DOI] [PubMed] [Google Scholar]
- 17.Tamarit J, Mulliez E, Meier C, Trautwein A, Fontecave M. The anaerobic ribonucleotide reductase from Escherichia coli. The small protein is an activating enzyme containing a [4Fe-4S]2+ center. J Biol Chem. 1999;274:31291–31296. doi: 10.1074/jbc.274.44.31291. [DOI] [PubMed] [Google Scholar]
- 18.Knappe J, Neugebauer F, Blaschkowski AHP, Gänzler M. Post-translational activation introduces a free radical into pyruvate formate-lyase. Proc Natl Acad Sci USA. 1984;81:1332–1335. doi: 10.1073/pnas.81.5.1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mulliez E, Fontecave M, Gaillard J, Reichard P. An iron-sulfur cluster and a free radical in the active anaerobic ribonucleotide reductase of Escherichia coli. J Biol Chem. 1993;268:2296–2299. [PubMed] [Google Scholar]
- 20.Sun X, Ollagnier S, Schmidt PP, Atta M, Mulliez E, Lepape L, Eliasson R, Gräslund A, Fontecave M, Reichard P, Sjöberg BM. The free radical of the anaerobic ribonucleotide reductase from Escherichia coli is at glycine 681. J Biol Chem. 1996;271:6827–6831. [PubMed] [Google Scholar]
- 21.Kessler D, Leibrecht I, Knappe J. Pyruvate-formate-lyase-deactivase and acetyl-CoA reductase activities of Escherichia coli reside on a polymeric protein particle encoded by adhE. FEBS Lett. 1991;281:59–63. doi: 10.1016/0014-5793(91)80358-a. [DOI] [PubMed] [Google Scholar]
- 22.Kessler D, Herth W, Knappe J. Ultrastructure and pyruvate formate-lyase radical quenching property of the multienzymic AdhE protein of Escherichia coli. J Biol Chem. 1992;267:18073–18079. [PubMed] [Google Scholar]
- 23.Janda M, Hemmerich P. Angew Chem Int Ed Engl. 1976;15:7. doi: 10.1002/anie.197604431. [DOI] [PubMed] [Google Scholar]
- 24.Ashton WT, Brown RD, Tolman RL. J Heterocyclic Chem. 1978;15:489–491. [Google Scholar]
- 25.Smit P. Recl Trav Chim Pays-Bays. 1986;105:538–544. [Google Scholar]
- 26.Krebs C, Henshaw TF, Cheek J, Huynh BH, Broderick JB. Conversion of 3Fe-4S to 4Fe-4S clusters in native pyruvate formate lyase activating enzyme: Mössbauer characterization and implications for mechanism. J Am Chem Soc. 2000;122:12497–12506. [Google Scholar]
- 27.Beinert H. Micro Methods for the quantitative determination of iron and copper in biological material. Methods Enzymol. 1978;54:435–445. doi: 10.1016/s0076-6879(78)54027-5. [DOI] [PubMed] [Google Scholar]
- 28.Knappe J, Sawers G. FEMS Microbiol Rev. 1990;75:383–398. doi: 10.1111/j.1574-6968.1990.tb04108.x. [DOI] [PubMed] [Google Scholar]
- 29.Wyborn NR, Messenger SL, Henderson RA, Sawers G, Roberts RE, Attwood MM, Green J. Expression of the Escherichia coli yfiD gene responds to intracellular pH and reduces the accumulation of acidic metabolic end products. Microbiol. 2002;148:1015–1026. doi: 10.1099/00221287-148-4-1015. [DOI] [PubMed] [Google Scholar]